

Abstract
The work described herein aims at finding new potential ligands for the brain imaging of 5-HT4 receptors using single-photon emission computed tomography (SPECT). Starting from the non-substituted phenanthridine compound 4a exhibiting a Ki value of 51 nM on 5-HT4R, we explored structure-affinity in this series. We found that substitution in position 4 of the tricycle with a fluorine atom gave the best result. Introduction of an additional nitrogen atom inside the tricyclic framework led to increase both the affinity and the selectivity for 5-HT4R suggesting the design of the antagonist 4v exhibiting a high affinity of 0.04 nM. Several iodinated analogues were then synthesized as potential SPECT tracers. The iodinated compound 11d was able to displace the reference radioiodinated 5-HT4R antagonist (1-butylpiperidin-4-yl)methyl-8-amino-7-iodo[123I]-2,3-dihydrobenzo[b][1,4]dioxine-5-carboxylate ([123I]1, [123I]SB 207710) both in vitro and in vivo in brain. Compound 11d was radiolabeled with [125I]iodine, providing a potential SPECT candidate for brain imaging of 5-HT4R.
INTRODUCTION
Since its discovery more than two decades ago,1,2 the serotonin 4 receptor subtype (5-HT4R) has emerged as a promising target for drug discovery, development and for medical applications.3,4 Pharmacological investigations coupled to the discovery of ligands exhibiting high affinity and selectivity for 5-HT4Rs have led to know their anatomical distribution and functional roles. 5-HT4Rs are found in the peripheral system, where they are implicated in gastrointestinal disorders5 and heart failures6. Brain 5-HT4Rs are mainly expressed in striatum, globus pallidus, nucleus accumbens and substantia nigra.7 Their distribution in the central nervous system and pharmacological studies using selective agonists and/or antagonists have shown that 5-HT4Rs are implicated in cognition,8 learning and memory processes,9 and more recently in neuropsychiatric disorders such as Alzheimer’s disease,10,11 food intake12 and depression.13 Although efforts have been made by both academics and pharmaceutical companies to develop 5-HT4R ligands with potential medical applications, only peripheral agonists have yet reached the market for gastrointestinal disorders.14 Brain 5-HT4 receptor ligands have entered clinical trials for the treatment of Alzheimer Disease but failed in phase IIb.15,16 Discovery of active 5-HT4Rs agonists and antagonists remains of great interest in clinical research. To this end, molecular imaging techniques using positron emission tomography (PET) or single photon emission computed tomography (SPECT) have emerged as valuable tools, both in clinical studies and drug discovery programs.17,18 These non-invasive techniques have found broad applications including diagnosis, imaging of neurotransmitter receptors, in vivo binding studies of new ligands and establishing treatment strategies. The bottleneck of these techniques remains the limited availability of suitable radioligands.
(1-butylpiperidin-4-yl)methyl-8-amino-7-iodo[123I]-2,3-dihydrobenzo[b][1,4]dioxine-5-carboxylate ([123I]1, [123I]SB 207710)19 and (1-methyl[11C]piperidin-4-yl)methyl-8-amino-7-chloro-2,3-dihydrobenzo[b][1,4]dioxine-5-carboxylate ([11C]2, [11C]SB 207145)20 have been described as potential radiotracers for respectively PET or SPECT imaging (Chart 1). [11C]2 has been successfully used in minipig for the determination of radioligand metabolism and binding kinetics.21 Further studies have shown that this radiotracer can be used for quantitative PET measurements of 5-HT4R in the human brain.22,23 Nevertheless, its short half-life (20.9 min) limits its use in facilities where both a cyclotron and a PET-camera are in proximity. Other analogs of 1 have recently been shown to exhibit pharmacological properties which could make them new promising 5-HT4R PET radiotracers.24 Among them, fluorinated compounds could serve as PET radiotracers of longer half-life. [123I]1 is to date the only SPECT tracer reported for brain imaging of 5-HT4R, but due to low brain penetration and rapid metabolism no further investigations with this product have been reported. We report here work aimed at developing new 5-HT4R radioligands for SPECT imaging. Based on previous works in the lab concerning the synthesis and evaluation of 5-HT4R ligands,25,26 we synthesized and evaluated new diversely substituted phenanthridine derivatives. As a result, we were able to design several selective and high-affinity 5-HT4R antagonists among which some iodinated compounds were identified and successfully radiolabeled with 125I, representing new potential radioligands for SPECT imaging studies of the 5-HT4R (Chart 1).
Chart 1.

Literature and prospective 5-HT4R radiotracers.
RESULTS
Chemistry
The starting (aza)phenanthridinones 3a-v were prepared according to the general route as previously described.27 Compounds 4a-v were obtained using a two-step procedure involving the formation of an imidoyl chloride intermediate in phosphorus oxychloride at 80 °C and the subsequent nucleophilic aromatic substitution of the chlorine atom with (1-propylpiperidin-4-yl)methanol, using conditions based on previous results.25,26 Compounds 4a-v were obtained in 42–91% overall yields except compound 4n which was prepared in 9% overall yield in a three-step procedure28 (Table 1).
Table 1.
Synthesis of (aza)phenanthridines 4a-v.a
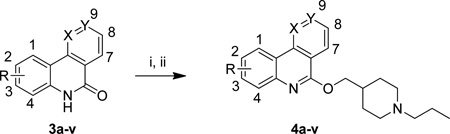 | |||||
|---|---|---|---|---|---|
| Starting Material | R | X | Y | Product | Yield (%)b |
| 3a | H | CH | CH | 4a | 76 |
| 3b | 2-F | CH | CH | 4b | 57 |
| 3c | 2-Cl | CH | CH | 4c | 70 |
| 3d | 2-Me | CH | CH | 4d | 49 |
| 3e | 2-MeO | CH | CH | 4e | 91 |
| 3f | 3-Cl | CH | CH | 4f | 87 |
| 3g | 3-MeO | CH | CH | 4g | 71 |
| 3h | 3-Me | CH | CH | 4h | 59 |
| 3i | 3-F | CH | CH | 4i | 65 |
| 3j | 4-Me | CH | CH | 4j | 47 |
| 3k | 4-MeO | CH | CH | 4k | 63 |
| 3l | 4-Cl | CH | CH | 4l | 62 |
| 3m | 4-F | CH | CH | 4m | 67 |
| 3n | 7-F | CH | CH | 4n | 9c |
| 3o | 8-NO2 | CH | CH | 4o | 57 |
| 3p | 8-MeO | CH | CH | 4p | 70 |
| 3q | 8-F | CH | CH | 4q | 44 |
| 3r | 9-Me | CH | CH | 4r | 71 |
| 3s | 9-F | CH | CH | 4s | 62 |
| 3t | H | CH | N | 4t | 42 |
| 3u | H | N | CH | 4u | 54 |
| 3v | 4-F | N | CH | 4v | 83 |
Reagents and conditions: (i) POCl3, 80 °C, overnight; (ii) (1-propylpiperidin-4-yl)methanol, NaH, DMF, 0 °C to rt, overnight.
Isolated yields.
Yield over 3 steps see experimental data for details.
For the design of potential SPECT ligands, we were interested in the introduction of iodine atoms either on the tricyclic framework or on the lateral chain. Iodinated phenanthridinone 8a and benzonaphthyridinone 8b were obtained in good overall yields starting from 2-trimethylsilylfluorobenzene by a four-step procedure involving: borylation, Suzuki cross-coupling reaction, iododesilylation and anionic ring closure (Scheme 1). For the iodinated side chain, we chose to add a terminal iodoaryl group to the propyl chain.25,26 Thus, 10 was prepared starting from 4-iododihydrocinnamic acid involving amidation with ethyl isonipecotate and reduction of both the carboxamide and ester function with diisobutylaluminium hydride (Scheme 2).
Scheme 1.

Synthesis of 4-iodo(aza)phenanthridin-6(5H)-ones 8a,b.a
aReagents and conditions: (i) s-BuLi 0.75 h, B(OMe)3 0.75 h, THF, −78 °C to rt; (ii) 2-BrPhCN or 2-Cl-3-CN-pyridine, Na2CO3, Pd(OAc)2, PPh3, DME/H2O, 14 h, 90 °C; (iii) ICl, DCM, 4.5 h, rt; (iv) KOH, t-BuOH, sealed tube, 1 h, 150 °C.
Scheme 2.

Synthesis of {1-[3-(4-iodophenyl)propyl]piperidin-4-yl}methanol 10.a
aReagents and conditions: (i) Ethyl isonipecotate, HOBt, EDCI, NEt3, 24 h, rt; (ii) DIBALH, THF, 3 h, −10 °C to rt.
The iodinated compounds 11a-d were obtained using the two-step chlorodehydroxylation/SNAr sequence starting from phenanthridinones 3a and 8a, benzonaphthyridinones 3v and 8b and the appropriate piperidine derivative (Table 2).
Table 2.
Synthesis of iodinated (aza)phenanthridines 11a–d.a
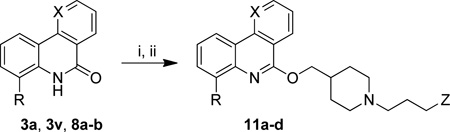 | |||||
|---|---|---|---|---|---|
| Starting Material | R | X | Z | Product | Yield (%)b |
| 8a | I | CH | H | 11a | 76 |
| 8b | I | N | H | 11b | 71 |
| 3a | H | CH | 4-iodophenyl | 11c | 39 |
| 3v | F | N | 4-iodophenyl | 11d | 41 |
Reagents and conditions: (i) POCl3, 80 °C, overnight; (ii) 1-propylpiperidin-4-ylmethanol or 10, NaH, DMF, 0 °C to rt, overnight.
Isolated yields.
Synthesis of radioligands
Stannylated precursors for radioiodination were prepared from iodinated compounds 11a,b,d using palladium-catalyzed tin-iodine exchange in the presence of PPh3 in toluene.29 Radioiodination from the stannylated compounds 12a,b,d was performed using Na125I as the source of radioactive iodine H2O2 (30%) as the oxidant in acidic medium (scheme 3). After HPLC purification, the radioiodinated compounds 13a,b,d were obtained in 56–85% radiochemical yields. The products 13a,b,d were found to be all carrier-free exhibiting apparent specific radioactivities of respectively 240, 110 and 280 Ci / mmol (11%, 5% and 13% of the carrier free specific activity).
Scheme 3.

Preparation of stannylated precursors 12a,b,d and 125I labeling.a
aReagents and conditions: (i) (Bu3Sn)2, Pd(OAc)2, PPh3, PhMe/H2O, 16 h, 90 °C. (ii) Na125I, 30% H2O2, EtOH/AcOH, rt, 20 min.
5-HT4R binding affinity and functional assays
Twenty six compounds (4a-v, 11a-d) were initially screened for their affinity toward 5-HT4R in Guinea Pig striatal membranes at 10−6 and 10−8M. Twenty compounds were selected for Ki determination in 5-HT4R Guinea Pig striatal membranes. These ligands show Ki values between 2.2 and 691 nM. Among them, ten were chosen for human 5-HT4R Ki determination and except 4t, all ligands showed better affinity for h-5-HT4R compared to guinea pig 5-HT4R, exhibiting Ki values between 0.04 and 33 nM (Table 3). All compounds were evaluated for their intrinsic activity and showed either an inverse agonist (4k-m,u) or a full antagonist profile (4v, 11a,b and 11d) as shown in Table 4.
Table 3.
Binding affinities of new 5-HT4R ligands.
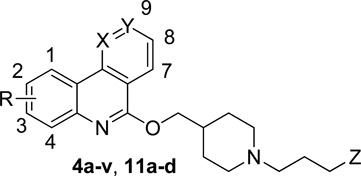 | |||||||
|---|---|---|---|---|---|---|---|
| compd | R | X | Y | Z | % inh (10−6M/10−8M)a |
5-HT4 Ki (nM)b |
h5-HT4 Ki (nM)c |
| 4a | H | CH | CH | H | 100/11 | 51.5 | n.m.d |
| 4b | 2-F | CH | CH | H | 47/0 | n.m. | n.m. |
| 4c | 2-Cl | CH | CH | H | 27/0 | n.m. | n.m. |
| 4d | 2-Me | CH | CH | H | 88/7 | 246 | n.m. |
| 4e | 2-MeO | CH | CH | H | 83/9 | 233 | n.m. |
| 4f | 3-Cl | CH | CH | H | 85/0 | n.m. | n.m. |
| 4g | 3-MeO | CH | CH | H | n.m. | 1900 | n.m. |
| 4h | 3-Me | CH | CH | H | 65/30 | 691 | n.m. |
| 4i | 3-F | CH | CH | H | 100/14 | 101 | 33.0e |
| 4j | 4-Me | CH | CH | H | 100/0 | 100 | n.m. |
| 4k | 4-MeO | CH | CH | H | 100/22 | 35.0 | 17.0e |
| 4l | 4-Cl | CH | CH | H | 100/24 | 21.9 | 5.0e |
| 4m | 4-F | CH | CH | H | 100/63 | 20.1 | 3.1e |
| 4n | 7-F | CH | CH | H | 100/0 | 21.6 | n.m. |
| 4o | 8-NO2 | CH | CH | H | 54/0 | n.m. | n.m. |
| 4p | 8-MeO | CH | CH | H | 85/0 | n.m. | n.m. |
| 4q | 8-F | CH | CH | H | 90/4 | 154 | n.m. |
| 4r | 9-Me | CH | CH | H | 28/0 | n.m. | n.m. |
| 4s | 9-F | CH | CH | H | 84/0 | 209 | n.m. |
| 4t | H | CH | N | H | 100/50 | 22.8 | 403e |
| 4u | H | N | CH | H | 96/64 | 13.1 | 7.5e |
| 4v | 4-F | N | CH | H | 100/98 | 2.2 | 0.04f |
| 11a | 4-I | CH | CH | H | 100/0 | 13.5 | 1.20f |
| 11b | 4-I | N | CH | H | 100/20 | 4.6 | 0.26f |
| 11c | H | CH | CH | 4-iodophenyl | 100/3 | 115 | n.m. |
| 11d | 4-F | N | CH | 4-iodophenyl | 100/94 | 2.5 | 0.23f |
Inhibition percentages were determined using guinea pig striatal membrane 5-HT4R.
Guinea pig striatal membrane 5-HT4R (n=3).
Human 5-HT4R (n=3).
n.m. = not measured.
Ki determinations performed at NIMH-PDSP – USA.
Ki determinations performed at CEREP- France. See experimental section for details.
Table 4.
Intrinsic activity and cLogD of 5-HT4R ligands 4k, 4l, 4m, 4u, 4v, 11a, 11b and 11d.
| 4k | 4l | 4m | 4u | 4v | 11a | 11b | 11d | |
|---|---|---|---|---|---|---|---|---|
| h5-HT4 | 17 | 5.0 | 3.1 | 7.5 | 0.04 | 1.20 | 0.26 | 0.23 |
| Ki(nM) efficacy |
Inv Aga | Inv Aga | Inv Aga | Inv Aga | Antagb | Antagb | Antagb | Antagb |
| EC50 (nM) | 79.4 | 251 | 63.1 | 10 | - | - | - | - |
| KB (nM) | - | - | - | - | 0.025 | 5.0 | 0.5 | 6.3 |
| cLogDc | 2.48 | 3.25 | 2.79 | 1.84 | 1.98 | 3.58 | 2.77 | 4.67 |
Functional assays performed at NIMH-PDSP – USA, Inv Ag = inverse agonist.
Functional assays performed at CEREP – France, Antag = Antagonist. See experimental section for details.
Calculated LogD (pH = 7.4) using MarvinSketch 5.2.6.
5-HTRs binding profile
Compounds 4k, 4l, 4m, 4u, 4v, 11a, 11b and 11d with the highest affinities for 5-HT4R were screened towards other 5-HTR subtypes. Results are shown in Table 5.
Table 5.
