

Abstract
Juxtanodin, also called ermin, is an F-actin-binding protein expressed by oligodendrocytes, the myelin-forming cells of the central nervous system. While juxtanodin carries a short conserved F-actin-binding segment at its C terminus, it otherwise shares no similarity with known protein sequences. We carried out a structural characterization of recombinant juxtanodin in solution. Juxtanodin turned out to be intrinsically disordered, as evidenced by conventional and synchrotron radiation CD spectroscopy. Small-angle X-ray scattering indicated that juxtanodin is a monomeric, highly elongated, unfolded molecule. Ensemble optimization analysis of the data suggested also the presence of more compact forms of juxtanodin. The C terminus was a strict requirement for co-sedimentation of juxtanodin with microfilaments, but juxtanodin had only mild effects on actin polymerization. The disordered nature of juxtanodin may predict functions as a protein interaction hub, although F-actin is its only currently known binding partner.
Within the vertebrate nervous system, myelin sheaths form compact and essential electrical insulators for axons, speeding up the transmission of nerve impulses. In the central nervous system, these highly specialized, lipid-rich structures are formed by oligodendrocytes that wrap their plasma membranes multiple times around axons. Myelin consists of two distinct compartments: tightly packed compact myelin and non-compact myelin, forming the abaxonal and adaxonal spaces as well as the paranodal loops. Non-compact myelin contains cytoskeletal elements, including microtubules and actin filaments, as well as their regulatory proteins. The ability of oligodendrocytes to mature and myelinate axons is highly dependent on the reorganization of the cell cytoskeleton1. The remodeling of the actin microfilament network is crucial in the initial stages of myelination, when oligodendrocyte processes are formed and branched in order to explore the cellular environment2. In oligodendrocytes, a number of actin-binding proteins, including Mayven3, actin-interacting protein-14, N-WASP and Arp2/35 regulate actin assembly.
Juxtanodin, also called ermin, is a 282-residue actin-binding protein, which co-localizes with filamentous actin (F-actin) and promotes the formation of cell protrusions during the differentiation of oligodendrocytes6,7,8. Juxtanodin shares no sequence similarity with other proteins, apart from its highly conserved C-terminal actin-binding segment that is almost identical to those found in the ERM (ezrin, radixin, moesin) proteins. Juxtanodin, however, lacks the typical membrane-associating FERM (4.1, ezrin, radixin, moesin) domain of ERM proteins8. The function of juxtanodin is regulated through the phosphorylation of Ser278 within the F-actin binding site, which diminishes its morphological effects in oligodendrocytes9. Thus far, the only known interaction partner of juxtanodin is F-actin, while an interaction with globular actin (G-actin) has not been observed9. Juxtanodin has been reported to increase the stability of actin filaments9, but its actual role in the regulation of actin dynamics at the molecular level, as well as its structural properties, are still unknown.
In the current work, using multiple biophysical techniques, we show that juxtanodin is an intrinsically disordered protein and that the conserved actin-binding segment is crucial for F-actin binding. Juxtanodin does not affect the organization of actin filaments, although a mild inhibitory effect on actin polymerization is observed.
Results
Bioinformatic analyses indicate multiple disordered regions in juxtanodin
The sequence of juxtanodin is unrelated to other known proteins, except for its C-terminal F-actin-binding region8, which is highly conserved between juxtanodin and the ERM proteins. Secondary structure predictions suggest that juxtanodin has long unfolded regions and only a few short helices (Figure 1a). The central part of juxtanodin (residues 172–198) also contains a highly acidic non-complex stretch. Typically, intrinsically disordered proteins (IDPs) have relatively large segments of charged and polar amino acids10. A plot of mean residue charge against hydropathy10 indicates that juxtanodin clearly locates in the disordered protein cluster (Figure 1c); it is unlikely to have a hydrophobic core.
Figure 1. Juxtanodin is predicted to be largely disordered.

(a) JPred secondary structure prediction. H-helix; E-strand. The acidic segment (red) and the conserved C terminal region (cyan) are coloured. (b) PONDR suggests the presence of two ordered segments in the middle region of juxtanodin. The coloured bars locate the acidic region (red) and the C terminus (cyan). (c) The charge/hydropathy plot clearly puts juxtanodin (green) into the cluster of disordered proteins. (d) Schematic view of full-length juxtanodin and its truncated variants produced in the current study. The F-actin binding site is coloured in cyan. Additional predictions are shown in Supplementary Information.
FoldIndex suggested juxtanodin to be completely disordered (Supplementary Figure 1) whereas PONDR-FIT predicted two ordered regions between residues 100–120 and 150–170 (Figure 1b). On the other hand, GlobPlot predicted a folded domain, and DisoPred gave results not necessarily indicative of a disordered protein (Supplementary Figure 1). However, all the used methods indicated the presence of extended disordered regions in juxtanodin. Experimental approaches were, thus, applied to gain more insight into juxtanodin folding and structure.
Juxtanodin is non-globular and monomeric in solution
To investigate the structural and functional properties of juxtanodin, we expressed and purified recombinant full-length juxtanodin (1–282) and three different truncated variants, containing residues 1–170 (N-terminal region before the acidic domain), 103–282 (C-terminal and acidic regions, plus segments before the acidic region predicted to form secondary structures) or 172–282 (C-terminal region including the acidic region) (Figure 1d). As previously shown8, the mobility of juxtanodin in SDS-PAGE is atypical. The same phenomenon was observed here with all untagged juxtanodin fragments (data not shown, see also Fig. 6a), which suggests the presence of unfolded regions. It is a common property of an unfolded protein, which lacks a hydrophobic core, to bind SDS anomalously and to have a lower mobility in SDS-PAGE than expected based on the molecular weight11.