Binding affinities of compounds 4k, 4l, 4m, 4u, 4v, 11a, 11b and 11d toward 5-HT receptors
| 5-HTR | 4k | 4l | 4m | 4u | 4v | 11a | 11b | 11d |
|---|---|---|---|---|---|---|---|---|
| h5-HT4 | 17 | 5.0 | 3.1 | 7.5 | 0.04 | 1.20 | 0.26 | 0.23 |
| h5-HT1A | > 104 | 8836 | > 104 | > 104 | 4987 | > 104 | 5629 | 680 |
| h5-HT1B | > 104 | > 104 | > 104 | > 104 | > 104 | > 104 | > 104 | > 104 |
| h5-HT1D | 2569 | 2360 | > 104 | > 104 | > 104 | > 104 | > 104 | > 104 |
| h5-HT1E | > 104 | > 104 | > 104 | > 104 | > 104 | > 104 | > 104 | n.m. |
| h5-HT2A | 5962 | 1780 | > 104 | > 104 | > 104 | 1019 | 1825 | 810 |
| h5-HT2B | 29.3 | 98.0 | 40.4 | 627 | 136 | 162 | > 104 | 340 |
| h5-HT2C | > 104 | 729 | 1049 | 2793 | 492 | 772 | 341 | 400 |
| h5-HT3 | > 104 | 2470 | 710 | 680 | 641 | 1314 | > 104 | 410 |
| h5-HT5A | > 104 | > 104 | > 104 | > 104 | > 104 | 5805 | > 104 | 1700 |
| h5-HT6 | > 104 | 6256 | > 104 | > 104 | > 104 | > 104 | > 104 | > 104 |
| h5-HT7 | 3473 | 1914 | 384 | > 104 | 1945 | 153 | 1018 | 150 |
Selectivity was performed at NIMH-PDSP – USA except for compound 11d for which the selectivity was performed at CEREP – France.
Discussion
Unlike PET imaging using 18F for which both a suitable fluorinated ligand and a precursor for radiofluorination have to be designed, for SPECT imaging an iodinated ligand can serve as both the non radioactive ligand for pharmacological studies and the precursor for the introduction of 123I via successive stannylation-iododestannylation reactions. A major challenge with SPECT imaging is to find a suitable iodinated ligand exhibiting high affinity and receptor selectivity. Whereas a hydrogen atom often can be replaced by a fluorine atom without significant decrease in biological activities, introduction of an iodine atom can lead to dramatic decrease in affinity due to its large atomic radius.
Starting from the unsubstituted phenanthridine 4a which exhibited good 5-HT4R binding affinity (Ki = 51 nM), we explored the influence of substitution of the tricyclic ring system in this series to find the best positions for the introduction of substituents, including an iodine atom. The chemical routes we have developed allowed the introduction of substituents in position 2, 3, 4, 7, 8, and 9 (table 3). The results show that substitution of the phenanthridine in position 2, 3, 8, 9 was detrimental for affinity compared to unsubstituted compound 4a, all compounds show Ki values above 100 nM, even with the small fluorine group. Better results were obtained when substituents were placed in position 4 and 7. The 7-fluoro compound 4n, showed a slight increase in affinity (Ki = 22 nM) whereas substituents in position 4 led to significant improvements in affinity (compounds 4k-m), except the 4-methyl substituted compound 4j. Interestingly, while compounds 4k-m exhibited 35, 22 and 20 nM Ki values respectively for guinea pig 5-HT4R, against human 5-HT4R they showed significant higher binding affinities with Ki values of respectively 17, 5 and 3 nM. Introduction of a nitrogen atom in position 9 or 10 led to benzonaphthyridines 4t and 4u, which exhibited increased activity compared to 4a with Ki 23 and 13 nM, respectively. When fluorine was introduced in position 4 to give benzonaphthyridine 4v, the affinity was significantly increased to 2.2 nM on guinea pig 5-HT4R and remarkably this compound exhibited a very high affinity on human 5-HT4R with a Ki value of 0.04 nM (pKi = 10.4, pKB=10.6). This compound represents one of the most active 5-HT4R antagonist reported to date, being in the same range as the reference antagonists [1-(2-methanesulfonamidoethyl)piperidin-4-yl]methyl 1-methylindole-3-carboxylate (GR 113808)30 or 1.31
With these structure-affinity results in hand, we investigated the design of iodinated compounds for potential development of SPECT radiotracers. As the position 4 seemed to give the best results in both the phenanthridine and benzonaphthyridine series, we chose to introduce an iodine atom in this position. Iodinated compounds 11a and 11b were synthesized and evaluated for their 5-HT4R binding affinity. Despite differences between fluorine and iodine in terms of size and electronic properties, 11a and 11b exhibited Ki values of 13 and 4.6 nM for guinea pig 5-HT4R and respectively, 1.2 and 0.26 nM for human 5-HT4R in the same range as their fluorinated analogues 4m and 4v. Further, we investigated compounds iodinated on the N-propyl group. In order to avoid the expected elimination of the iodine atom if bonded to a saturated carbon, it was not directly attached to the N-propyl chain but to an aromatic group using the N-3-(4-iodophenyl)propyl chain. Previous results have shown that some 5-HT4R ligands bearing bulky aromatic group bonded to the lateral chain can exhibit high affinities.3 Accordingly, compounds 11C and 11d were synthesized. Despite the introduction of the bulky iodoaryl group, the iodinated 4-fluorobenzonaphthyridine 11d was found to exhibit high affinity for both guinea pig 5-HT4R (Ki = 2.5 nM) and human 5-HT4R (Ki = 0.23 nM) in the similar order of magnitude as compound 4v. Along with high binding affinities, intrinsic activity and binding selectivity are other key parameters for the development of suitable ligands for imaging. The best 5-HT4R ligands 4k-m, 4u-v, 11a-b and 11d were evaluated for both intrinsic activity and selectivity toward other 5-HTRs (Table 4 and 5). Compounds 4k-m and 4u appeared to be inverse agonists with pEC50 ranging from 6.6 to 8.0 whereas compounds 4v, 11a-b, and 11d were found to be full antagonists with pKB values ranging from 8.2 to 10.6 in accordance with their Ki values. The difference observed in intrinsic activity could be due to high constitutive activity of the cloned receptors used in the functional tests. Some ligands have been found to behave either as antagonists or as inverse agonists depending on the functional models used.32 These compounds were then evaluated for their selectivity toward other 5-HT receptors. All compounds showed high selectivity toward other 5-HT receptors except the 5-HT2BR for which compounds 4k-m exhibited almost the same potency as for 5-HT4R. Interestingly, the additional nitrogen atom in position 10 not only led to increased affinities for 5-HT4R but also contributes to decreased affinity for 5-HT2BR, resulting in compounds with higher selectivity (>100). In order to try to rationalize this result we performed docking studies using a model of 5-HT4R based on the crystal structures of compounds 4m, 4v and 11d assuming that the crystalline conformations are the most energetically stable (see supporting information). The homology model of the human 5-HT4 receptor was constructed using the β2-adrenergic receptor crystal structure, one of the available GPCR structures that exhibit a sequence identity of about 40% with the 5-HT4R.33 The two crystallographic conformers of compound 4m were docked into the 5-HT4R model (see supporting information). In the selected pose (Figure 1A, mean Chem Score Fit = 31.18), the basic piperidine nitrogen interacted with Asp100(consistent with the constraint used during the docking) and an additional polar interaction could form through this basic nitrogen and the Tyr302 hydroxyl group.34 The tricyclic framework was oriented toward the transmembrane helix 5 (TM5, colored in yellow in Figure 1), in a position analogous to that observed for beta adrenergic receptor ligands in solved X-ray structures.33,35 In the 4m docking position, the tricycle is oriented parallel to the TM5 helix axis and is surrounded by several aromatic residues (Phe186, Tyr192, Phe275, Phe276), oriented approximately perpendicularly to the tricyclic framework, indicating possible π stacking interactions. The fluorine atom pointed inside the receptor. The addition of a supplementary nitrogen atom in derivative 4v could lead to a new hydrogen bond with Ser197 hydroxyl group as seen in Figure 1B (mean ChemScore Fit = 33.10). While this additional interaction could explain the increased affinity for the 5-HT4R of 4v versus 4m, it does not explain its lower affinity for the 5-HT2BR. Indeed, a careful comparison of these receptors sequences showed that Ser197 is present in both the 5-HT4 and 5-HT2B receptors; therefore it is not probable that the additional nitrogen interacts with this serine. Comparison of amino acid sequences in the TM5 part of the binding cavity shows three differences: Tyr192(5-HT4R)/Phe217(5-HT2BR), Cys196(5-HT4R)/Gly221(5-HT2BR) and Ala193(5-HT4R)/Met218(5-HT2BR). We reasoned that the supplementary nitrogen atom in 4v could interact with the hydroxyl group of the 5-HT4R Tyr192. Therefore, the docking studies of 4v were carried out by taking into account a supplementary hydrogen bonding constraint between this additional nitrogen atom and the Tyr192 hydroxyl group. In the best scoring docking pose of this study (mean ChemScore Fit = 41.55), the tricycle was oriented perpendicularly to the TM5 helix axis (Figure 1C), the N-1 nitrogen atom formed the H-bond with Tyr192 as set during the docking and was located far from the Ser197. Furthermore, in this docking pose, the fluorine, the N-6 nitrogen atom and the oxygen atom were placed near to the 5-HT4R Trp294 (TM7), so that electrostatic interactions can occur with this amino acid. Binding interactions with this Trp294 has been previously shown to be essential for some 5-HT4R ligands.36 The docking of compound 11d showed that it can be positioned in the same manner as 4v (mean ChemScore Fit = 33.54), the 4-iodophenyl group going up toward the extracellular entrance (Figure 1D), as it was observed for some co-crystallized extended ligands such as the full agonist carmoterol in β1-adrenergic receptor37 or for the antagonist JDTic in κ-opioid receptor.38
Figure 1.

Docking poses of compounds 4m (A), 4v (B and C) and 11d (D).
Among the iodinated compounds, 11a, 11b and 11d could be considered as promising candidates for the development of a SPECT tracer owing to their subnanomolar binding affinities, high selectivity over other 5-HTRs including 5-HT2BR and computed lipophilicities within a range adequate for brain penetration.39 Compound 11d, which exhibits the highest Ki value and selectivity was chosen for further evaluation. In order to address its 5-HT4R specific binding capacity, in vitro competition experiments with the selective and specific antagonist radioligand [125I]1 were performed. Increasing concentrations of 11d co-administered with [125I]1 show a decrease in the 5-HT4R-specific radioactivity at 10 pM of 11d while increasing the concentration to 1 nM led to the almost complete abolishment of the signal (Figure 2). The same experiment was performed ex vivo. The specific binding of [125I]1 is slightly noticeable in the olfactory tubercles but is markedly reduced compared to a reference experiment (Figure 3). The activity between the hemispheres in the co-injection experiment is due to the presence of blood in the brain sections. Taken together, these two experiments show that: 1) 11d is able to compete with the specific radioligand [125I]1 both in vitro and in vivo at very low concentration; 2) 11d is able to cross the blood brain barrier, making 11d a suitable candidate for use in SPECT imaging studies.
Figure 2.

Autoradiograms obtained by incubation of sections with 100 pM of [125I]1 in the presence of growing concentrations of 11d (1, 10, 100 and 1000 pM from left to right).
Figure 3.

Ex vivo autoradiogram after intravenous injection of [125I]1 alone (left) and after co-injection with 50 µg/kg of 11d to a mouse (right).
The radioiodinated compound 13d was successfully prepared from 11d by a two-step sequence involving stannylation and Sn-125I exchange (Scheme 3). 13d was obtained in a 85% radiochemical yield and the specific radioactivity was measured at 280 Ci/mmol (13% of the carrier free specific activity). The two other iodinated compounds 11a and 11b were radioiodinated following the same strategy affording 13a and 13b in respectively 56 and 70% RCY and with a specific activity of respectively 240 and 110 Ci/mmol (11 and 5% of the carrier free specific activity).
CONCLUSION
Our studies aimed at the development of new potential radiotracers for SPECT imaging of brain 5-HT4 receptors. Starting from a phenanthridine scaffold we designed new antagonists exhibiting high affinity and selectivity toward this receptor. The fluorinated compounds 4v has shown the best profile with a low subnanomolar Ki value, and a high selectivity toward other 5-HT receptor subtypes. Compared to the phenanthridine analog 4m, the additional nitrogen atom in compound 4v led to a significant improvement of both affinity and selectivity for the 5-HT4R. Having established the structure-affinity relationships for this series, we have successfully introduced iodine atoms without negatively affecting either affinities or selectivities. Among these iodinated compounds, 11d was chosen for further evaluation as a potential radiotracer. This compound was able to displace a specific 5-HT4R ligand both in vitro and in vivo at subnanomolar concentrations, thereby demonstrating receptor specificity and capacity to cross the blood brain barrier. Three iodinated compounds were successfully radiolabelled with [125I] and owing to their favourable pharmacological properties represent novel candidates for further evaluation as SPECT radiotracers.
EXPERIMENTAL SECTION
All chemical reagents and solvents were purchased from commercial sources and used without further purification except THF which was distilled from Na/benzophenone. Thin-layer chromatography (TLC) was performed on silica gel plates. Silica gel 0.06–0.2 mm, 60 Å was used for all column chromatography. Melting points were determined on a Kofler melting point apparatus. IR spectra were recorded as neat films on a Nicolet 380 FT-IR or on KBr discs using a PerkinElmer BX-FT-IR. 1H and 13C NMR spectra were recorded on a Jeol Lambda 400 spectrometer with chemical shifts expressed in parts per million (in DMSO-d6 or CDCl3). High Resolution Mass Spectra (EI) were performed on a Jeol GC-Mate Spectrometer. High Resolution Mass Spectra (ESI) were performed on a Bruker APEX III FT-ICR-MS system. Elemental Analyses were performed at the “Institut de Recherche en Chimie Organique Fine” (Rouen, France). The purities of all tested compounds were analyzed by LC-MS, with the purity all being higher than 95%. Analyses were performed using a Waters alliance 2695 using the following gradient: A (95%)/B (5%) to A (5%)/B(95%) in 10 min. This ratio held for 3 min before return to initial conditions in 1 min. Initial conditions were then maintained for 5 min (A: H2O, B: MeCN; each containing HCOOH: 0.1%; Column: C18 Xterra MSC118/2.1_50 mm). MS detection was performed with a Micromass ZMD 2000. Suitable crystals of solved structures were obtained by slow evaporation from MeCN solution. Data for crystal structures analysis were collected at 296 K with a Bruker–Nonius Kappa CCD area detector diffractometer with graphite–monochromatized Mo Kλ radiation (λ=0.71073 Å). The structures were solved using direct methods and refined by full-matrix least-squares analysis on F2. Program(s) used to solve structures: SHELXS–97 (Sheldrick). Program(s) used to refine structures: SHELXL–97. Software used to prepare material for publication: SHELXL–97 (Sheldrick). Crystallographic data for compounds 4c, 4d, 4g, 4h, 4j, 4m, 4p, 4q, 4r, 4v and 11d have been deposited at the Cambridge Crystallographic Data Centre, CCDC No 889427, 889429-889437 and 889548. Copies of this information may be obtained free of charge from the Director, CCDC, 12 Union Road, Cambridge, CB2 1EZ, UK (+44-1223-336408; E-mail: deposit@ccdc.cam.ac.uk or http://www.ccdc.cam.ac.uk).
7-Fluorobenzo[h]-1,6-naphthyridin-5(6H)-one 3v
In a sealed tube were introduced, KOH (1.94 g, 34.6 mmol), 2-(2,3-difluorophenyl)nicotinonitrile40 (1.5 g, 6.9 mmol) and t-BuOH (40 mL). The tube was heated to 150 °C for 0.5 h. Water (30 mL) was added and the obtained precipitate was filtered. The solid was then dried under vacuum to afford 3v (1.3 g) as a white powder. Yield: 87%. Mp > 260 °C. IR (KBr) ν (cm−1) 3022, 1675 (CO), 1587, 1419, 763. 1H NMR (400 MHz, DMSO-d6) δ 7.28 (m, 1H), 7.49 (m, 1H), 7.70 (dd, 1H, 3J = 7.8 Hz, 3J = 3.9 Hz), 8.43 (d, 1H, 3J = 7.8 Hz), 8.68 (dd, 1H, 3J = 7.8 Hz, 4J = 1.9 Hz), 9.07 (d, 1H, 3J = 3.9 Hz), 11.87 (brs, 1H). 13C NMR (100 MHz, DMSO-d6) δ 116.5 (d, 2J = 17 Hz), 119.7 (d, J = 3 Hz), 121.0 (d, J = 2 Hz), 121.6, 122.2 (d, J = 7 Hz), 123.9, 126.5 (d, 2J = 14 Hz), 135.9, 149.4 (d, 1J = 243 Hz), 149.9 (d, J = 3 Hz), 154.3, 160.7. HRMS (EI) calcd. for C12H7FN2O 214.0542, found 214.0548.
General procedure A for the synthesis of compounds 4a-v, and 11a-d
The chosen (aza)phenanthridin-6(5H)-one (3a-v, 8a-b) and POCl3 (5 mL.mmol−1) were heated to 90 °C overnight in a round bottom flask. After cooling, the mixture was poured carefully on cold water and crushed ice. The pH was carefully adjusted to 12 using a 28% ammonia solution. The product was extracted using EtOAc (3 times). The organic phase was dried with MgSO4, filtered and evaporated. The crude material was added at 0 °C to a solution of either (1-propylpiperidin-4-yl)methanol or {1-[3-(4-iodophenyl)propyl]piperidin-4-yl}methanol 10 (1 equiv.) and NaH (4 equiv.) in anhydrous DMF (10 mL.mmol−1). The solution was allowed to reach room temperature, stirred overnight, hydrolyzed with water, and extracted with AcOEt (3 times). The combined organic phases were washed with water (3 times), dried over MgSO4, filtered, evaporated and purified by silica gel chromatography.
6-(1-Propylpiperidin-4-yl)methyloxyphenanthridine 4a
Starting from 3a (213 mg, 1.1 mmol) using general procedure A and cyclohexane/ethyl acetate 8/2 with 5% of NEt3 as the eluent for the chromatography, 4a was obtained as a white powder (277 mg). Yield: 76%. Mp = 74–76 °C. IR (KBr) ν (cm−1) 2924, 1590, 1461, 1343, 1319. 1H NMR (400 MHz, CDCl3) δ 0.91 (t, 3H, 3J = 7.6 Hz), 1.50-1.62 (m, 4H), 1.91-2.03 (m, 5H), 2.30-2.34 (m, 2H), 3.00-3.03 (m,2H), 4.50 (d, 2H, 3J = 6.0 Hz), 7.47 (ddd, 1H, 3J = 8.0 Hz, 3J = 6.8 Hz, 4J = 1.2 Hz), 7.59-7.64 (m, 2H), 7.80 (ddd, 1H, 3J = 8.4 Hz, 3J = 7.2 Hz, 4J = 1.6 Hz), 7.86 (dd, 1H, 3J = 8.4 Hz, 4J = 1.6 Hz), 8.38 (dd, 1H, 3J = 8.0 Hz, 4J = 1.2 Hz), 8.40-8.42 (m, 1H), 8.48-8.51 (m, 1H). 13C NMR (100 MHz, CDCl3) δ 12.3, 20.4, 29.5 (2C), 36.1, 53.8 (2C), 61.4, 70.6, 120.4, 122.0, 122.2, 122.6, 124.4, 125.2, 127.3, 127.9, 128.9, 130.9, 134.9, 143.5, 159.1. LC-MS (ESI): tR = 5.02 min; [M+H]+: 335.58. HRMS (EI) calcd. for C22H26N2O 334.2046, found 334.2046.
2-Fluoro-6-(1-propylpiperidin-4-yl)methyloxyphenanthridine 4b
Starting from 3b (180 mg, 0.84 mmol), using general procedure A and cyclohexane/ethyl acetate 7/3 with 5% of NEt3 as the eluent for the chromatography, 4b was obtained as a white powder (169 mg). Yield: 57%. Mp = 115-116 °C. IR (KBr) ν (cm−1) 3076, 2927, 2764, 1618, 1591, 1495, 1453, 1436, 1348, 1315, 1243. 1H NMR (400 MHz, CDCl3) δ0.91 (t, 3H), 1.53-1.59 (m, 4H), 1.90-2.02 (m, 5H), 2.29-2.33 (m, 2H), 2.99-3.02 (m, 2H), 4.45 (d, 2H, 3J = 6.1 Hz), 7.33 (ddd, 1H, 3J = 8.8 Hz, 3J = 8.0 Hz, 4J = 2.8 Hz), 7.64 (ddd, 1H, 3J = 8.0 Hz, 3J = 6.8 Hz, 4J = 1.2 Hz), 7.77 (ddd, 1H, 3J = 8.0 Hz, 3J = 7.2 Hz, 4J = 1.6 Hz), 7.81 (dd, 1H, 3J = 8.8 Hz, 3J = 5.2 Hz), 7.98 (dd, 1H, 3J = 10.0 Hz, 4J = 2.8 Hz), 8.33 (d, 1H, 3J = 8.4 Hz), 8.36 (dd, 1H, 3J = 8.0 Hz, 4J = 1.2 Hz). 13C NMR (100 MHz, CDCl3) δ 12.1, 20.2, 29.3 (2C), 35.9, 53.6 (2C), 61.2, 70.5, 107.1 (d, 2J = 23 Hz), 116.9 (d, 2J = 24 Hz), 120.2, 121.9, 123.3 (d, J = 9 Hz), 125.1, 127.7, 129.4 (d, J = 5 Hz), 130.8, 134.0 (d, J = 4Hz), 139.8, 158.3 (d, J = 2Hz), 159.7 (d, 1J = 241 Hz). LC-MS (ESI): tR = 5.23 min; [M+H]+: 353.34. HRMS (EI) calcd. for C22H25FN2O 352.1950, found 352.1938.
2-Chloro-6-(1-propylpiperidin-4-yl)methyloxyphenanthridine 4c
Starting from 3c (670 mg, 2.9 mmol), using general procedure A and cyclohexane/ethyl acetate 8/2 with 5% of NEt3 as the eluent for the chromatography, 4c was obtained as a white powder (753 mg). Yield: 70%. Mp = 110-111 °C. IR (KBr) ν (cm−1) 2930, 1589, 1345, 1317, 1096, 820. 1H NMR (400 MHz, CDCl3) δ 0.92 (t, 3H, 3J = 6.8 Hz), 1.50-1.61 (m, 4H), 1.91-2.02 (m, 5H), 2.30-2.34 (m, 2H), 3.00-3.03 (m, 2H), 4.47 (d, 2H, 3J = 5.9 Hz), 7.54 (dd, 1H, 3J = 8.8 Hz, 4J = 1.9 Hz), 7.65 (t, 1H, 3J = 6.8 Hz), 7.76-7.81 (m, 2H), 8.33 (d, 1H, 4J = 2.0 Hz), 8.35-8.39 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 12.1, 20.2, 29.3 (2C), 35.9, 53.6 (2C), 61.2, 70.6, 120.3, 121.8, 121.9, 123.5, 125.1, 127.8, 129.0, 129.1, 129.8, 131.0, 133.7, 141.8, 159.1. LC-MS (ESI): tR = 5.58 min; [M+H]+: 369.26, 371.26. HRMS (EI) calcd. for C22H25ClN2O 368.1655, found 368.1659.
2-Methyl-6-(1-propylpiperidin-4-yl)methyloxyphenanthridine 4d
Starting from 3d (400 mg, 1.9 mmol), using general procedure A and cyclohexane/ethyl acetate 8/2 with 5% of NEt3 as the eluent for the chromatography, 4d was obtained as a white powder (326 mg). Yield: 49%. Mp = 87-89 °C. IR (KBr) ν (cm−1) 2951, 2928, 2798, 2761, 1589, 1344, 1317, 1302, 1148, 1088, 820, 772. 1H NMR (400 MHz, CDCl3) δ 0.91 (t, 3H, 3J = 7.8 Hz), 1.52-1.60 (m, 4H), 1.92-2.02 (m, 5H), 2.30-2.34 (m, 2H), 2.56 (s, 3H), 3.00-3.03 (m, 2H), 4.48 (d, 2H, 3J = 5.9 Hz), 7.43 (d, 1H, 3J = 7.8 Hz), 7.61 (t, 1H, 3J = 6.8 Hz), 7.75-7.80 (m, 2H), 8.20 (s, 1H), 8.36 (d, 1H, 3J = 7.8 Hz), 8.48 (d, 1H, 3J = 7.8 Hz). 13C NMR (100 MHz, CDCl3) δ 12.1, 20.2, 21.7, 29.3 (2C), 36.0, 53.7 (2C), 61.3, 70.4, 120.2, 121.8, 121.9, 122.2, 125.0, 127.0, 127.5, 130.3, 130.6, 133.8, 134.6, 141.4, 158.4. LC-MS (ESI): tR = 5.53 min; [M+H]+: 349.35. Anal. Calcd. for C22H26N2O: C, 79.01; H, 7.84; N, 8.38. found: C, 79.27; H, 7.91; N, 8.01.
2-Methoxy-6-(1-propylpiperidin-4-yl)methyloxyphenanthridine 4e
Starting from 3e (300 mg, 1.33 mmol), using general procedure A and cyclohexane/ethyl acetate 9/1 with 5% of NEt3 as the eluent for the chromatography, 4e was obtained as a colorless oil (440 mg). Yield: 91%. IR (KBr) ν (cm−1) 2935, 1620, 1591, 1498, 1345, 1316, 1243. 1H NMR (400 MHz, DMSO-d6) δ 0.84 (t, 3H, 3J = 7.8 Hz), 1.38-1.45 (m, 4H), 1.81-1.91 (m, 5H), 2.19-2.22 (m, 2H), 2.86-2.89 (m, 2H), 3.94 (s, 3H), 4.36 (d, 2H, 3J = 5.9 Hz), 7.27 (dd, 1H, 3J = 8.8 Hz, 4J = 2.9 Hz), 7.70-7.76 (m, 2H), 7.90 (t, 1H, 3J = 6.8 Hz), 8.04 (d, 1H, 4J = 2.9 Hz), 8.28 (d, 1H, 3J = 7.8 Hz), 8.76 (d, 1H, 3J = 8.8 Hz). 13C NMR (100 MHz, DMSO-d6) δ 11.9, 19.7, 28.8 (2C), 35.5, 53.1 (2C), 55.6, 60.3, 70.0, 104.3, 118.3, 119.2, 122.8, 122.9, 124.3, 127.9, 128.6, 131.1, 133.9, 137.1, 156.5, 156.7. LC-MS (ESI): tR = 5.31 min; [M+H]+: 365.38. HRMS/ESI calcd. for C23H29N2O2 [M+H]+ 365.2229, found 365.2211.
3-Chloro-6-(1-propylpiperidin-4-yl)methyloxyphenanthridine 4f
Starting from 3f (124 mg, 0.54 mmol), using general procedure A and cyclohexane/ethyl acetate 95/5 with 5% of NEt3 as the eluent for the chromatography, 4f was obtained as a white powder (173 mg ). Yield: 87%. Mp = 84-86 °C. IR (KBr) ν (cm−1) 3400, 2939, 1590, 1482, 1352, 1317, 1081, 765. 1H NMR (400 MHz, DMSO-d6) δ 0.90 (t, 3H, 3J = 6.4 Hz), 1.70-1.80 (m, 4H), 2.02-2.04 (m, 5H), 2.31-2.34 (m, 2H), 2.93-3.00 (m, 2H), 4.48 (s, 2H), 7.57 (d, 1H, 3J = 8.8 Hz), 7.76-7.81 (m, 2H), 7.96 (t, 1H, 3J = 6.8 Hz), 8.37 (d, 1H, 3J = 6.8 Hz), 8.67 (d, 1H, 3J = 8.8 Hz), 8.75 (d, 1H, 3J = 7.8 Hz). 13C NMR (100 MHz, CDCl3) δ 12.0, 20.5, 28.9 (2C), 36.2, 53.1 (2C), 61.2, 70.6, 119.7, 121.8, 122.2, 122.9, 125.1, 127.8, 128.7, 129.1, 129.8, 131.0, 133.7, 141.8, 159.1. LC-MS (ESI): tR = 5.79 min; [M+H]+: 369.32, 371.33. HRMS (EI) calcd. for C22H25ClN2O 368.1655, found 368.1666.
3-Methoxy-6-(1-propylpiperidin-4-yl)methyloxyphenanthridine 4g
Starting from 3g (291 mg, 1.3 mmol), using general procedure A and cyclohexane/ethyl acetate 9/1 with 5% of NEt3 as the eluent for the chromatography, 4g was obtained as a white powder (334 mg). Yield: 71%. Mp = 76-77 °C. IR (KBr) ν (cm−1) 2932, 1615, 1587, 1484, 1350, 1315, 1174, 1087, 983, 770. 1H NMR (400 MHz, CDCl3) δ 0.91 (t, 3H, 3J = 7.8 Hz), 1.50-1.62 (m, 4H), 1.92-2.03 (m, 5H), 2.30-2.34 (m, 2H), 3.01-3.03 (m, 2H), 3.96 (s, 3H), 4.48 (d, 2H, 3J = 6.8 Hz), 7.09 (dd, 1H, 3J = 8.8 Hz, 4J = 2.0 Hz), 7.30 (d, 1H, 4J = 2.0 Hz), 7.55 (t, 1H, 3J = 6.8 Hz), 7.75 (t, 1H, 3J = 8.8 Hz), 8.28 (d, 1H, 3J = 8.8 Hz), 8.34 (d, 1H, 3J = 7.8 Hz), 8.37 (d, 1H, 3J = 7.8 Hz). 13C NMR (100 MHz, CDCl3) δ 12.1, 20.2, 29.3 (2C), 36.0, 53.7 (2C), 55.5, 61.3, 70.5, 108.5, 114.6, 116.2, 119.1, 121.3, 123.3, 125.1, 126.0, 130.9, 134.9, 145.0, 159.5, 160.3. LC-MS (ESI): tR = 5.25 min; [M+H]+: 365.38. HRMS (EI) calcd. for C23H28N2O2 364.2150, found 364.2140.
3-Methyl-6-(1-propylpiperidin-4-yl)methyloxyphenanthridine 4h
Starting from 3h (120 mg, 0.57 mmol), using general procedure A and cyclohexane/ethyl acetate 8/2 with 5% of NEt3 as the eluent for the chromatography, 4h was obtained as a white powder (118 mg). Yield: 59%. Mp = 107-108 °C. IR (KBr) ν (cm−1) 3430, 2939, 1590, 1344, 1314, 1090, 774. 1H NMR (400 MHz, CDCl3) δ 0.91 (t, 3H, 3J = 6.8 Hz), 1.53-1.60 (m, 4H), 1.92-2.03 (m, 5H), 2.30-2.34 (m, 2H), 2.53 (s, 3H), 3.00-3.03 (m, 2H), 4.49 (d, 2H, 3J = 5.8 Hz), 7.30 (d, 1H, 3J = 8.8 Hz), 7.59 (t, 1H, 3J = 7.8 Hz), 7.68 (s, 1H), 7.78 (t, 1H, 3J = 7.8 Hz), 8.29 (d, 1H, 3J = 7.8 Hz), 8.36 (d, 1H, 3J = 8.8 Hz), 8.46 (d, 1H, 3J = 7.8 Hz). 13C NMR (100 MHz, CDCl3) δ 12.1, 20.2, 21.5, 29.4 (2C), 36.0, 53.7 (2C), 61.3, 70.4, 119.9, 120.0, 121.6, 121.9, 125.0, 125.9, 126.7, 127.6, 130.7, 134.9, 138.9, 143.4, 159.0. LC-MS (ESI): tR = 5.64 min; [M+H]+: 349.33. HRMS/ESI calcd. for C23H29N2O [M+H]+ 349.2280, found 349.2266.