Juxtanodin co-localizes with actin filaments in oligodendrocytes9. A great portion of the known F-actin-binding proteins are dimeric, which enables them to crosslink filaments12. We studied the oligomeric state of juxtanodin in solution using size exclusion chromatography and static light scattering (SLS) in combination. Full-length juxtanodin eluted as a single, sharp peak with a relatively small elution volume (Figure 2). As the elution volume does not provide accurate information about the molecular weight of the protein, but rather, about its hydrodynamic radius, we additionally used SLS to determine the molecular weight of juxtanodin. Interestingly, the SLS analysis clearly illustrates that juxtanodin is monomeric in solution, with a molecular weight of 32 kDa (Figure 2), which is essentially identical to the expected value of 32.2 kDa for a monomer. The small elution volume indicates a large hydrodynamic radius, i.e. juxtanodin has an extended and non-globular shape.
Figure 2. Determination of the oligomeric status of juxtanodin.

Size exclusion chromatography of full-length juxtanodin displays a single peak. The molecular weight of the peak calculated using static light scattering, shown as a dashed line, represents a juxtanodin monomer.
Circular dichroism spectra of juxtanodin fragments exhibit characteristics of disordered proteins
Synhrotron radiation circular dichroism (SRCD) spectra were recorded for full-length juxtanodin and truncated variants. All spectra exhibited a negative minimum close to 200 nm and a relatively low ellipticity above 210 nm (Figure 3a,b), a typical observation for disordered proteins13,14. However, for the two longer juxtanodin fragments (1–282 and 103–282), the negative peak shifted towards higher wavelengths and the negative shoulder at 220 nm was more pronounced compared to the shorter fragments (Figure 3b). This shoulder diminished slowly, when an increasing concentration of urea was added to the full-length juxtanodin (data not shown). Thus, the existence of a negative shoulder at 222 nm with full-length juxtanodin and the 103–282 fragment suggests that there are minor α-helical regions in these constructs, while the shorter constructs (1–170 and 172–282) are to a larger extent unfolded. This result may reflect interactions between different regions of juxtanodin, required for local secondary structure formation. Also the synthetic peptide representing the C-terminal actin binding site was disordered (Figure 3b). Compared to the well-characterized IDP from myelin, the myelin basic protein (MBP)15, the negative minimum in juxtanodin is approximately at the same position, but the shoulder around 210–220 nm is more pronounced, indicating a higher secondary structure content than for MBP in solution.
Figure 3. CD spectroscopic analysis of juxtanodin folding.

(a) High-resolution SRCD spectrum of full-length juxtanodin in phosphate buffer. (b) Comparison of SRCD spectra for the different juxtanodin variants. (c) Titration of juxtanodin with TFE followed by SRCD. The concentrations range from 10% to 89% (solid lines), with intermediate concentrations of 20, 30, 40, 60, and 70% (dashed lines). (d) SRCD spectra of different juxtanodin variants in 30% TFE. Colours as in (b). (e) Temperature scan (25–90°C) of full-length juxtanodin in phosphate buffer. (f) Temperature scan of full-length juxtanodin in 50% TFE. The direction of spectral change upon heating in (e) and (f) is indicated by the arrows. Spectra are shown at intervals of 2°C.
We also tested the tendency of juxtanodin to form secondary structures in the presence of trifluoroethanol (TFE), which lowers the dielectric constant of the solution. TFE is a cosolvent commonly used for stabilizing protein secondary structures and for studying protein folding16,17,18. Full-length juxtanodin was titrated with TFE; the protein folded into a helical conformation with increasing TFE concentrations (Figure 3c). In 30% (v/v) TFE solution, the spectra of all juxtanodin fragments showed clear α-helical characteristics (Figure 3d). This predicts that, under some conditions, juxtanodin may fold; for example, upon binding to an interaction partner. Other additives, including ions or lipid mixtures, did not induce significant conformational changes (data not shown).
Temperature scans for juxtanodin were recorded in the absence and presence of 50% of TFE. In the temperature scan without TFE, no large changes were observed in the secondary structure composition of juxtanodin, which supports the disordered nature of the protein (Figure 3e). In the presence of TFE, a slow disappearance of the helical structure was observed upon heating, without a sharp transition (Figure 3f). Similar non-cooperative unfolding behaviour has been observed, for example, for lysozyme in 50% TFE19.
Small-angle X-ray scattering illustrates the extended and flexible properties of juxtanodin
In order to obtain more detailed 3D structural information on juxtanodin, we performed synchrotron small-angle X-ray scattering (SAXS) experiments for full-length juxtanodin as well as the truncated fragments. The 1–170, 172–282, and full-length constructs behaved well in SAXS; the 103–282 construct, instead, was problematic and will not be analyzed in detail here. The scattering profiles of all fragments were smooth and featureless (Figure 4a). The Rg values derived from the Guinier plot, the distance distribution functions and Debye functions (Table I) showed highly extended particle shapes for all juxtanodin fragments. The large maximum particle dimensions (Dmax) of juxtanodin fragments (Table I) also support a disordered nature for juxtanodin (Figure 4d). The molecular mass, estimated based on the forward scattering intensity, for all juxtanodin constructs is in good corroboration with the expected one, indicating a monomeric state. Furthermore, especially the Rg values correspond well to those predicted using random coil assumptions20, while the estimated Dmax is even larger than that expected for a random coil (Table I).
Figure 4. Small-angle X-ray scattering.