3-Fluoro-6-(1-propylpiperidin-4-yl)methyloxyphenanthridine 4i
Starting from 3i (100 mg, 0.47 mmol), using general procedure A and cyclohexane/ethyl acetate 7/3 with 5% of NEt3 as the eluent for the chromatography, 4i was obtained as a colorless oil (107 mg). Yield: 65%. IR (KBr) ν (cm−1) 2995, 2852, 1588, 1463, 1260, 1088, 1019, 799, 770. 1H NMR (400 MHz, CDCl3) δ 0.91 (t, 3H, 3J = 7.8 Hz), 1.54-1.64 (m, 9H), 1.90-2.02 (m, 5H), 2.30-2.34 (m, 2H), 3.01-3.04 (m, 2H), 4.48 (d, 2H, 3J = 5.8 Hz), 7.19-7.28 (m, 1H), 7.52 (dd, 1H, 3J = 10.8 Hz, 3J = 2.9 Hz), 7.62 (t, 1H, 3J = 7.8 Hz), 7.80 (t, 1H, 3J = 6.8 Hz), 8.34-8.43 (m, 3H). 13C NMR (100 MHz, CDCl3) δ 12.1, 20.2, 29.3 (2C), 35.9, 53.7 (2C), 61.3, 70.3, 112.7 (d, 2J = 22 Hz), 112,9 (d, 2J = 23 Hz), 119,0 (d, J = 1 Hz), 119.6, 121.7, 123,7 (d, J = 10 Hz), 125.2, 127,0, 131.1, 134.5, 144,8 (d, J = 12 Hz), 159,7, 163,0 (d, 1J = 246 Hz). LC-MS (ESI): tR = 5.47 min; [M+H]+: 353.28. HRMS/EI calcd. for C22H25FN2O 352.1950, found 352.1941.
4-Methyl-6-(1-propylpiperidin-4-yl)methyloxyphenanthridine 4j
Starting from 3j (217 mg, 1.03 mmol), using general procedure A and cyclohexane/ethyl acetate 9/1 with 5% of NEt3 as the eluent for the chromatography, 4j was obtained as a white powder (170 mg). Yield: 47%. Mp = 93-95 °C. IR (KBr) ν (cm−1) 2938, 2764, 1589, 1312, 760. 1H NMR (400 MHz, CDCl3) δ 0.91 (t, 3H, 3J = 7.5 Hz,), 1.51-1.62 (m, 4H), 1.92-2.04 (m, 5H), 2.30-2.33 (m, 2H), 2.73 (s, 3H), 3.01 (m, 2H), 4.52 (d, 2H, 3J = 6.5 Hz), 7.37 (dd, 1H, 3J = 8.0 Hz, 3J = 7.0 Hz), 7.48-7.50 (m, 1H), 7.61 (ddd, 1H, 3J = 8.0 Hz, 3J = 7.5 Hz, 4J = 1.5 Hz), 7.78 (ddd, 1H, 3J = 8.0 Hz, 3J = 7.0 Hz, 4J = 1.5 Hz), 8.27-8.29 (m, 1H), 8.37 (dd, 1H, 3J = 8.1 Hz, 4J = 1.4 Hz), 8.49-8.51 (m, 1H). 13C NMR (100 MHz, CDCl3) δ 12.1, 18.3, 20.2, 29.4 (2C), 35.9, 53.7 (2C), 61.2, 70.3, 119.8, 119.9, 122.0, 122.1, 123.7, 124.9, 126.9, 129.4, 130.6, 135.2, 135.7, 141.8, 157.6. LC-MS (ESI): tR = 5.65 min; [M+H]+: 349.57. HRMS (EI) calcd. for C23H28N2O 348.2201, found 348.2215.
4-Methoxy-6-(1-propylpiperidin-4-yl)methyloxyphenanthridine 4k
Starting from 3k (150 mg, 0.66 mmol), using general procedure A and cyclohexane/ethyl acetate 9/1 with 5% of NEt3 as the eluent for the chromatography, 4k was obtained as a white powder (153 mg). Yield: 47%. Mp = 99-100 °C. IR (KBr) ν (cm−1) 3433, 2951, 2768, 1590, 1321, 1256, 748. 1H NMR (400 MHz, CDCl3) δ 0.92 (t, 3H, 3J = 7.8 Hz), 1.52-1.67 (m, 4H), 1.94-2.04 (m, 5H), 2.31-2.35 (m, 2H), 3.01-3.04 (m, 2H), 4.08 (s, 3H), 4.56 (d, 2H, 3J = 5.9 Hz), 7.10 (d, 1H, 3J = 7.8 Hz), 7.42 (t, 1H, 3J = 8.8 Hz), 7.64 (t, 1H, 3J = 7.8 Hz), 7.80 (t, 1H, 3J = 8.8 Hz), 8.03 (d, 1H, 3J = 7.8 Hz), 8.40 (d, 1H, 3J = 7.8 Hz), 8.49 (d, 1H, 3J = 7.8 Hz). 13C NMR (100 MHz, CDCl3) δ 12.1, 20.2, 29.3 (2C), 36.1, 53.7 (2C), 56.5, 61.3, 70.4, 109.4, 114.4, 120.2, 122.4, 123.6, 124.3, 125.1, 127.3, 130.8, 133.8, 134.9, 154.5, 158.4. LC-MS (ESI): tR = 5.24 min; [M+H]+: 365.38. HRMS (EI) calcd. for C23H28N2O2 364.2150, found 364.2139.
4-Chloro-6-(1-propylpiperidin-4-yl)methyloxyphenanthridine 4l
Starting from 3l (150 mg, 0.65 mmol), using general procedure A and cyclohexane/ethyl acetate 9/1 with 5% of NEt3 as the eluent for the chromatography, 4l was obtained as a white powder (149 mg). Yield: 62%. Mp = 75-77 °C. IR (KBr) ν (cm−1) 3434, 2919, 2852, 1591, 1458, 1399, 1342, 1097, 747. 1H NMR (400 MHz, CDCl3) δ 0.91 (t, 3H, 3J = 6.8 Hz), 1.52-1.62 (m, 4H), 1.92-2.03 (m, 5H), 2.30-2.34 (m, 2H), 3.00-3.03 (m, 2H), 4.60 (d, 2H, 3J = 6.8 Hz), 7.38 (t, 1H, 3J = 7.8 Hz), 7.67 (t, 1H, 3J = 6.8 Hz), 7.73 (d, 1H, 3J = 7.8 Hz), 7.83 (t, 1H, 3J = 6.8 Hz), 8.33 (d, 1H, 3J = 7.8 Hz), 8.40 (d, 1H, 3J = 7.8 Hz), 8.49 (d, 1H, 3J = 8.8 Hz). 13C NMR (100 MHz, CDCl3) δ 12.1, 20.2, 29.3 (2C), 35.9, 53.7 (2C), 61.2, 70.8, 120.1, 120.8, 122.2, 124.0, 124.1, 125.2, 127.8, 129.1, 131.2, 132.0, 134.7, 139.8, 159.2. LC-MS (ESI): tR = 5.64 min; [M+H]+: 369.35, 371.30. HRMS/ESI calcd. for C23H26ClN2O [M+H]+ 369.1734, found 365.1729.
4-Fluoro-6-(1-propylpiperidin-4-yl)methyloxyphenanthridine 4m
Starting from 3m (260 mg, 1.22 mmol), using general procedure A and cyclohexane/ethyl acetate 8/2 with 5% of NEt3 as the eluent for the chromatography, 4m was obtained as a white powder (288 mg). Yield: 67%. Mp = 105-106 °C. IR (KBr) ν (cm−1) 2925, 1591, 1348, 1317, 1234, 753. 1H NMR (400 MHz, CDCl3) δ 0.91 (t, 3H, 3J = 7.5 Hz), 1.51-1.64 (m, 4H), 1.93-2.03 (m, 5H), 2.31-2.34 (m, 2H), 3.02-3.04 (m, 2H), 4.55 (d, 2H, 3J = 6.0 Hz), 7.33-7.41 (m, 2H), 7.67 (ddd, 1H, 3J = 8.0 Hz, 3J = 7.0 Hz, 4J = 1.0 Hz), 7.82 (ddd, 1H, 3J = 8.2 Hz, 3J = 7.2 Hz, 4J = 1.4 Hz), 8.17-8.18 (m, 1H), 8.39-8.41 (m, 1H), 8.46(d, 1H, 3J = 8.5 Hz). 13C NMR (100 MHz, CDCl3) δ 12.1, 20.2, 29.3 (2C), 36.0, 53.6 (2C), 61.2, 70.7, 113.9 (d, 2J = 20 Hz), 117.5 (d, J = 4 Hz), 120.3, 122.2, 123.8 (d, J = 8 Hz), 124.5 (d, J = 2 Hz), 125.2, 127.7, 131.2, 132.7 (d, 2J = 11 Hz), 134.2 (d, J = 3 Hz), 157.5 (d, 1J = 250 Hz), 159.1. LC-MS (ESI): tR = 5.32 min; [M+H]+: 353.30. HRMS (EI) calcd. for C22H25FN2O 352.1951, found 352.1943.
7-Fluoro-6-(1-propylpiperidin-4-yl)methyloxyphenanthridine 4n
In sealed tube were introduced, KOH (1.07 g, 19 mmol), 2’-fluoro-3-fluorobiphenyl-2-carbonitrile41 (0.82 g, 3.8 mmol) and t-BuOH (30 mL). The tube was heated to 150 °C for 0.5 h. Water (20 mL) was added and the resulting precipitate was filtered. The solid was then dried under vacuum to afford a mixture of the expected 7-fluorophenanthridin-6(5H)-one along with side products. The mixture was then subjected to general procedure A using cyclohexane/ethyl acetate 9/1 as the eluent for the chromatography. 4n was obtained as yellow oil (0.12 g). Yield: 9%. IR (KBr) ν (cm−1) 3432, 2931, 2765, 1595, 1338, 758. 1H NMR (400 MHz, CDCl3) δ 0.91 (t, 3H, 3J = 6.8 Hz), 1.52-1.54 (m, 4H), 1.99-2.02 (m, 5H), 2.30-2.34 (m, 2H), 3.00-3.04 (m, 2H), 4.47 (d, 2H, 3J = 6.8 Hz), 7.28-7.31 (m, 1H), 7.47 (t, 1H, 3J = 6.8 Hz), 7.63 (t, 1H, 3J = 6.8 Hz), 7.72 (m, 1H), 7.83 (d, 1H, 3J = 9.8 Hz), 8.29 (d, 1H, 3J = 8.7 Hz), 8.36 (d, 1H, 3J = 6.8 Hz). 13C NMR (100 MHz, CDCl3) δ 12.1, 20.2, 29.3 (2C), 35.9, 53.7 (2C), 61.3, 70.3, 109.7 (d, J = 9 Hz), 111.7 (d, J = 5 Hz), 114.1 (d, 2J = 23 Hz), 121.2, 122.5, 124.5, 127.6, 129.4, 131.4 (d, 2J = 10 Hz), 137.7, 143.4, 157.6 (d, J = 7 Hz), 160.23 (d, 1J = 262 Hz). LC-MS (ESI): tR = 5.38 min; [M+H]+: 353.47. HRMS (EI) calcd. for C22H25FN2O 352.1950, found 352.1935.
8-Nitro-6-(1-propylpiperidin-4-yl)methyloxyphenanthridine 4o
Starting from 3o (0.80 g, 3.3 mmol), following general procedure A and using cyclohexane/ethyl acetate (9/1) as eluent for the chromatography, 4o was obtained as a white powder (0.72 g). Yield: 57%. Mp = 123-125 °C. IR (KBr) ν (cm−1) 2940, 2765, 1604, 1344. 1H NMR (400 MHz, CDCl3) δ 0.92 (t, 3H, 3J = 7.8 Hz), 1.53-1.58 (m, 4H), 1.95-2.05 (m, 5H), 2.31-2.35 (m, 2H), 3.02-3.05 (m, 2H), 4.52 (d, 2H, 3J = 6.8 Hz), 7.54 (t, 1H, 3J = 6.8 Hz), 7.73 (t, 1H, 3J = 6.8 Hz), 7.90 (d, 1H, 3J = 7.8 Hz), 8.42 (d, 1H, 3J = 7.8 Hz), 8.55 (m, 2H), 9.19 (d, 1H, 4J = 1.9 Hz). 13C NMR (100 MHz, CDCl3) δ 12.1, 20.2, 29.3 (2C), 35.9, 53.5 (2C), 61.3, 70.3, 119.8, 121.0, 121.4, 123.0, 123.5, 124.5, 125.1, 128.2, 130.9, 138.9, 144.7, 146.2, 158.4. LC-MS (ESI): tR = 5.39 min; [M+H]+: 380.36. HRMS (EI) calcd. for C22H25N3O3 379.1895, found 379.1913.
8-Methoxy-6-(1-propylpiperidin-4-yl)methyloxyphenanthridine 4p
Starting from 3p (0.80 g, 3.55 mmol), following general procedure A and using cyclohexane/ethyl acetate (9/1) as the eluent for the chromatography, 4p was obtained as a white powder (0.90 g). Yield: 70%. Mp = 92-94 °C. IR (KBr) ν (cm−1) 3430, 2765, 2940, 1590, 1462, 1219. 1H NMR (400 MHz, CDCl3) δ 0.86 (t, 3H, 3J = 6.8 Hz), 1.51-1.57 (m, 4H), 1.93-2.03 (m, 5H), 2.30-2.34 (m, 2H), 3.00-3.03 (m, 2H), 3.98 (s, 3H), 4.51 (d, 2H, 3J = 6.8 Hz), 7.43 (m, 2H), 7.45 (t, 1H, 3J = 6.8 Hz), 7.71 (d, 1H, 3J = 2.9 Hz), 7.84 (d, 1H, 3J = 8.8 Hz), 8.33 (d, 1H, 3J = 7.8 Hz), 8.42 (d, 1H, 3J = 8.8 Hz). 13C NMR (100 MHz, CDCl3) δ 12.1, 20.2, 29.3 (2C), 35.8, 53.7 (2C), 55.5, 61.3, 70.3, 105.3, 120.8, 121.3, 121.4, 122.4, 123.5, 124.3, 127.6, 127.7, 128.9, 142.2, 158.2, 158.7. LC-MS (ESI): tR = 5.47 min; [M+H]+: 365.33. HRMS (EI) calcd. for C23H28N2O2 364.2150, found 364.2135.
8-Fluoro-6-(1-propylpiperidin-4-yl)methyloxyphenanthridine 4q
Starting from 3q (0.26 g, 1.22 mmol), following general procedure A and using cyclohexane/ethyl acetate (9/1) as the eluent for the chromatography, 4q was obtained as a white powder (0.19 g). Yield: 44%. Mp = 70-71 °C. IR (KBr) ν (cm−1) 2939, 755. 1H NMR (400 MHz, CDCl3) δ 0.92 (t, 3H, 3J = 7.0 Hz), 1.53-1.60 (m, 4H), 1.92-2.02 (m, 5H), 2.31-2.34 (m, 2H), 3.01-3.03 (m, 2H), 4.47 (d, 2H, 3J = 6.8 Hz), 7.48 (t, 1H, 3J = 7.5 Hz), 7.52 (dt, 3J = 8.5 Hz, 4J = 2.5 Hz, 1H), 7.61 (t, 1H, 3J = 7.5 Hz), 7.86 (d, 1H, 3J = 8.0 Hz), 7.97 (dd, 1H, 3J = 9.0 Hz, 4J = 2.5 Hz), 8.34 (d, 1H, 3J = 8.0 Hz), 8.48 (dd, 1H, 3J = 9.0 Hz, 4J = 5.5 Hz). 13C NMR (100 MHz, CDCl3) δ 12.1, 20.2, 29.3 (2C), 35.9, 53.7 (2C), 61.3, 70.3, 110.1 (d, 2J = 22 Hz), 119.8 (d, 2J = 22 Hz), 121.6 (d, J = 7 Hz), 121.9 (d, J = 11 Hz), 122.0, 124.5 (d, J = 7 Hz), 124.7, 128.0, 128.7, 131.5 (d, J = 2 Hz), 143.0, 158.2 (d, J = 2 Hz), 161.7 (d,1J = 246 Hz). LC-MS (ESI): tR = 5.41 min; [M+H]+: 353.31. HRMS (EI) calcd. for C22H25FN2O 352.1950, found 352.1958.
9-Methyl-6-(1-propylpiperidin-4-yl)methyloxyphenanthridine 4r
Starting from 3r (1.00 g, 4.77 mmol), following general procedure A and using cyclohexane/ethyl acetate (9/1) as the eluent for the chromatography 4r was obtained as a white powder (1.18 g). Yield: 71%. Mp = 90-91 °C. IR (KBr) ν (cm−1) 3411, 3064, 2933, 1596, 1336, 758. 1H NMR (400 MHz, CDCl3) δ 0.90 (t, 3H, 3J = 6.8 Hz), 1.50-1.61 (m, 4H), 1.92-2.01 (m, 5H), 2.30-2.32 (m, 2H), 2.60 (s, 3H), 2.99-3.02 (m, 2H), 4.48 (d, 2H, 3J = 6.8 Hz), 7.42-7.46 (m, 2H), 7.59 (t, 1H, 3J = 6.8 Hz), 7.83 (d, 1H, 3J = 8.8 Hz’), 8.23-8.26 (m, 2H), 8.37 (d, 1H, 3J = 7.8 Hz). 13C NMR (100 MHz, CDCl3) δ 12.1, 20.2, 22.3, 29.3 (2C), 35.9, 53.7 (2C), 61.3, 70.3, 118.1, 121.6, 122.0, 122.3, 124.0, 124.9, 127.6, 128.6, 128.7, 134.8, 141.1, 143.6, 159.0. LC-MS (ESI): tR = 5.61 min; [M+H]+: 349.32. HRMS (EI) calcd. for C23H28N2O 348.2201, found 348.2202.
9-Fluoro-6-(1-propylpiperidin-4-yl)methyloxyphenanthridine 4s
Starting from 3s (0.20 g, 0.94 mmol), following general procedure A and using cyclohexane/ethyl acetate (9/1) as the eluent for the chromatography, 4s was obtained, after recrystalisation in MeCN, as a white powder (0.20 g). Yield: 62%. Mp = 79-80 °C. IR (KBr) ν (cm−1) 3399, 2944, 2765, 1593, 1336, 762. 1H NMR (400 MHz, CDCl3) δ 0.90 (t, 3H, 3J = 7.2 Hz), 1.52-1.59 (m, 4H), 1.89-1.99 (m, 5H), 2.30-2.33 (m, 2H), 3.00 (m, 2H), 4.48 (d, 2H, 3J = 6.8 Hz), 7.33 (ddd, 1H, 3J = 8.9 Hz, 3J = 8.2 Hz, 4J = 2.5 Hz), 7.47 (ddd, 1H, 3J = 8.1 Hz, 3J = 7.1 Hz, 4J = 1.3 Hz), 7.64 (ddd, 1H, 3J = 8.1 Hz, 3J = 7.1 Hz, 4J = 1.4 Hz), 7.86 (dd, 1H, 3J = 8.1 Hz, 4J = 1.4 Hz), 8.04 (dd, 1H, 3J = 10.7 Hz, 4J = 2.9 Hz), 8.28 (dd, 1H, 3J = 8.3 Hz, 4J = 1.4 Hz), 8.39 (dd, 1H, 3J = 8.8 Hz, 3J = 5.8 Hz). 13C NMR (100 MHz, CDCl3) δ 12.1, 20.2, 29.3 (2C), 35.9, 53.7 (2C), 61.3, 70.3, 107.2 (d, 2J = 22 Hz), 115.8 (d, 2J = 23 Hz), 116.9 (d, 4J = 1 Hz), 121.9 (d, 4J = 3 Hz), 122.3, 124.3, 127.8, 127.9 (d, 3J = 10 Hz), 129.3, 137.1 (d, 3J = 11 Hz), 143.8, 158.5, 164.3 (d, 1J = 249 Hz). LC-MS (ESI): tR = 5.45 min; [M+H]+: 353.28. HRMS/EI calcd. for C22H25FN2O 352.1950, found 352.1938.
5-(1-Propylpiperidin-4-yl)methyloxybenzo[c]-2,6-naphthyridine 4t
Starting from 3t (0.1 g, 0.5 mmol), following general procedure A and using cyclohexane/ethyl acetate (4/1) and cyclohexane/ethyl acetate (1/1) as the eluents for the chromatography, 4t was obtained as a white powder (72 mg). Yield: 42%. Mp = 113-115 °C. IR (KBr) ν (cm−1) 2953, 2932, 1613, 1585, 1459, 1337, 1235, 1128, 1985, 844, 768, 673. 1H NMR (400 MHz, CDCl3) δ 0.92 (t, 3H, 3J = 7.4 Hz), 1.50-1.61 (m, 4H), 1.91-2.03 (m, 5H), 2.32 (m, 2H), 3.02 (d, 2H, J = 11.7 Hz), 4.51 (d, 2H, 3J = 6.5 Hz), 7.55 (t, 1H, J = 7.6 Hz), 7.69 (t, 1H, J = 7.5 Hz), 7.90 (d, 1H, 3J = 8.3 Hz), 8.12 (d, 1H, 3J = 5.3 Hz), 8.54 (d, 1H, 3J = 8.3 Hz), 8.83 (d, 1H, 3J = 5.3 Hz), 9.92 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 12.0, 20.1, 29.1 (2C), 35.7, 53.5 (2C), 61.1, 70.8, 117.1, 120.2, 121.4, 124.1, 125.2, 127.9, 128.6, 129.5, 143.5, 146.1, 143.4, 157.5. LC-MS (ESI): tR = 4.59 min; [M+H]+: 336.25. HRMS/ESI calcd. for C21H26N3O [M+H]+ 336.2076, found 336.2067.
5-(1-Propylpiperidin-4-yl)methyloxybenzo[h]-1,6-naphthyridine 4u
Starting from 3u (0.1 g, 0.5 mmol), following general procedure A and using cyclohexane/ethyl acetate (1/1) and ethyl acetate with 5% of NEt3 as the eluents for the chromatography, 4u was obtained as a white powder (92 mg). Yield: 54%. Mp = 114-115 °C. IR (KBr) ν (cm−1) 2931, 1605, 1590, 1458, 1328, 767, 733. 1H NMR (400 MHz, CDCl3) δ 0.84 (t, 3H, 3J = 7.3 Hz), 1.18 (brs, 1H), 1.43-1.56 (m, 3H), 1.18-1.96 (m, 5H), 2.23-2.27 (m, 2H), 2.95 (d, 2H, J = 11.2 Hz), 4.44 (d, 2H, 3J = 6.3 Hz), 7.45-7.50 (m, 2H), 7.62-7.65 (m, 1H), 7.80 (d, 1H, 3J = 7.5 Hz), 8.55 (dd, 1H, 3J = 8.2 Hz, 4J = 1.4 Hz), 8.88 (dd, 1H, 3J = 8.1 Hz, 4J = 1.4 Hz), 9.03 (dd, 1H, 3J = 4.4 Hz, 4J = 1.2 Hz). 13C NMR (100 MHz, CDCl3) δ 12.1, 20.2, 29.3 (2C), 35.9, 53.6 (2C), 61.2, 70.8, 115.4, 122.3, 123.5, 123.7, 124.9, 127.2, 130.4, 133.0, 144.9, 150.8, 152.9, 158.4. LC-MS (ESI): tR = 4.84 min; [M+H]+: 336.28. HRMS/EI calcd. for C21H25N3O 335.1997, found 335.1998.
7-Fluoro-5-(1-propylpiperidin-4-yl)methyloxybenzo[h]-1,6-naphthyridine 4v
Starting from 3v (1 g, 4.6 mmol), following general procedure A and using ethyl acetate and ethyl acetate/NEt3 98/2 as the eluents for the chromatography, 4v was obtained as a white powder (1.37 g). Yield: 83%. Mp = 119-120 °C. IR (KBr) ν (cm−1) 2952, 2932, 1605, 1589, 1329, 1235, 1152, 774. 1H NMR (400 MHz, CDCl3) δ 0.91 (t, 3H, 3J = 6.8 Hz), 1.50-1.64 (m, 4H), 1.77 (brs, 1H), 1.90-2.04 (m, 4H), 2.30-2.34 (m, 2H), 3.02 (d, 2H, J = 11.7 Hz), 4.56 (d, 2H, 3J = 6.8 Hz), 7.41-7.49 (m, 2H), 7.59 (dd, 1H, 3J = 8.3 Hz, 3J = 4.8 Hz), 8.64 (dd, 1H, 3J = 7.8 Hz, 4J = 1.9 Hz), 8.72 (dd, 1H, 3J = 8.3 Hz, 4J = 1.9 Hz), 9.11 (dd, 1H, 3J = 4.8 Hz, 4J = 1.9 Hz). 13C NMR (100 MHz, CDCl3) δ 12.1, 20.2, 29.2 (2C), 35.8, 53.6 (2C), 61.2, 71.1, 115.6, 115.6 (d, 2J = 19 Hz), 119.2 (d, J = 4 Hz), 122.8, 124.4 (d, J = 7 Hz), 125.5 (d, J = 2 Hz), 133.2, 134.1 (d, 2J = 11 Hz), 150.3 (d, J = 3 Hz), 153.3, 157.0 (d, 1J = 250 Hz), 158.6. LC-MS (ESI): tR = 6.00 min; [M+H]+: 354.37. HRMS/EI calcd. for C21H24FN3O 353.1903, found 353.1915.
2-Fluoro-3-(trimethylsilanyl)phenylboronic acid 5
In a three necks round-bottom flask under N2 at −80 °C were introduced (2-fluorophenyl)trimethylsilane42 (7.4 g, 44 mmol), THF (90 mL) and s-BuLi 1.3 M in n-hexane/cyclohexane 98/2 (33.82 mL, 44 mmol). The yellow solution was stirred for 0.75 h, trimethyl borate (5.49 mL, 48.4 mmol) was added and the solution stirred again for 0.75 h. The mixture was allowed to reach room temperature, hydrolyzed with water (200 mL), washed with Et2O (3 × 150 mL), acidified to pH = 1 using HCl 1 M and extracted with Et2O (3 × 150 mL). The combined organic layers were dried over MgSO4, filtered and evaporated in vacuo to afford 5 (6.4 g) as white crystals. Yield: 69%. Mp = 93-94 °C. IR (KBr) ν (cm−1) 3512, 3351, 2952, 1423, 1351, 844, 760, 605. 1H NMR (400 MHz, CDCl3) δ 0.32 (s, 9H), 5.29 (s, 1H), 5.31 (s, 1H), 7.19 (td, 1H, 3J = 7.2 Hz, J = 1.5 Hz), 7.52 (m, 1H), 7.84 (td, 1H, 3J = 7.2 Hz, J = 1.6 Hz). 13C NMR (100 MHz, CDCl3) δ −1.0, 124.3 (d, 4J = 2 Hz), 125.6 (d, 2J = 36 Hz), 138.2 (d, 3J = 7 Hz), 138.8 (d, 3J = 13 Hz), 172.7 (d, 1J = 235 Hz), signal of the carbon bonded to the boron is missing.
2’-Fluoro-3’-(trimethylsilanyl)biphenyl-2-carbonitrile 6a
In a round-bottom flask under N2, were introduced DME (15 mL) and water (15 mL). The solution was degazed by bubbling N2 for 15 min and Pd(OAc)2 (62 mg, 0.27 mmol) and PPh3 (144 mg, 0.55 mmol) were added. The solution was heated to 50 °C for 10 min and 2-bromobenzonitrile (1 g, 5.5 mmol), 5 (1.75 g, 8.25 mmol) and Na2CO3 (2.33 g, 22 mmol) were added. The solution was heated to 90 °C for 14h, cooled to room temperature, filtered over a pad of celite and extracted with EtOAc (3 × 50 mL). The combined organic layers were dried over MgSO4, filtered, evaporated in vacuo and purified by flash chromatography using cyclohexane/AcOEt 9/1 as the eluent to afford 6a (1.45 g) as a colorless oil. Yield: 98%. IR (KBr) ν (cm−1) 2957, 2227 (CN), 1637, 1412, 585, 842, 761. 1H NMR (400 MHz, CDCl3) δ 0.35 (s, 9H), 7.25 (t, 1H, 3J = 7.8 Hz), 7.40-7.53 (m, 4H), 7.65 (t, 1H, 3J = 7.8 Hz), 7.77 (d, 1H, 3J = 7.8 Hz). 13C NMR (100 MHz, CDCl3) δ −1.0, 112.9, 118.1, 124.1 (d, J = 3 Hz), 125.0 (d, 2J = 19 Hz), 127.5 (d, 3J = 36 Hz), 127.9, 130.9, 132.4, 132.4, 133.2, 136.1 (d, J = 13 Hz), 140.1, 163.5 (d, 1J = 242 Hz). HRMS/EI calcd. for C16H16FNSi 269.1036, found 269.1034.
2-(2-Fluoro-3-trimethylsilanylphenyl)nicotinonitrile 6b
Same procedure as for 6a, starting from 2-chloronicotinonitrile (1 g, 7.2 mmol), Pd(OAc)2 (81 mg, 0.36 mmol), PPh3 (189 mg, 0.72 mmol), 5 (2.29 g, 10.8 mmol) and Na2CO3 (3.06 g, 28.8 mmol). Cyclohexane/AcOEt 8/2 was used as the eluent for chromatography to afford 6b (1.85 g) as a colorless oil. Yield: 95%. IR (KBr) ν (cm−1) 2957, 2231 (CN), 1604, 1428, 1414, 1251, 862, 842, 762. 1H NMR (400 MHz, CDCl3) δ 0.36 (s, 9H), 7.29 (m, 1H), 7.43 (dd, 1H, 3J = 8.3 Hz, 3J = 4.9 Hz), 7.53-7.60 (m, 2H), 8.08 (d, 1H, 3J = 7.8 Hz), 8.90 (dd, 1H, 3J = 4.9 Hz, 4J = 1.9 Hz). 13C NMR (100 MHz, CDCl3) δ −1.0, 110.6, 116.5, 122.0, 124.2 (d, J = 3 Hz), 124.7 (d, 2J = 19 Hz), 127.5 (d, 2J = 31 Hz), 132.3 (d, J = 2 Hz), 137.2 (d, J = 11 Hz), 140.6, 152.5, 157.9, 163.8 (d, 1J = 243 Hz). HRMS/EI calcd. for C15H15FN2Si 270.0988, found 270.0979.
2’-Fluoro-3’-iodobiphenyl-2-carbonitrile 7a
In a round-bottom flask were introduced 6a (1.05 g, 3.7 mmol), DCM (30 mL) and ICl (279 µL, 5.6 mmol). The solution was allowed to stir for 4.5 h and a saturated Na2S2O3 solution in water (40 mL) was added. The aqueous layer was then extracted with DCM (3 × 30 mL) and the combined organic layers were dried MgSO4, filtered and evaporated in vacuo to afford 7a (1.11 g) as a white powder. Yield: 88%. Mp = 123-125 °C. IR (KBr) ν (cm−1) 2222 (CN), 1443, 1425, 788, 759. 1H NMR (400 MHz, CDCl3) δ 7.03 (t, 1H, 3J = 7.8 Hz), 7.40 (m, 1H), 7.49-7.53 (m, 2H), 7.67 (td, 1H, J = 7.8 Hz, J = 1.9 Hz), 7.67 (d, 1H, 3J = 7.8 Hz), 7.84 (m, 1H). 13C NMR (100 MHz, CDCl3) δ 82.2 (d, 2J = 25 Hz), 112.6, 117.7, 125.8 (d, J = 5 Hz), 126.3 (d, 2J = 17 Hz), 128.6, 130.8 (d, J = 2 Hz), 131.4 (d, J = 2 Hz), 132.6, 133.3, 138.6, 140.2 (d, J = 2 Hz), 158.2 (d, 1J = 246 Hz). HRMS/EI calcd. for C13H7FIN 322.9607, found 322.9601.