(a) Scattering curves for juxtanodin fragments 1–170 (black), 1–282 (green) and 172–282 (red). (b) Kratky plot, (c) Debye-Porod plot and (d) distance distribution functions for the same fragments. (e) Experimental scattering profile for full-length juxtanodin (green spheres) overlaid with the calculated scattering pattern (black line) of the ab initio model of full-length juxtanodin represented in panel (f).
Table 1. SAXS parameters for juxtanodin constructs.
| Sample | Rg (Guinier), nm | Rg (GNOM), nm | Rg (Debye), nm | Dmax, nm | Rg/Dmax (predicted), nm | MM, kDa | Expected MM, kDa |
|---|---|---|---|---|---|---|---|
| 1–282 | 5.59 | 6.00 | 5.97 | 21 | 5.9/15.2 | 33.5 | 32.2 |
| 1–170 | 4.26 | 4.27 | 4.41 | 14 | 4.4/11.8 | 28.8 | 19.7 |
| 172–282 | 3.15 | 3.40 | 3.47 | 12 | 3.4/9.5 | 15.0 | 12.9 |
| 103–282 | 3.74 | − | 3.93 | − | 4.5/12.1 | 20.7 | 21.0 |
The 103–282 fragment could only be measured at a very low concentration and was not fully analyzed. The predicted Rg/Dmax values were calculated based on sequence length, assuming random coil structure20. Molecular masses (MM) were determined by way of forward scattering comparison to a standard sample of bovine serum albumin.
Traditionally, IDPs can be recognized using the Kratky plot (I(s)s2 vs s), where they show non-bell-shaped curves21. The Kratky plot for all juxtanodin fragments revealed the typical behavior of disordered proteins (Figure 4b), with a short plateau followed by a monotonic increase, and the lack of a clear maximum. Typically, unfolded proteins do not reach a plateau in the Porod-Debye plot within the low-resolution region of SAXS data22. To investigate this aspect, the low-resolution parts of the SAXS data were plotted as I(s)s4 vs s4 (Figure 4c); none of the measured fragments reached a plateau in this Porod-Debye presentation, again confirming the unfolded nature of juxtanodin.
A chain-like ab initio model was reconstructed for full-length juxtanodin using GASBOR23. The fit of the model against the raw SAXS data is very good with a chi value of 0.59 (Figure 4e). The resulting model shows a highly extended conformation of juxtanodin without globular domains (Figure 4f).
Ensemble optimization analysis reveals a compact subpopulation of full-length juxtanodin structures
Based on the relatively large Rg and Dmax values, characteristic Kratky and Porod-Debye plots, as well as the ab initio model, the SAXS results confirm that juxtanodin is intrinsically disordered. In order to get an idea of possible structural heterogeneity, we further used the ensemble optimization method (EOM)24 to analyze the SAXS data of the 1–282, 172–282 and 1–170 fragments (Figure 5). EOM creates an ensemble of models that together represent the observed scattering profile. All constructs gave good fits against the scattering data with chi values of 0.63, 0.93 and 1.26 for the 1–282, 172–282 and 1–170 constructs, respectively.
Figure 5. Analysis of juxtanodin conformations by EOM.
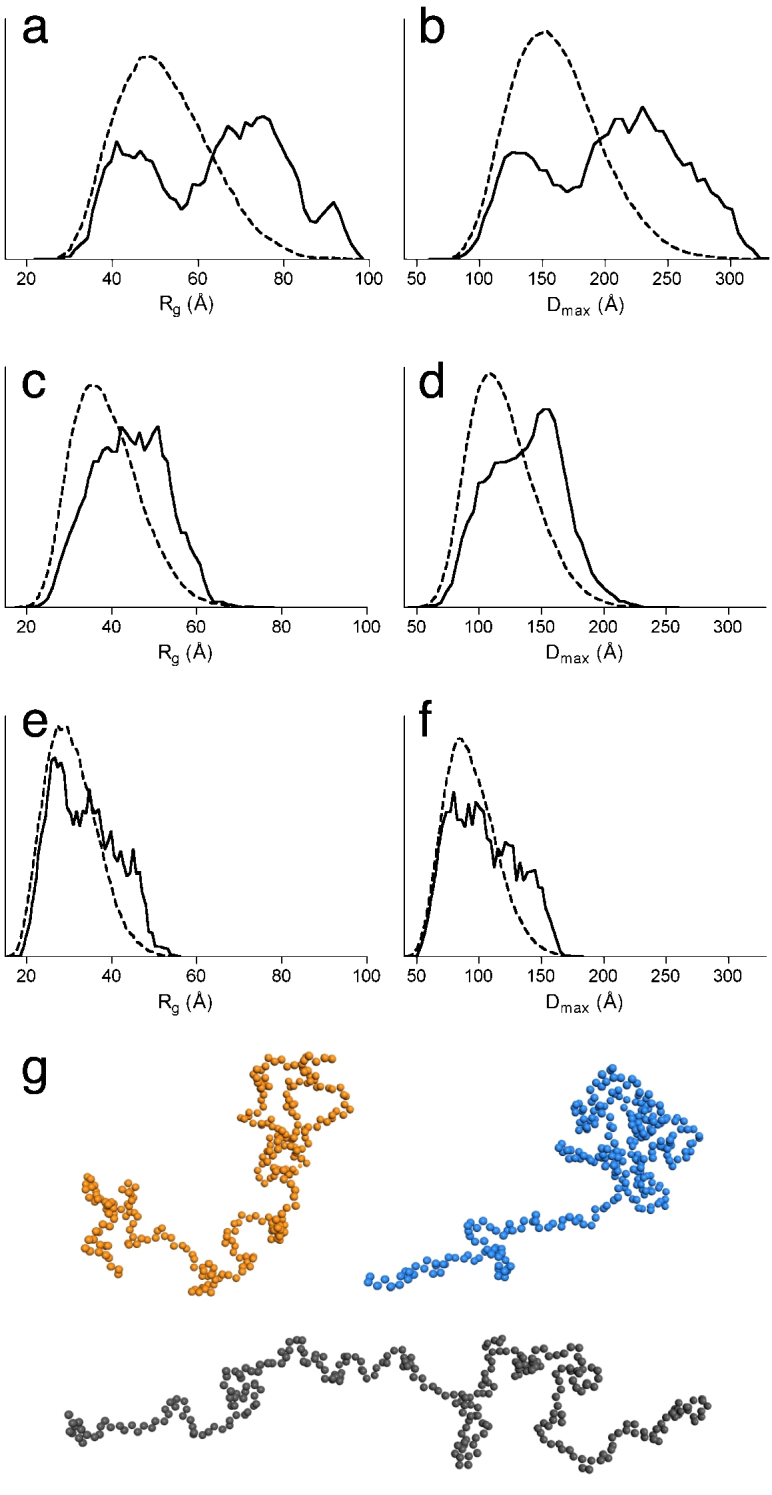
The Rg and Dmax distributions from EOM analysis for juxtanodin constructs 1–282 (a,b), 1–170 (c,d), and 172–282 (e,f). The solid lines represent the Rg and Dmax distributions of the selected ensembles that fit the experimental scattering profiles, and the dashed curves show the distributions of random ensembles. (g) Three selected dummy residue models from the ensemble of full-length juxtanodin with Dmax values of 130 (orange), 152 (blue), and 274 Å (black).