2-(2-Fluoro-3-iodophenyl)nicotinonitrile 7b
Same procedure as for 7a, starting from 6b (0.82 g, 3 mmol) and ICl (228 µL, 4.5 mmol). The crude was purified by flash chromatography using cyclohexane/AcOEt 8/2 as the eluent to afford 7b (0.82 g) as a white powder. Yield: 84%. Mp = 100–101 °C. IR (KBr) ν (cm−1) 2228 (CN), 1424, 1227, 805, 761, 725. 1H NMR (400 MHz, CDCl3) δ 7.08 (t, 1H, 3J = 7.8 Hz), 7.48 (dd, 1H, 3J = 7.8 Hz, 3J = 4.9 Hz), 7.56 (m, 1H), 7.92 (m, 1H), 8.11 (dd, 1H, 3J = 7.8 Hz, 4J = 1.9 Hz), 8.92 (dd, 1H, 3J = 4.9 Hz, 4J = 1.9 Hz). 13C NMR (100 MHz, CDCl3) δ 82.3 (d, 2J = 26 Hz), 110.4, 116.1, 122.6, 126.0 (d, J = 5 Hz), 126.1 (d, 2J = 16 Hz), 131.4 (d, J = 2 Hz), 140.8, 141.4 (d, J = 2 Hz), 152.6, 156.5, 158.5 (d, 1J = 247 Hz). HRMS/EI calcd. for C12H6FIN2 323.9568, found 323.9559.
4-Iodophenanthridin-6(5H)-one 8a
Same procedure as for compound 3v, using 7a (1.11 g, 3.4 mmol), KOH (0.96 g, 14 mmol) and t-BuOH (30 mL), heating at 150 °C for 1 h. 8a (0.73 g) was obtained as a white powder. Yield: 67%. Mp = 240–242 °C. IR (KBr) ν (cm−1) 3274, 1654 (CO), 1603, 1351, 752, 709, 625. 1H NMR (400 MHz, DMSO-d6) δ 7.04 (t, 1H, 3J = 7.8 Hz), 7.66 (t, 1H, 3J = 7.8 Hz), 7.86 (t, 1H, 3J = 7.8 Hz), 8.00 (d, 1H, 3J = 6.8 Hz), 8.33 (d, 1H, 3J = 6.8 Hz), 8.42 (d, 1H, 3J = 7.8 Hz), 8.50 (d, 1H, 3J = 8.8 Hz), 9.29 (brs, 1H). 13C NMR (100 MHz, DMSO-d6) δ 105.1, 119.0, 122.6, 123.6, 125.9, 127.5, 128.2, 132.5, 134.0, 137.3, 137.7, 139.3, 172.9. HRMS/EI calcd. for C13H8INO 320.9651, found 320.9665.
7-Iodobenzo[h]-1,6-naphthyridin-5(6H)-one 8b
Same procedure as for compound 3v, using 7b (0.7 g, 2.16 mmol), KOH (0.6 g, 10.8 mmol) and t-BuOH (35 mL), heating at 150 °C for 1 h. 8b (0.51 g) was obtained as a white powder. Yield: 73%. Mp = 230–231 °C. IR (KBr) ν (cm−1) 3325, 1660 (CO), 1594, 1424, 765, 621. 1H NMR (400 MHz, CDCl3) δ 7.11 (t, 1H, 3J = 7.8 Hz), 7.56 (dd, 1H, 3J = 7.8 Hz, 3J = 4.8 Hz), 8.01 (d, 1H, 3J = 7.8 Hz), 8.74 (dd, 1H, 3J = 7.8 Hz, 4J = 1.9 Hz), 8.80 (d, 1H, 3J = 6.8 Hz), 8.90 (brs, 1H), 9.04 (dd, 1H, 3J = 4.8 Hz, 4J = 1.9 Hz). 13C NMR (100 MHz, CDCl3) δ 84.4, 121.1, 121.3, 123.5, 124.6, 125.6, 136.4, 136.8, 141.0, 150.8, 154.4, 161.4. HRMS/EI calcd. for C12H7IN2O 321.9603, found 321.9592.
Ethyl 1-[3-(4-iodophenyl)propanoyl]piperidine-4-carboxylate 9
To a solution of 4-iododihydrocinamic acid43 (5 g, 18.11 mmol) in DCM (50 mL) was added at room temperatue HOBt (3.67 g, 27.17 mmol), EDCI (5.21 g, 27.17 mmol), NEt3 (3.79 mL, 27.17 mmol) and ethyl isonipecotate (3.07 mL, 19.92 mmol). The solution was allowed to stir at room temperature for 24 h, evaporated to dryness and purified by silica gel chromatography using cyclohexane/EtOAc 9/1 and 7/3 as the eluents to afford 9 (6.31 g) as white powder. Yield: 84%. Mp = 73–74 °C. IR (KBr) ν (cm−1) 2953, 2930, 1729 (CO), 1644 (CO), 1447, 1178, 1040, 1006, 811. 1H NMR (400 MHz, CDCl3) δ 1.26 (t, 3H, 3J = 6.8 Hz), 1.55-1.65 (m, 2H), 1.90 (m, 2H), 2.49 (m, 1H), 2.58 (d, 2H, 3J = 7.8 Hz), 2.79 (m, 1H), 2.91 (d, 2H, 3J = 7.8 Hz), 3.03 (m, 1H), 3.74 (m, 1H), 4.14 (t, 2H, 3J = 6.8 Hz), 4.42 (m, 1H), 6.97 (d, 2H, 3J = 8.3 Hz), 7.60 (d, 2H, 3J = 8.3 Hz). 13C NMR (100 MHz, CDCl3) δ 14.2, 27.8, 28.3, 30.8, 34.7, 40.9, 41.0, 44.7, 60.6, 91.2, 130.5 (2C), 137.5 (2C), 140.9, 170.1, 174.1. HRMS/EI calcd. for C17H22INO3 415.0644, found 415.0634.
{1-[3-(4-iodophenyl)propyl]piperidin-4-yl}methanol 10
To a solution of 9 (1.31 g, 3.15 mmol) in anhydrous THF (20 mL) at −5 °C was added DIBALH 1M solution in n-hexane (12.62 mL, 12.62 mmol). The mixture was allowed to stir at room temperature for 15 h, carefully hydrolyzed with water (30 mL), extracted with EtOAc (3× 50 mL), dried with MgSO4, filtered and evaporated. The crude product was then purified by silica gel chromatography using cyclohexane/EtOAc 9/1 and 7/3 as the eluents to afford 10 (0.96g) as a colorless oil. Yield: 85%. IR (KBr) ν (cm−1) 3400 (OH), 2922, 1728, 1483, 1042, 1006, 797. 1H NMR (400 MHz, CDCl3) δ 1.26 (d, 1H, 3J = 5.9 Hz), 1.26 (m, 2H), 1.70-1.80 (m, 5H), 1.92 (m, 2H), 2.32 (t, 2H, 3J = 7.8 Hz), 2.56 (t, 2H, 3J = 7.8 Hz), 2.92 (m, 2H), 3.49 (d, 2H, 3J = 6.8 Hz), 6.93 (d, 2H, 3J = 8.3 Hz), 7.58 (d, 2H, 3J = 8.3 Hz). 13C NMR (100 MHz, CDCl3) δ 28.6, 28.8 (2C), 33.3, 38.6, 53.5 (2C), 58.2, 67.9, 92.0, 130.5 (2C), 137.3 (2C), 141.9. HRMS/EI calcd. for C15H22INO 359.0746, found 359.0759.
4-Iodo-6-(1-propylpiperidin-4-yl)methyloxyphenanthridine 11a
Starting from 8a (0.47 g, 1.46 mmol), using general procedure A and cyclohexane/ethyl acetate 8/2 as the eluent for the chromatography, 11a was obtained as a yellow powder (0.51 g). Yield: 76%. Mp = 87–89 °C. IR (KBr) ν (cm−1) 2926, 1591, 1396, 1339, 1136, 751. 1H NMR (400 MHz, CDCl3) δ 0.91 (t, 3H, 3J = 7.3 Hz), 1.51-1.64 (m, 4H), 1.93-2.09 (m, 5H), 2.31 (t, 2H, J = 7.8 Hz), 3.02 (d, 2H, J = 10.7 Hz), 4.63 (d, 2H, 3J = 6.8 Hz), 7.19 (t, 1H, J = 6.8 Hz), 7.67 (t, 1H, 3J = 7.8 Hz), 7.82 (t, 1H, 3J = 7.8 Hz), 8.20 (d, 1H, 3J = 7.8 Hz), (t, 2H, 3J = 7.3 Hz), 8.49 (d, 1H, 3J = 8.8 Hz). 13C NMR (100 MHz, CDCl3) δ 12.1, 20.2, 29.2 (2C), 35.6, 53.5 (2C), 61.1, 71.2, 101.8, 120.2, 122.0, 122.6, 123.0, 125.2, 125.5, 127.8, 131.1, 132.9, 134.9, 138.9, 159.5. LC-MS (ESI): tR = 5.84 min; [M+H]+: 461.28. HRMS/EI calcd. for C22H25IN2O 460.1012, found 460.1008.
7-Iodo-5-(1-propylpiperidin-4-yl)methyloxybenzo[h]-1,6-naphthyridine 11b
Starting from 8b (0.16 g, 0.5 mmol), using general procedure A and cyclohexane/ethyl acetate 8/2 with 5% of NEt3 as the eluent for the chromatography, 11b was obtained as a yellow powder (0.16 g). Yield: 71%. Mp = 120–122 °C. IR (KBr) ν (cm−1) 2917, 1603, 1588, 1341, 1093, 762. 1H NMR (400 MHz, CDCl3) δ 0.91 (t, 3H, 3J = 7.3 Hz), 1.51-1.59 (m, 4H), 1.91-2.08 (m, 5H), 2.32 (m, 2H), 3.02 (d, 2H, J = 8.7 Hz), 4.63 (d, 2H, 3J = 5.8 Hz), 7.27 (t, 1H, J = 7.8 Hz), 7.59 (dd, 1H, 3J = 7.8 Hz, 3J = 4.9 Hz), 8.28 (d, 1H, 3J = 8.7 Hz), 8.64 (dd, 1H, 3J = 7.8 Hz, 4J = 1.9 Hz), 8.96 (m, 1H), 9.11 (dd, 1H, 3J = 4.9 Hz, 4J = 1.9 Hz). 13C NMR (100 MHz, CDCl3) δ 12.0, 20.0, 29.1 (2C), 35.5, 53.5 (2C), 61.0, 71.5, 100.6, 115.2, 122.7, 124.1, 124.3, 125.9, 133.1, 140.4, 144.1, 150.7, 153.2, 158.8. LC-MS (ESI): tR = 5.22 min; [M+H]+: 462.22. HRMS/EI calcd. for C21H24IN3O 461.0964, found 461.0970.
6-{1-[3-(4-Iodophenyl)propyl]piperidin-4-yl}methyloxyphenanthridine 11C
Starting from 3a (0.5 g, 2.56 mmol), using general procedure A and cyclohexane/ethyl acetate 8/2 as the eluent for the chromatography, 11C was obtained as a colorless oil (0.54 g). Yield: 39%. IR (KBr) ν (cm−1) 2925, 1637, 1620, 1486, 1317, 759, 727. 1H NMR (400 MHz, CDCl3) δ 1.58 (m, 2H), 1.79-2.05 (m, 7H), 2.37 (t, 2H, 3J = 7.8 Hz), 2.59 (t, 2H, 3J = 6.7 Hz), 3.00 (d, 2H, 3J = 10.7 Hz), 4.50 (d, 2H, 3J = 5.8 Hz), 6.95 (d, 2H, 3J = 8.8 Hz), 7.48 (d, 1H, 3J = 6.8 Hz), 7.59 (d, 2H, 3J = 8.8 Hz), 7.63 (m, 2H), 7.82 (d, 1H, 3J = 6.8 Hz), 7.86 (d, 1H, J = 8.8 Hz), 8.38 (d, 1H, 3J = 6.8 Hz), 8.42 (d, 1H, 3J = 7.8 Hz), 8.51(d, 1H, 3J = 7.8 Hz). 13C NMR (100 MHz, CDCl3) δ 28.6, 29.3 (2C), 33.3, 35.8, 53.6 (2C), 58.3, 70.4, 90.7, 120.1, 121.8, 122.1, 122.3, 124.3, 125.0, 127.1, 127.7, 128.7, 130.5 (2C), 130.8, 134.7, 137.3 (2C), 141.8, 143.3, 158.9. LC-MS (ESI): tR = 6.33 min; [M+H]+: 537.25. HRMS/ESI calcd. for C28H30IN2O 537.1397, found 537.1401.
7-Fluoro-5-{1-[3-(4-iodophenyl)propyl]piperidin-4-yl}methyloxybenzo[h]-1,6-naphthyridine 11d
Starting from 3v (0.5 g, 2.33 mmol), using general procedure A and cyclohexane/ethyl acetate 8/2 as the eluent for the chromatography, 11d was obtained as a white powder (0.53 g). Yield: 41%. Mp = 142–144 °C. IR (KBr) ν (cm−1) 2925, 1603, 1589, 1354, 1330, 1315, 1106, 794, 770. 1H NMR (400 MHz, CDCl3) δ 1.59 (m, 2H), 1.78-2.05 (m, 7H), 2.37 (t, 2H, 3J = 7.8 Hz), 2.59 (t, 2H, 3J = 6.7 Hz), 2.99 (d, 2H, 3J = 10.7 Hz), 4.56 (d, 2H, 3J = 5.8 Hz), 6.95 (d, 2H, 3J = 8.8 Hz), 7.41-7.49 (m, 2H), 7.56-7.62 (m, 3H), 8.64 (dd, 1H, J = 7.8 Hz, J = 1.9 Hz), 8.73 (dd, 1H, 3J = 8.3 Hz, 4J = 1.9 Hz), 9.11 (dd, 1H, 3J = 4.9 Hz, 4J = 1.9 Hz). 13C NMR (100 MHz, CDCl3) δ 28.5, 29.2 (2C), 33.3, 35.8, 53.5 (2C), 58.2, 71.0, 90.7, 115.6, 115.7 (d, 2J = 19 Hz), 119.2, 122.7, 124.4 (d, J = 7 Hz), 125.4 (d, J = 1 Hz), 130.5 (2C), 133.1, 134.1 (d, 2J = 11 Hz), 137.2 (2C), 141.8, 150.2 (d, J = 3 Hz), 153.2, 157.0 (d, 1J = 250 Hz), 158.5. LC-MS (ESI): tR = 6.04 min; [M+H]+: 556.38. HRMS/ESI calcd. for C27H28FIN3O 556.1256, found 556.1260.
General procedure B for the synthesis of stannylated compounds 12a,b,d
In a schlenk flask under nitrogen, were introduced, toluene (4 mL), water (0.4 mL), Pd(OAc)2 (0.025 mmol) and PPh3 (0.05 mmol). The mixture was heated at 50 °C for 0.3 h and the iodinated derivative (11a, 11b or 11d ; 0.5 mmol in 7 mL of toluene) and hexa-n-butylditin (0.75 mmol) were added. The mixture was heated at 90 °C for 16 h, filtered on celite and evaporated in vacuo. The obtained crude oil was then purified by chromatography on silica gel using Et2O and Et2O/NEt3 (99/1) as the eluents.