EOM analysis of full-length juxtanodin exhibits clearly bimodal distributions for both Rg and Dmax. This is typical behavior for some IDPs and may indicate the existence of two different subpopulations of conformations21. The Rg and Dmax distributions for full-length juxtanodin demonstrate that there is also a relatively compact subpopulation of juxtanodin with an Rg of 40–45 Å and Dmax of 130 Å (Figure 5a,b,g). On the other hand, the more extended, and probably mainly disordered subpopulation, with an Rg of approximately 76 Å and Dmax of 230 Å, is more pronounced.
EOM analysis of N-terminal juxtanodin (1–170) displays relatively wide distributions for Rg and Dmax, with an average Rg of 45.5 Å and Dmax of 140 Å (Figure 5c,d). The distributions of these parameters, peaking at higher values than a random ensemble, suggest that the N-terminal part of juxtanodin is highly disordered and elongated in solution. A wide, somewhat bimodal distribution was observed for the 172–282 construct, with average values for Dmax of 106 Å and Rg of 35 Å; the distribution in this case indicates more compact structures than for the N-terminal construct (Figure 5e,f). The EOM analyses for juxtanodin are in agreement with the CD spectroscopy results, where some secondary structure was detected in the longer constructs (see above). These findings together clearly confirm the presence of some compactness and nascent secondary structures mainly in the C-terminal part of juxtanodin, while the N-terminal part, containing residues 1–170, is disordered in solution.
The C-terminal segment of juxtanodin binds to F-actin
Juxtanodin has a conserved actin-binding region, of the ERM type, at its C terminus, and it affects actin dynamics in oligodendrocytes9. Using tagged recombinant juxtanodin, a stabilizing effect on F-actin has been detected in vitro9. To test the functionality of our juxtanodin constructs and the ability of the untagged juxtanodin fragments to interact with F-actin, we performed classical actin co-sedimentation assays. Both fragments (103–282 and 172–282) that contain the C-terminal actin-binding region co-sedimented with actin filaments during ultracentrifugation, similarly to the full-length protein (Figure 6a). However, the degradation products of these fragments did not associate with F-actin. Mass spectrometric analysis confirmed that these degradation products lack the C-terminal end of the construct (data not shown). The essence of the C-terminal region for F-actin binding was further confirmed with the N-terminal fragment of juxtanodin (1–170), which was found in the soluble fraction in the co-sedimentation assay. None of the juxtanodin fragments had detectable effects on the amount of polymerized actin in pellets. Importantly, this experiment shows that our recombinant, intrinsically disordered juxtanodin fragments bind to F-actin efficiently and are, thus, functional.
Figure 6. Interaction of juxtanodin with actin.

(a) Co-sedimentation assay with F-actin. S – supernatant, P – pellet. Left: truncated variants; right: full-length juxtanodin. For the full-length protein, both Coomassie staining (top) and immunoblotting (bottom) results are shown. Positions of molecular weight markers (kDa) are shown beside the figures. (b) An example of raw data from actin polymerization assays. Shown are increasing concentrations (0–20 µM) of full-length juxtanodin (direction of the arrow). (c) Quantification of polymerization rates as a function of juxtanodin concentration. (d) Negatively stained EM micrographs of 5 µM actin in F-buffer without (left) and with (right) full-length juxtanodin (5 µM) after 16 h incubation show no changes in actin organization upon addition of juxtanodin. Scale bars represent 200 nm.
The effect of juxtanodin fragments on the rate of actin polymerization was further studied using a fluorescence-based polymerization assay. Full-length juxtanodin weakly inhibited actin polymerization in a concentration-dependent manner (Figure 6b,c). The effect of juxtanodin is already observable when its molar concentration is half (2.5 µM) of the actin concentration, and the inhibitory effect is rather clear with a 4-fold excess of juxtanodin. A slowdown of actin polymerization was not detected with the N- (1–170) or C-terminal (172–282) constructs alone. The results indicate that although the C terminus of juxtanodin is required for direct F-actin interaction, the whole protein may be essential for juxtanodin function.
In order to study the effects of juxtanodin on supramolecular actin structures, we incubated juxtanodin with polymerized muscle actin in a 1:1 molar ratio and used electron microscopy (EM) to observe possible changes in actin organization. The negatively stained EM grids did not show any remarkable changes in actin organization in the presence of juxtanodin (Figure 6d). The resolution of EM is not high enough to detect the juxtanodin interaction with the actin filaments. Nevertheless, juxtanodin neither induced F-actin bundle formation or crosslinking, nor a complete depolymerization of microfilaments.
Discussion
In the current study, we have unequivocally shown that both full-length juxtanodin, as well as shorter fragments of the protein, behave like intrinsically disordered molecules in solution. Subtle differences between the constructs were observed in CD spectroscopy, effectively dividing them into two groups. Full-length juxtanodin and the 103–282 construct contain somewhat more secondary structure than the 1–170 and 172–282 fragments. The result implies that in the presence of both the C-terminal half of the protein and the region 103–170, more folding occurs. This could imply interactions between different parts of juxtanodin, and may also be linked to the subtle effects on actin polymerization observed with the different constructs.
SAXS data and ab initio model building for juxtanodin clearly demonstrate the disordered nature of juxtanodin. Interestingly, while the N-terminal fragment is largely disordered, the EOM analysis revealed the presence of a more compact subpopulation of full-length juxtanodin. This kind of behavior is typically seen with proteins that, at least partially, fold upon binding to their ligands. Most likely, the C-terminal part of juxtanodin mediates ligand binding and adopts some secondary structures induced by the interactions. Considered together with the CD spectroscopic data, it seems further possible that there is conformational flexibility in juxtanodin, resulting in open-close motions, whereby the region 103–170 interacts with the C-terminal part and secondary structures are induced. Although the only currently known interaction partner of juxtanodin is F-actin, these results suggest juxtanodin, as an IDP, could have a role as an adaptor protein that links the actin cytoskeleton to other proteins, enabling the formation of oligodendrocytic protrusions during their maturation.
Disordered proteins are by definition flexible, also having large maximum dimensions. On the other hand, IDPs are often known to be able to interact with a number of different ligands, enabling them to act as interaction hubs and molecular rulers. Therefore, disordered proteins are rather common among cytoskeletal regulators, where multiple binding sites and high flexibility are essential properties. For example, synaptopodins are intrinsically disordered actin-binding proteins that interact with several actin regulators and actin itself and induce actin polymerization in different cell types25. Another IDP, myristoylated alanine-rich C kinase substrate (MARCKS), is an F-actin-binding protein that is especially enriched in the brain26. MARCKS crosslinks actin filaments, accelerates actin polymerization, and participates in the formation of neuronal processes. Its interactions are regulated by post-translational modifications, autoinhibition, phosphorylation, and Ca2+/calmodulin binding27. The cytoskeletal adaptor protein dematin has a disordered core domain and a folded headpiece. Dematin is a trimeric actin-bundler found mainly in junctional complexes28. It participates in RhoA signaling and, thus, regulation of actin dynamics29. Other disordered actin-binding proteins include spinophilin30 and cortactin31. However, it still remains unclear whether or not these disordered proteins fold upon binding to actin or other ligands.
The only sequence similarity to other proteins in juxtanodin comprises its very C terminus, where the F-actin binding site is located, similarly to other members of the ERM family32. In comparison to other members of the family, juxtanodin lacks the N-terminal FERM domain that mediates autoinhibition through interactions with the C terminus. Upon classical ERM protein activation, the C terminus is released while the protein becomes extended and binds F-actin. The FERM domain has also been suggested to link the protein to the membrane through interactions with membrane proteins. The lack of this domain implies that juxtanodin is unlikely to bind to the membrane, and that it is constitutively in an ‘active’ conformation, with its C terminus free to interact with F-actin. Instead of autoinhibition through FERM domain binding, the actin binding of juxtanodin is presumably regulated by the phosphorylation of Ser278 located at the conserved actin-binding region9.
In FERM family proteins, a helical domain links the FERM domain and the C-terminal domain33; in juxtanodin, some helical structure is predicted, but our spectroscopic data indicate only little secondary structure is present. All in all, juxtanodin appears an outlier in the ERM family, only having the F-actin binding site conserved. Interestingly, in another ERM protein, merlin/schwannomin, the conserved regions are present except for the F-actin binding site.
Juxtanodin lacks an N-terminal FERM domain, which is common to other ERM proteins and regulates their activity by masking the actin-binding segment. Since the C terminus of juxtanodin is nearly identical to that of moesin and other ERM proteins (Figure 7), it is possible that juxtanodin interacts with FERM domains from other proteins. A crystal structure for human moesin has been determined (Figure 7), where the binding mode of its C-terminal tail to its FERM domain is elucidated34; due to the high sequence conservation, the juxtanodin C terminus could bind similarly to FERM domains. Such interactions have been proposed to be involved in homo- and heterotypic interactions between ERM family members35,36, and interactions between juxtanodin and FERM domains could be possible since ERM proteins have been detected in myelin37. In addition to merlin, ezrin, moesin and radixin, a potential FERM domain-containing interaction partner for juxtanodin is the 4.1G protein that also participates in promoting the formation of oligodendrocytic processes during their maturation38.
Figure 7. The conserved C terminus in juxtanodin.