6-(1-Propylpiperidin-4-yl)methyloxy-4-(tributylstannyl)phenanthridine 12a
Starting from 11a and following general procedure B, 12a was obtained as a colorless oil (140 mg). Yield: 45%. IR (KBr) ν (cm−1) 2955, 2922, 2870, 2851, 1589, 1459, 1339, 1311, 761. 1H NMR (400 MHz, CDCl3) δ 0.85 (t, 6H, J = 6.3 Hz), 0.92 (t, 6H, J = 6.8 Hz), 1.16-1.20 (m, 4H), 1.27-1.43 (m, 10H), 1.50-1.62 (m, 10H), 1.97 (d, 2H, J = 9.7 Hz), 2.06 (t, 1H, J = 11.2 Hz), 2.36 (t, 2H, J = 7.8 Hz), 3.05 (d, 2H, J = 10.7 Hz), 4.48 (d, 2H, J = 5.8 Hz), 7.46 (t, 1H, 3J = 7.8 Hz), 7.61 (t, 1H, 3J = 7.8 Hz), 7.78 (m, 2H), 8.38 (t, 2H, J = 7.8 Hz), 8.50 (d, 1H, 3J = 8.7 Hz). 13C NMR (100 MHz, CDCl3) δ 10.2 (3C), 12.0, 13.7 (3C), 20.0, 27.4 (3C), 29.1 (2C), 29.4 (3C), 35.7, 53.5 (2C), 61.1, 70.6, 119.9, 121.4, 121.9, 122.3, 124.1, 124.9, 126.9, 130.6, 135.4, 136.9, 143.6, 148.0, 157.7. HRMS/ESI calcd. for C34H53N2OSn 625.3181, found 625.3186.
7-(1-Propylpiperidin-4-yl)methyloxy-5-(tributylstannyl)benzo[h]-1,6-naphthyridine 12b
Starting from 11b and following general procedure B, 12b was obtained as a colorless oil (139 mg). Yield: 44%. IR (KBr) ν (cm−1) 2956, 2924, 2871, 2852, 1603, 1460, 1328, 1153, 773. 1H NMR (400 MHz, CDCl3) δ 0.85 (t, 6H, J = 7.3 Hz), 0.92 (t, 6H, J = 6.8 Hz), 1.16-1.21 (m, 4H), 1.25-1.43 (m, 10H), 1.51-1.63 (m, 10H), 1.95 (d, 2H, J = 10.7 Hz), 2.06 (t, 1H, J = 11.2 Hz), 2.36 (t, 2H, J = 7.8 Hz), 3.06 (d, 2H, J = 10.7 Hz), 4.48 (d, 2H, J = 5.8 Hz), 7.51-7.56 (m, 2H), 7.86 (dd, 1H, 3J = 6.8 Hz, 4J = 1.9 Hz), 8.62 (dd, 1H, 3J = 7.8 Hz, 4J = 1.9 Hz), 8.92 (d, 1H, 3J = 7.8 Hz), 9.09 (dd, 1H, 3J = 4.9 Hz, 4J = 1.9 Hz). 13C NMR (100 MHz, CDCl3) δ 10.2 (3C), 12.0, 13.5 (3C), 20.0, 27.4 (3C), 29.1 (3C), 29.3 (2C), 35.6, 53.4 (2C), , 61.1, 70.9, 115.1, 122.0, 122.7, 123.9, 124.7, 132.9, 138.6, 142.9, 149.7, 151.3, 152.8, 157.3. HRMS/ESI calcd. for C33H52N3OSn 626.3133, found 626.3136.
7-Fluoro-5-{1-[3-(4-tributylstannylphenyl)propyl]piperidin-4-yl}methyloxybenzo[h]-1,6-naphthyridine 12d
Starting from 11d and following general procedure B, 12d was obtained as a colorless oil (147 mg). Yield: 41%. IR (KBr) ν (cm−1) 2954, 2925, 1608, 1591, 1462, 1331, 1314, 1240, 1151, 1083, 769. 1H NMR (400 MHz, CDCl3) δ 0.85 (t, 9H, J = 7.3 Hz), 0.92 (t, 6H, J = 8.1 Hz), 1.18-1.29 (m, 8H), 1.42-1.52 (m, 6H), 1.79-1.96 (m, 7H), 2.34 (t, 2H, J = 7.8 Hz), 2.06 (t, 2H, J = 7.8 Hz), 2.94 (d, 2H, J = 11.2 Hz), 4.48 (d, 2H, J = 6.1 Hz), 7.10 (d, 2H, 3J = 7.8 Hz), 7.30 (d, 2H, 3J = 7.8 Hz), 7.34-7.41 (m, 2H), 7.52 (dd, 1H, 3J = 8.2 Hz, 3J = 4.58 Hz), 8.57 (dd, 1H, 3J = 8.2 Hz, 4J = 1.8 Hz), 8.64 (m, 1H), 9.04 (dd, 1H, 3J = 4.5 Hz, 4J = 1.8 Hz). 13C NMR (100 MHz, CDCl3) δ 9.5 (3C), 13.6 (3C), 27.3 (3C), 28.7, 29.1 (3C), 29.2 (2C), 33.8, 35.8, 53.5 (2C), 58.6, 71.0, 115.6, 115.6 (d, 2J = 20 Hz), 119.2 (d, J = 4 Hz), 122.8, 124.4 (d, J = 7 Hz), 125.5 (d, J = 2 Hz), 128.1 (2C), 133.2, 134.1 (d, 2J = 11 Hz), 136.4 (2C), 138.5, 141.8, 150.3 (d, J = 4 Hz), 153.3, 157.0 (d, 1J = 250 Hz), 158.6. HRMS/ESI calcd. for C39H55FN3OSn 720.3353, found 720.3356.
Pharmacological Assay and Screen
Binding of all compounds to native 5-HT4R from guinea pig was determined using the method of Grossman.44 For membrane preparations male guinea pigs (300–350 g, Charles River) were subjected to euthanasia by cervical dislocation and decapitated. Brains were rapidly removed at 4 °C and striatal regions carefully dissected and pooled. The tissues were then suspended in 10 volumes of HEPES buffer 50 mM pH 7.4 at 4 °C. After homogenization at 4 °C (Ultra-Turrax, maximal speed, 15 sec), and ultracentrifugation (23,000 × g, 60 min, 4 °C), the pellet was resuspended in 10 volumes of HEPES buffer 50 mM pH 7.4 at 4 °C in order to obtain a tissue concentration of about 100 mg protein/mL. The protein concentration was determined by the method of Lowry45 using bovine serum albumin as standard. For radioligand binding studies, 600 µg of membrane were incubated in duplicate at 37 °C for 30 min with [3H]GR 113808 (Perkin Elmer), fixed concentration of compound and HEPES buffer 50 mM pH 7.4 at 37 °C. Incubation was terminated by rapid vacuum filtration through 0.5% polyethylenimine-presoaked Whatman GF/B filters (Alpha Biotech) using a Brandel Cell Harvester. Filters were subsequently washed three times with 4 ml of HEPES buffer 50 mM pH 7.4 at 4 °C. The method was validated from saturation studies: 6 concentrations of [3H]GR 113808 were used to give final concentrations of 0.02–0.8 nM, non-specific binding of [3H]GR 113808 was defined in the presence of 30 µM serotonin to determine the Kd and the Bmax. For competition studies, [3H]GR 113808 was used to give a final concentration of 0.1 nM. Percentages of inhibition of the binding of [3H]GR 113808 were obtained for concentrations of 10−6 and 10−8 M of the ligands tested. For some of these compounds, affinity constants were calculated from 5-point inhibition curves using the EBDA-Ligand software, and expressed as Ki ± SD.
Ligands 4i, 4k, 4l, 4m, 4t, and 4u were submitted to the National Institute of Mental Health Psychoactive Drug Screening Program (NIMH/PDSP) for assessment of binding affinity to human recombinant 5-HT4 receptors and to other serotonin receptors (5-HT1A-E, 5-HT2A-C, 5-HT3, 5-HT5A, 5-HT6, 5-HT7). For h5-HT4R Ki determinations, [3H]GR 113808 was used as hot ligand and GR 113808 as reference. 5-HT4R membrane was made with HEK T cells transiently transfected with human 5-HT4 DNA. Binding protocol is the same as other 5-HT subtypes in the PDSP assay protocol book. Selected ligands 4k, 4l, 4m, and 4u were also assessed for agonist/partial agonist activity and for antagonist activity. For 5-HT4R mediated Gs activation, cAMP was measured using GloSensor tech from Promega, with serotonin as a reference agonist according to a literature procedure.46 For other experimental details please refer to the PDSP web site http://pdsp.med.unc.edu/ and click on "Binding Assay".
Other selected ligands 4v, 11a, 11b and 11d were evaluated for binding to human 5-HT4 and other serotonin receptors (5-HT1A-E, 5-HT2A-C, 5-HT3, 5-HT5A, 5-HT6, 5-HT7) as well as for intrinsic activity at CEREP. Detailed assay protocols are available at the CEREP web site (http://www.cerep.com).
Radiosynthesis of 13a
0.05 M NaOH solution containing 165 µCi of carrier free Na125I (PerkinElmer – 1 µL) was added to a mixture made of 12a (3 µg) in EtOH (1.5 µL), glacial acetic acid (5 µL) and 30% hydrogen peroxide solution (5 µL). After incubation at room temperature for 20 min, HPLC phase (MeCN/water 80/20, 10 mM AcOH buffer pH5 - 350 µL) was added and 13a was isolated by an isocratic HPLC using a Bondclone C18 10 µm 300X7.8 mm, Phenomenex column at 3 mL.min−1. The apparent specific activity was determined thanks to a calibration curve measured using growing amounts of the cold reference compound and to UV monitoring during the HPLC purification step. The overall radiochemical yield was 56%. The apparent specific radioactivity was 240 Ci/mmol (11% of the carrier free specific activity).
Radiosynthesis of 13b
0.05 M NaOH solution containing 128 µCi of carrier free Na125I (PerkinElmer – 1 µL) was added to a mixture made of 12b (3 µg) in EtOH (1.5 µL), glacial acetic acid (5 µL) and 30% hydrogen peroxide solution (5 µL). After incubation at room temperature for 20 min, HPLC phase (MeCN/water 80/20, 10 mM AcOH buffer pH5 - 350 µL) was added and 13b was isolated by an isocratic HPLC using a Bondclone C18 10 µm 300X7.8 mm, Phenomenex column at 3 mL.min−1. The apparent specific activity was determined thanks to a calibration curve measured using growing amounts of the cold reference compound and to UV monitoring during the HPLC purification step. The overall radiochemical yield was 70%. The apparent specific radioactivity was 110 Ci/mmol (5% of the carrier free specific activity).
Radiosynthesis of 13d
0.05 M NaOH solution containing 210 µCi of carrier free Na125I (PerkinElmer – 1 µL) was added to a mixture made of 12b (10 µg) in EtOH (1 µL), glacial acetic acid (5 µL) and 30% hydrogen peroxide solution (5 µL). After incubation at room temperature for 20 min, HPLC phase (MeCN/water 80/20, 10 mM AcOH buffer pH5 - 350 µL) was added and 13d was isolated by an isocratic HPLC using a Bondclone C18 10 µm 300X7.8 mm, Phenomenex column at 3 mL.min−1. The apparent specific activity was determined thanks to a calibration curve measured using growing amounts of the cold reference compound and to UV monitoring during the HPLC purification step. The overall radiochemical yield was 85%. The apparent specific radioactivity was 280 Ci/mmol (13% of the carrier free specific activity).
Radiosynthesis of [125I]1
0.05 M NaOH (1 µL) solution containing 210 µCi of carrier free Na125I (PerkinElmer) was added to a mixture made of 1 trimethylstannyl precursor (ERAS Labo, France 100 µg) in a mixture containing EtOH (10 µL), glacial acetic acid (5 µL) and 30% hydrogen peroxide solution (5 µL). After an incubation at room temperature for 20 min, HPLC phase (MeCN/water 30/70, 10 mM H3PO4 350 µL) was added and [125I]1 was isolated by an isocratic HPLC using a Bondclone C18 10 µm 300X7.8 mm, Phenomenex column at 3 mL.min−1. The apparent specific activity was determined thanks to a calibration curve measured using growing amounts of the cold reference compound and to UV monitoring during the HPLC purification step. After collection, pooling and evaporation of the corresponding fractions, the overall radiochemical yield of iodination with 125I was 70%. The apparent specific radioactivity was 146 Ci/mmol (6.6% of the carrier free specific activity).
In vitro competition experiments
For each in vitro autoradiographic experiment, frozen rat brain coronal sections at the level of the striatum and accumbens nucleus were thawed and dried, preincubated in 50 mM Tris-HCl pH = 7.4 for 5 min at room temperature, and then incubated in the same buffer with given concentrations of the radioligand for 30 min at room temperature. During this step, competition experiments were done by incubating the sections with 100 pM of [125I]1 and various concentrations of 11d. After incubation, sections were washed three times for 5 minutes in ice-cold buffer, dipped in ice-cold deionised water, and rapidly dried under a cold air stream. The labelled sections were exposed on phosphorimaging plates overnight before detection with Fujifilm BAS scanner.
In vivo competition experiments
For the in vivo competition experiment, a mouse was injected intravenously with a saline solution containing a dose of 50 µg/kg of 11d and 30 µCi [125I]1 (146 Ci/mmol specific activity, 6.6% of the carrier free). The animal was then sacrificed with CO2 and 20 µm thin cryosections were collected in 5HT4R-rich regions (striatum, accumbens nucleus, olfactive tubercles), and finally exposed overnight on phosphorimaging plates before detection with Fujifilm BAS scanner.
Molecular Modeling study
Receptor Model
First, the sequence of the human 5-HT4R was retrieved from the UniProt Knowledgebase (UniProtKB)47 (ID : Q712M9_HUMAN). Using screening methods like FUGUE,48 SP3,49 PSIBLAST,50,51 HHSEARCH52 and the @tome-2 server53, the β2 adrenergic receptor has been identified as the best 3D experimental template for the homology modeling of the 5-HT4R (Sequence identity = 40%). The high-resolution (2.4 Å) crystal structure of the human β2 adrenergic receptor (β2AR)-T4 lysozyme fusion protein bound to the carazolol (PDB: 2RH1)33 was used as the 3D template. The alignment between the two sequences was manually optimized to avoid insertions and deletions in secondary structure elements (see supporting info for final sequence alignment). Disulfide bond: C93-C184 between the transmembrane helix 3 (TM3) and the extracellular loop (ECL2) was conserved. This alignment was used as the basis for the homology modeling with the Modeller software.54 The resulting model was then evaluated by methods like verify3D55 and Eval23D.56
Docking studies
The docking of the compounds into the 5-HT4R was carried out with the GOLD program (v5.0) using the default parameters.57,58 This program applies a genetic algorithm to explore conformational spaces and ligand binding modes. To evaluate the proposed ligand positions, the ChemScore fitness function was applied in these docking studies. The binding site in the 5-HT4R model was defined as a 10Å sphere centred on the aspartic acid residue Asp100. Because the mutagenesis studies have shown that the interaction between the positively ionisable amine of ligands and Asp100 of 5-HT4R is crucial for ligand binding, a hydrogen bond constraint between positively ionisable amine ligand and OD atom of Asp100 was used during the docking.36 Furthermore, special attention was paid during the docking procedure to the following amino acids in the binding site, which were kept flexible: Arg96, Asp100, Thr104, Tyr192, Ser197 and Trp294. For 4v docking a second hydrogen bond constraint between the tricycle nitrogen and the hydroxyle group of Tyr192 was added. For the two crystal conformers available for each compound, a dozen separate docking procedures were carried out and analyzed.
Supplementary Material
ACKNOWLEDGMENT
We thank the National Institute of Mental Health's Psychoactive Drug Screening Program, Contract # HHSN-271-2008-00025-C (NIMH PDSP) which generously provided Ki determinations and functional assays. The NIMH PDSP is Directed by Bryan L. Roth MD, PhD at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscol at NIMH, Bethesda MD, USA. We thank Aurélien Lesnard for helpful management of biological results. This work was in part supported by the Swiss National Science Foundation (grant no. 310030_120369).