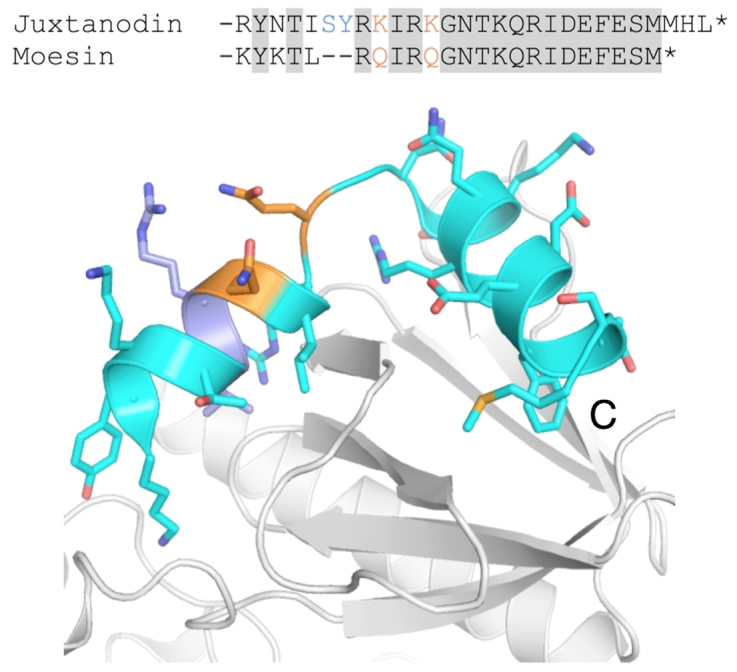
Top: Alignment of F-actin binding domains from juxtanodin and human moesin, for which the crystal structure in complex with the FERM domain is known34. Bottom: The crystal structure of the C terminus of moesin (cyan) bound to the FERM domain (white). The segment shown is the same as in the alignment above; two single-amino-acid changes between moesin and juxtanodin are indicated in orange. The C-terminal helix is fully conserved in juxtanodin.
Does the disordered structure of juxtanodin tell us anything about its function? Juxtanodin is expressed by the myelin-forming cells, oligodendrocytes, of the central nervous system6,8. The best characterized IDP in myelin is myelin basic protein (MBP). We and others have shown it to be unfolded in solution, while secondary structure formation is observed upon ligand binding15,39. In addition to membrane surfaces, one of the MBP ligands is actin40,41,42. MBP induces actin polymerization, in a process that may be regulated by calmodulin, and it is likely that the actin binding sites become ordered upon complex formation. Many of the IDPs that bind to actin filaments act as adaptor proteins with multiple interaction partners. Since juxtanodin has no actin-bundling activity, its function may be to serve as an interaction hub for cytoskeletal regulatory proteins and other proteins that participate in the maturation of myelinating oligodendocytes. For example, juxtanodin colocalizes with the myelin-specific enzyme 2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNPase) and promotes its trafficking to oligodendrocytic processes8. Since CNPase interacts also with microtubules43, juxtanodin could be involved in providing links between microfilaments and microtubules.
To conclude, we have shown here that the oligodendrocytic F-actin-binding protein juxtanodin is intrinsically disordered. However, juxtanodin possesses the ability to form secondary structures and more compact conformations, which may be crucial to its physiological function. The function of juxtanodin at the moment is unknown; while it does interact with F-actin, its effects on microfilaments at the molecular level are unclear. Further detailed structural and functional studies will be required to fully understand the dynamics and possible ligand-induced folding of juxtanodin. In fact, we cannot be certain how disordered juxtanodin is in vivo, in the intracellular conditions of the myelinating oligodendrocyte. Recently, α-synuclein – long thought to be intrinsically disordered – was suggested to be folded in vivo44, and it is possible that similar phenomena occur with other proteins characterized as IDPs in vitro.
Methods
Bioinformatics
Secondary structure predictions for rat juxtanodin were done using Jpred345. Disorder predictions were additionally carried out with FoldIndex46, GlobPlot47, DisoPred248, and PONDR-FIT49. FoldIndex is based on average residue hydrophobicity and overall net charge, and is designed to predict if a given sequence is folded or not. GlobPlot uses a running sum of residue propensities to be in an ordered or disordered state, and is optimal for finding domain boundaries in folded proteins. DisoPred is a disorder predictor trained on disordered regions of crystal structures from the PDB, and hence, perhaps not optimal for analyzing truly disordered sequences. PONDR-FIT, on the other hand, combines a number of different prediction methods, including FoldIndex.
Molecular cloning
Full-length rat juxtanodin (amino acids 1–282) cDNA and three truncated fragments, coding for amino acids 1–170, 103–282 and 172–282, were amplified together with a Tobacco Etch Virus (TEV) protease cleavage site by PCR and cloned into the pGGWA vector50 using Gateway cloning technology (Invitrogen). The sequences were confirmed by nucleotide sequencing at the Biocenter Oulu DNA Sequencing Core Facility.
Protein expression and purification
Glutathione S-transferase -juxtanodin fusion proteins were expressed in Escherichia coli Rosetta (DE3) pLysS cells grown in Luria-Bertani medium with chloramphenicol and ampicillin. Cells were cultured at 37°C until OD600 was 0.6–0.8. Protein expression was induced with 0.4 mM isopropyl β-D-1-thiogalactopyranoside. After 4 h, the cells were harvested, resuspended in buffer A (50 mM Tris-HCl pH 7.5, 300 mM NaCl, 1 mM DTT), and stored at −70°C.
After thawing, the cells were broken by sonication, and debris was removed by centrifugation at 48000 g for 30 min at 4°C. The sample was then loaded onto a glutathione agarose 4B column (Macherey-Nagel) and incubated for 2–4 h at 4°C. After washing with buffer A, TEV protease was added into the column, and the sample was incubated for 16 h at 4°C. Untagged juxtanodin fragments were then eluted with buffer A and further purified by size exclusion chromatography on a HiLoad 16/60 Superdex 75 pg column (GE Healthcare), using 20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM DTT as running buffer. Fractions containing juxtanodin were pooled, concentrated, and frozen in liquid nitrogen. The identity of all juxtanodin fragments was verified with MALDI-TOF mass spectrometry at the Biocenter Oulu Proteomics Core Facility.
TEV protease was expressed and purified as previously described51. A synthetic 28-mer peptide comprising the actin binding site of juxtanodin (RYNTISYRKIRKGNTKQRIDEFESMMHL, with acetylated N terminus) was purchased from TAG Copenhagen.
Molecular weight determination by static light scattering
Analytical size exclusion chromatography was performed for 50 µg of juxtanodin in 20 mM Tris-HCl, pH 8, 150 mM NaCl, and 1 mM DTT, using a Superdex 75 pg 10/300 GL column (GE Healthcare) coupled to a Shimadzu liquid chromatography system. The setup also included a mini-DAWN TREOS multi-angle static light scattering detector (Wyatt) and a Shimadzu refractive index detector for absolute molecular weight determination. The molecular weight of juxtanodin was determined based on the measured light scattering and refractive index and/or UV absorbance using the ASTRA software (Wyatt).
Synchrotron radiation and conventional CD spectroscopy
SRCD spectra were recorded at the CD1 beamline of the ASTRID storage ring, ISA, Århus (Denmark). Before measurements, all juxtanodin fragments were dialyzed into 20 mM potassium phosphate (pH 7.0) containing 150 mM NaF. SRCD spectra were collected in the wavelength range of 170–280 nm using 10- and 100-µm pathlength quartz cuvettes. The measurements were also carried out in the presence of 10–89% (v/v) TFE, 10 mM CaCl2, and a 1:1 mixture of dimyristoylphosphatidylcholine and dimyristoylphosphatidylglycerol (DMPC and DMPG, respectively) at 4 mM lipid concentration.
Conventional CD spectroscopy was carried out using a Chirascan Plus instrument (Applied Photophysics), in a 0.5-mm quartz cuvette. Continuous ramping temperature scans in the presence and absence of 50% TFE were also carried out between 25–90°C.
Small-angle X-ray scattering
Synchrotron SAXS data were collected on beamline I911-4 at MAX-Lab, Lund (Sweden), beamline X33 at EMBL/DESY (Hamburg), and on the BM29 beamline at ESRF (Grenoble, France). X-ray scattering patterns were recorded with protein concentrations between 1–10 mg/ml in 20 mM Tris-HCl pH 7.5, 150–300 mM NaCl, 1 mM DTT.
Programs of the ATSAS suite52 were used for SAXS data analysis. PRIMUS53 was utilized for SAXS data processing and analysis, including Rg determination with the Guinier method. The radius of gyration (Rg) and maximum distance (Dmax) were evaluated using GNOM54, which was also used to calculate the distance distribution functions. An ab initio model of full-length juxtanodin was built using GASBOR23, and the ensemble optimization method24 was used to further analyze the ensembles of juxtanodin structures in solution. Molecular weights were estimated based on the forward scattering intensity I(0), comparing to a fresh standard solution of monomeric bovine serum albumin.
For Rg determination, in addition to Guinier analysis, the data were fitted to the Debye function, which is known to be more accurate for molecules behaving as random polymers, such as intrinsically disordered proteins. Briefly, the appoximation I(0)/I(q) = 1 + 0.359(qRg)2.206 was used to obtain the Rg, essentially as previously described55. For comparison with experimental values, predicted values for Rg and Dmax based on sequence length and random coil conformation were calculated using the parameters described20.
Preparation of muscle actin
Skeletal muscle actin was prepared from the loin of pig and further purified as described previously56,57. The pig loins were gifts from the University of Oulu Laboratory Animal Centre and were from animals used in other animal experiments with separate licenses from the Finnish National Animal Experiment Board. The experiments were approved by the local ethics committee of the University of Oulu Laboratory Animal Centre (decision number 096/11).
Actin co-sedimentation assays
2 and 4 μM purified full-length juxtanodin and 4 μM pig muscle actin were used in actin co-sedimention assays. With truncated juxtanodin fragments (1–170, 103–282 and 172–282) 10 µM juxtanodin and 5 μM actin were used. Actin was first polymerized in F-buffer (5 mM Tris-HCl pH 7.5, 0.1 mM CaCl2, 0.2 mM ATP, 1 mM DTT, 0.1 M KCl, 1 mM MgCl2) for 1 h at 23°C and then incubated with juxtanodin for 45 min at 23°C. Polymerized actin was pelleted by ultracentrifugation (400000 g, at 20°C, for 45 min). F-actin and full-length juxtanodin alone were used as controls. The supernatants and pellets were analyzed by SDS-PAGE and Coomassie Brilliant Blue staining. The fractions from co-sedimentation assays with full-length juxtanodin were also analyzed by western blotting using a rabbit polyclonal anti-juxtanodin (1:300)8 as the primary antibody and a 1:2500 dilution of horseradish peroxidase-conjugated anti-rabbit IgG (W401B, Promega) as the secondary antibody. Immunodetection was performed using Pierce ECL western blotting substrate (Thermo Scientific).
Actin polymerization assays
Actin polymerization rates were studied by fluorescence spectroscopy by measuring the change in fluorescence intensity (excitation 365 nm, emission 406 nm) with a Synergy H4 Hybrid Multi-Mode Microplate Reader (BioTek). 5 µM of unlabeled pig muscle actin containing 5% pyrene-labeled rabbit muscle actin (Cytoskeleton Inc.) were mixed with 0–20 µM juxtanodin. Actin was polymerized at 26°C in the presence of 50 mM KCl, 2 mM MgCl2, 0.1 mM CaCl2, 1 mM ATP, 1 mM DTT, 5 mM Tris-HCl (pH 8), and the fluorescence intensity was measured every 20 s for 35 min. Values from 3–5 separate measurements were used in the analysis.