Footnotes
X-ray crystallographic data of compounds 4c, 4d, 4g, 4h, 4j, 4m, 4p, 4q, 4r, 4v and 11d. Amino-acid sequences alignment of 5-HT4R and human β2-adrenergic receptor. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Dumuis A, Bouhelal R, Sebben M, Bockaert J. A 5-HT receptor in the central nervous system, positively coupled with adenylate cyclase, is antagonized by ICS 205930. Eur. J. Pharmacol. 1988;146:187–188. doi: 10.1016/0014-2999(88)90503-1. [DOI] [PubMed] [Google Scholar]
- 2.Dumuis A, Bouhelal R, Sebben M, Cory R, Bockaert J. A non-classical 5-hydroxytryptamine receptor positively coupled with adenylate cyclase in the central nervous system. Mol. Pharmacol. 1988;34:880–887. [PubMed] [Google Scholar]
- 3.Bureau R, Boulouard M, Dauphin F, Lezoualc’h F, Rault S. Review of 5-HT4R ligands: state of arts and clinical applications. Curr. Top. Med. Chem. 2010;10:527–553. doi: 10.2174/156802610791111551. [DOI] [PubMed] [Google Scholar]
- 4.Lucas G. Serotonin receptors type 4: a new hope ? Curr. Drug Targets. 2009;10:1085–1095. doi: 10.2174/138945009789735200. [DOI] [PubMed] [Google Scholar]
- 5.Beattie DT, Smith JAM. Serotonin pharmacology in the gastrointestinal tract: a review. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2008;377:181–203. doi: 10.1007/s00210-008-0276-9. [DOI] [PubMed] [Google Scholar]
- 6.Qvigstad E, Brattelid T, Sjaastad I, Andressen KW, Krobert KA, Birkeland JA, Sejersted OM, Kaumann AJ, Skomedal T, Osnes JB, Levy FO. Appearance of a ventricular 5-HT4 receptor-mediated inotropic response to serotonin in heart failure. Cardiovasc. Res. 2005;65:869–878. doi: 10.1016/j.cardiores.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 7.Eglen RM, Wong EHF, Dumuis A, Bockaert J. Central 5-HT4 receptors. TIPS. 1995;16:391–398. doi: 10.1016/s0165-6147(00)89081-1. [DOI] [PubMed] [Google Scholar]
- 8.Matsumoto M, Togashi H, Mori K, Hueno K, Ohashi S, Kogima T, Yoshioka M. Evidence for involvement of central 5-HT4 receptors in cholinergic function associated with cognitive processes: behavioural, electrophysiological, and neurochemical studies. J. Pharmacol. Exp. Ther. 2001;296:676–682. [PubMed] [Google Scholar]
- 9.Meneses A, Hong E. Effects of 5-HT4 receptor agonists and antagonists in learning. Pharmacol., Biochem. Behav. 1997;56:347–351. doi: 10.1016/s0091-3057(96)00224-9. [DOI] [PubMed] [Google Scholar]
- 10.Lezoualc’h F. 5-HT4 receptor and Alzheimer’s disease: the amyloid connection. Exp. Neurol. 2007;205:325–329. doi: 10.1016/j.expneurol.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 11.Cho S, Hu Y. Activation of 5-HT4 receptors inhibits secretion of beta-amyloid peptides and increases neuronal survival. Exp. Neurol. 2007;203:274–278. doi: 10.1016/j.expneurol.2006.07.021. [DOI] [PubMed] [Google Scholar]
- 12.Jean A, Conductier G, Manrique C, Bouras C, Berta P, Hen R, Charnay Y, Bockaert J, Compan V. Anorexia induced by activation of serotonin 5-HT4 receptors is mediated by increases in CART in the nucleus accumbens. Proc. Natl. Acad. Sci. USA. 2007;104:16335–16340. doi: 10.1073/pnas.0701471104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lucas G, Rymar W, Du J, Mnie-Filali O, Bisgaard C, Manta S, Lambas-Senas L, Wiborg O, Haddjeri N, Pineyro G, Sadikot AF, Debonnel G. Serotonin(4) (5-HT(4)) receptor agonists putative antidepressant with a rapid onset of action. Neuron. 2007;55:712–725. doi: 10.1016/j.neuron.2007.07.041. [DOI] [PubMed] [Google Scholar]
- 14.Manabe N, Wong BS, Camilleri M. New generation 5-HT4 recptor agonists: potential for for treatment of gastrointestinal motility disorders. Expert Opin. Investig. Drugs. 2010;19:765–775. doi: 10.1517/13543784.2010.482927. [DOI] [PubMed] [Google Scholar]
- 15.Moser PC, E, Bergis O, Jegham S, Lochead A, Duconseille E, Terranova J-P, Caille D, Berque-Bestel I, Lezoualc’h F, Fischmeister R, Dumuis A, Bockaert J, George P, Soubrie P, Scatton B. SL65.0155, a novel 5-hydroxytryptamine4 receptor partial agonist with potent cognition-enhancing properties. J. Pharmacol. Exp. Ther. 2002;302:731–742. doi: 10.1124/jpet.102.034249. [DOI] [PubMed] [Google Scholar]
- 16.Mohler EG, Shacham S, Noiman S, Lezoualc’h F, Robert S, Gastineau M, Rutkowski J, Marantz Y, Dumuis A, Bockaert J, Gold PE, Ragozzino ME. VRX-03011, a novel 5-HT4 agonist, enhances memory and hippocampal acetylcholine efflux. Neuropharmacology. 2007;53:563–573. doi: 10.1016/j.neuropharm.2007.06.016. [DOI] [PubMed] [Google Scholar]
- 17.Wong DF, Gründer G, Brasić JR. Brain imaging research: does the science serve clinical practice ? Int. Rev. Psychiatry. 2007;19:541–558. doi: 10.1080/09540260701564849. [DOI] [PubMed] [Google Scholar]
- 18.Gibson RE, Burns HD, Hamill TG, Eng WS, Francis BE, Ryan C. Non-invasive radiotracer imaging as a tool for drug development. Curr. Radiopharm. Des. 2000;6:973–989. doi: 10.2174/1381612003399987. [DOI] [PubMed] [Google Scholar]
- 19.Pike VW, Halldin C, Nobuhara K, Hiltunen J, Mulligan RS, Swahn C-G, Karlsson P, Olsson H, Hume SP, Hirani E, Whalley J, Pilowsky LS, Larson S, Schnell P-O, Ell PJ, Farde L. Radioiodinated SB 207710 as a radioligand in vivo: imaging of brain 5-HT4 receptors with SPET. Eur. J. Nucl. Med. Mol. Imaging. 2003;30:1520–1528. doi: 10.1007/s00259-003-1307-x. [DOI] [PubMed] [Google Scholar]
- 20.Gee AD, Martarello L, Passchier J, Wishart M, Parker C, Matthews J, Comley R, Hopper R, Gunn R. Synthesis and evaluation of [11C]SB207145 as the first in vivo serotonin 5-HT4 receptor radioligand for PET imaging in man. Curr. Radiopharm. 2008;1:110–114. [Google Scholar]
- 21.Kornum BR, Lind NM, Gillings N, Marner L, Andersen F, Knudsen GM. J. Cereb. Blood Flow Metab. 2009;29:186–196. doi: 10.1038/jcbfm.2008.110. [DOI] [PubMed] [Google Scholar]
- 22.Madsen K, Haahr MT, Marner L, Keller SH, Baaré WF, Svarer C, Hasselbalch SG, Knudsen GM. Age and sex effects on 5-HT(4) receptors in the human brain: a [(11)C]SB207145 PET study. J. Cereb. Blood Flow Metab. 2011;31:1475–1481. doi: 10.1038/jcbfm.2011.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marner L, Gillings N, Madsen K, Erritzoe D, Baaré WF, Svarer C, Hasselbalch SG, Knudsen GM. Brain imaging of serotonin 4 receptors in humans with [11C]SB207145-PET. Neuroimage. 2010;50:855–861. doi: 10.1016/j.neuroimage.2010.01.054. [DOI] [PubMed] [Google Scholar]
- 24.Xu R, Hong J, Morse CL, Pike VW. Synthesis, Structure-Affinity Relationships, and Radiolabeling of Selective High-Affinity 5-HT4 Receptor Ligands as Prospective Imaging Probes for Positron Emission Tomography. J. Med. Chem. 2010;53:7035–7047. doi: 10.1021/jm100668r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hinschberger A, Butt S, Lelong V, Boulouard M, Dumuis A, Fauphin F, Bureau R, Pfeiffer B, Renard P, Rault S. New Benzo[h]-1,6-naphthyridine and Azepino[3,2-c]quinoleine Derivatives as selective Antagonists of 5-HT4 Receptors: Binding Profile and Pharmacological Characterization. J. Med. Chem. 2003;46:138–147. doi: 10.1021/jm020954v. [DOI] [PubMed] [Google Scholar]
- 26.Lemaitre S, Lepailleur A, Bureau R, Butt-Gueulle S, Lelong-Boulouard V, Duchatelle P, Boulouard M, Dumuis A, Daveu C, Lezoualc'h F, Pfeiffer B, Dauphin F, Rault S. Novel antagonists of serotonin-4 receptors: Synthesis and biological evaluation of pyrrolothienopyrazines. Bioorg. Med. Chem. 2009;17:2607–2622. doi: 10.1016/j.bmc.2008.11.045. [DOI] [PubMed] [Google Scholar]
- 27.Dubost E, Magnelli R, Cailly T, Legay R, Fabis F, Rault S. General method for the synthesis of substituted phenanthridin-6(5H)-ones using a KOH-mediated anionic ring closure as the key step. Tetrahedron. 2010;66:5008–5016. [Google Scholar]
- 28.See experimental data for details.
- 29.Donohue SR, Varnäs K, Jia Z, Gulyás B, Pike VW, Halldin C. Synthesis and in vitro autoradiographic evaluation of a novel high affinity radioiodinated ligand for imaging brain cannabinoid subtype-1 receptors. Bioorg. Med. Chem. Lett. 2009;19:6209–6212. doi: 10.1016/j.bmcl.2009.08.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaumann AJ. Blockade of human atrial 5-HT4 receptors by GR 113808. Br. J. Pharmacol. 1993;110:1172–1174. doi: 10.1111/j.1476-5381.1993.tb13937.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaumann AJ, Gaster LM, King FD, Brown AM. Blockade of human atrial 5-HT4 receptors by SB 207710, a selective and high affinity 5-HT4 receptor antagonist. Naunyn-Schmiedeberg's Arch. Pharmacol. 1994;349:546–548. doi: 10.1007/BF00169146. [DOI] [PubMed] [Google Scholar]
- 32.Kenakin T. Efficacy as a vector: the relative prevalence and paucity of inverse agonism. Mol. Pharmacol. 2004;66:2–11. doi: 10.1124/mol.65.1.2. [DOI] [PubMed] [Google Scholar]
- 33.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC, Kobilka BK. GPCR Engineering Yields High-Resolution Structural Insights into β2 Adrenergic Receptor Function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 35.Moukhametzianov R, Warne T, Edwards PC, Serrano-Vega MJ, Leslie AG, Tate CG, Schertler GF. Two distinct conformations of helix 6 observed in antagonist-bound structures of a beta1-adrenergic receptor. Proc. Natl, Acad. Sci. USA. 2011;108:8228–8232. doi: 10.1073/pnas.1100185108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rivail L, Giner M, Gastineau M, Berthouze M, Soulier J-L, Fischmeister R, Lezoualc’h F, Maigret B, Sicsic S, Berque-Bestel I. New insights into the human 5-HT4 receptor binding site: exploration of a hydrophobic pocket. Br. J. Pharmacol. 2004;143:361–370. doi: 10.1038/sj.bjp.0705950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Warne A, Moukhametzianov R, Baker JG, Nehme R, Edwards PC, Leslie AGW, Schertler GFX, Tate CG. The structural basis for agonist and partial agonist action on a β1-adrenergic receptor. Nature. 2011;469:241–244. doi: 10.1038/nature09746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu H, Wacker D, Katritch V, Mileni M, Han GW, Vardy E, Liu W, Thompson AA, Huang XP, Carroll FI, Mascarella SW, Westkaemper RB, Mosier PD, Roth BL, Cherezov V, Stevens RC. Structure of the human κ-opioid receptor in complex with JDTic. Nature. 2012;485:327–332. doi: 10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Waterhouse RN. Determination of lipophilicity and its use as a predictor of blood brain barrier penetration of molecular imaging agents. Mol. Imaging Biol. 2005;5:376–389. doi: 10.1016/j.mibio.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 40.Bedjeguelal K, Rabot R, Kaloun El B, Mayer P, Marchand A, Rahier N, Schambel P, Bienayme H. Pyrazolopyridine derivatives as anticancer agent. PCT Int. Appl. WO 2011045344 (A1) 2011 [Google Scholar]
- 41.Kristensen JL, Vedsø V, Begtrup M. Synthesis of Pentacyclic 13-Azadibenzo[a,de]anthracenes via Anionic Cascade Ring Closure. J. Org. Chem. 2003;68:4091–4092. doi: 10.1021/jo0300340. [DOI] [PubMed] [Google Scholar]
- 42.Heiss C, Marzi E, Mongin F, Schlosser M. Remote trimethylsilyl groups interfering with the ortho deprotonation of fluoroarenes and chloroarenes. Eur. J. Org. Chem. 2007:669–675. [Google Scholar]
- 43.Kawaksaki M, Goto M, Kawabata S, Kometani T. The effect of vinyl esters on the enantioselectivity of the lipase-catalysed transesterification of alcohols. Tetrahedron: Asymmetry. 2001;12:585–596. [Google Scholar]
- 44.Grossman CJ, Kilpatrick GJ, Bunc KT. Development of a radioligand binding assay for 5-HT4 receptors in guinea pig and rat brain. Br. J. Pharmacol. 1993;109:618–624. doi: 10.1111/j.1476-5381.1993.tb13617.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the folin phenol reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 46.Allen JA, Yost JM, Setola V, Chen X, Sassano MF, Chzn M, Peterson S, Yadav PN, Huang XP, Feng B, Jensen NH, Che X, Bai X, Frye SV, Wetsel WC, Caron MG, Javitch JA, Roth BL, Jin J. Discovery of β-arrestin-biased dopamine D2 ligands for probing signal transduction pathways essential for antipsychotic efficacy. Proc. Natl, Acad. Sci. USA. 2011;108:18488–18493. doi: 10.1073/pnas.1104807108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jain E, Bairoch A, Duvaud S, Phan I, Redaschi N, Suzek BE, Martin MJ, McGarvey P, Gasteiger E. Infrastructure for the life sciences: design and implementation of the UniProt website. BMC Bioinformatics. 2009;10:136–155. doi: 10.1186/1471-2105-10-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shi J, Blundell TL, Mizuguchi K. FUGUE: sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J. Mol. Biol. 2001;310:243–257. doi: 10.1006/jmbi.2001.4762. [DOI] [PubMed] [Google Scholar]
- 49.Zhou H, Zhou Y. Fold recognition by combining sequence profiles derived from evolution and from depth-dependent structural alignment of fragments. Proteins. 2005;58:321–328. doi: 10.1002/prot.20308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucl. Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schäffer AA, Aravind L, Madden TL, Shavirin S, Spouge JL, Wolf YI, Koonin EV, Altschul SF. Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements. Nucl. Acids Res. 2001;29:2994–3005. doi: 10.1093/nar/29.14.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Soding J. Protein homology detection by HMM-HMM comparison. Bioinformatics. 2005;21:951–960. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- 53.Pons JL, Labesse G. @TOME-2: a new pipeline for comparative modeling of protein–ligand complexes. Nucleic Acids Research. 2009;37(Web Server issue):W485–W491. doi: 10.1093/nar/gkp368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eswar N, Eramian D, Webb B, Shen M, Sali A. Protein structure modeling with MODELLER. Methods Mol. Biol. 2008;426:145–159. doi: 10.1007/978-1-60327-058-8_8. [DOI] [PubMed] [Google Scholar]
- 55.Eisenberg D, Lüthy R, Bowie JU. VERIFY3D: assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997;277:396–404. doi: 10.1016/s0076-6879(97)77022-8. [DOI] [PubMed] [Google Scholar]
- 56.Gracy J, Chiche L, Sallantin J. Improved alignment of weakly homologous protein sequences using structural information. Protein Eng. 1993;6:821–829. doi: 10.1093/protein/6.8.821. [DOI] [PubMed] [Google Scholar]
- 57.Jones G, Willett P, Glen RC. Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J. Mol. Biol. 1995;245:43–53. doi: 10.1016/s0022-2836(95)80037-9. [DOI] [PubMed] [Google Scholar]
- 58.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.