Electron microscopy
Polymerized pig muscle actin was incubated with juxtanodin fragments in 100 mM KCl, 2 mM MgCl2, 0.1 mM CaCl2, 0.5 mM ATP, 1 mM DTT, 5 mM Tris-HCl (pH 8) for 16 h at 23°C. For negative staining, samples were placed on freshly glow-discharged copper grids, washed, and incubated for 30 s in 1% uranyl acetate. Excess staining solution was removed, and the grids were air-dried. The grids were imaged with a Tecnai G2 Spirit 120 kV transmission electron microscope with Veleta and Quemesa CCD cameras. Both the grid preparation and imaging were done at the Biocenter Oulu EM Core facility.
Author Contributions
PK and FL conceived of the study. SR and MC produced juxtanodin. SR, MC, IK, and PK carried out structural studies. SR, JV, and SPB purified pig actin. SR, MC, JV, and SPB did actin binding assays. FL provided juxtanodin cDNA and antibody. SR, MC, and PK wrote the manuscript and prepared figures. All authors provided intellectual input to the manuscript and approved its submission.
Supplementary Material
Supplementary Figure 1
Acknowledgments
Synchrotron beamtime and excellent user support at MAX-Lab, EMBL/DESY, ESRF, and ISA are gratefully acknowledged. We wish to thank Moon Chatterjee for SRCD and Gitte Meriläinen for SAXS data collection, Arne Raasakka for carrying out mass spectrometry, as well as Ilkka Miinalainen and Raija Sormunen for helping with the EM experiments. This study was financially supported by grants from the Academy of Finland, the Sigrid Jusélius Foundation, and the Research and Science Foundation of the City of Hamburg.
References
- Rumsby M., Afsari F., Stark M. & Hughson E. Microfilament and microtubule organization and dynamics in process extension by central glia-4 oligodendrocytes: evidence for a microtubule organizing center. Glia 42, 118–129 (2003). [DOI] [PubMed] [Google Scholar]
- Bauer N. G., Richter-Landsberg C. & Ffrench-Constant C. Role of the oligodendroglial cytoskeleton in differentiation and myelination. Glia 57, 1691–1705 (2009). [DOI] [PubMed] [Google Scholar]
- Williams S. K. et al. Role of Mayven, a kelch-related protein in oligodendrocyte process formation. J Neurosci Res 81, 622–631 (2005). [DOI] [PubMed] [Google Scholar]
- Doukhanine E., Gavino C., Haines J. D., Almazan G. & Richard S. The QKI-6 RNA binding protein regulates actin-interacting protein-1 mRNA stability during oligodendrocyte differentiation. Mol Biol Cell 21, 3029–3040 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Clainche C. & Carlier M. F. Regulation of actin assembly associated with protrusion and adhesion in cell migration. Physiol Rev 88, 489–513 (2008). [DOI] [PubMed] [Google Scholar]
- Brockschnieder D., Sabanay H., Riethmacher D. & Peles E. Ermin, a myelinating oligodendrocyte-specific protein that regulates cell morphology. J Neurosci 26, 757–762 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T. et al. Human Ermin (hErmin), a new oligodendrocyte-specific cytoskeletal protein related to epileptic seizure. Brain Res 1367, 77–84 (2011). [DOI] [PubMed] [Google Scholar]
- Zhang B. et al. Juxtanodin: an oligodendroglial protein that promotes cellular arborization and 2′,3′-cyclic nucleotide-3′-phosphodiesterase trafficking. Proc Natl Acad Sci U S A 102, 11527–11532 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng J., Xia W., Tang J., Tang B. L. & Liang F. Dephosphorylation-dependent inhibitory activity of juxtanodin on filamentous actin disassembly. J Biol Chem 285, 28838–28849 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V. N., Gillespie J. R. & Fink A. L. Why are "natively unfolded" proteins unstructured under physiologic conditions? Proteins 41, 415–427 (2000). [DOI] [PubMed] [Google Scholar]
- Tompa P. Intrinsically unstructured proteins. Trends Biochem Sci 27, 527–533 (2002). [DOI] [PubMed] [Google Scholar]
- Winder S. J. & Ayscough K. R. Actin-binding proteins. J Cell Sci 118, 651–654 (2005). [DOI] [PubMed] [Google Scholar]
- Kelly S. M., Jess T. J. & Price N. C. How to study proteins by circular dichroism. Biochim Biophys Acta 1751, 119–139 (2005). [DOI] [PubMed] [Google Scholar]
- Ranjbar B. & Gill P. Circular dichroism techniques: biomolecular and nanostructural analyses- a review. Chem Biol Drug Des 74, 101–120 (2009). [DOI] [PubMed] [Google Scholar]
- Wang C. et al. Charge isomers of myelin basic protein: structure and interactions with membranes, nucleotide analogues, and calmodulin. PLoS One 6, e19915 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck M. Trifluoroethanol and colleagues: cosolvents come of age. Recent studies with peptides and proteins. Q Rev Biophys 31, 297–355 (1998). [DOI] [PubMed] [Google Scholar]
- Gast K., Zirwer D., Muller-Frohne M. & Damaschun G. Trifluoroethanol-induced conformational transitions of proteins: insights gained from the differences between alpha-lactalbumin and ribonuclease A. Protein Sci 8, 625–634 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H., Buck M., Radford S. E. & Dobson C. M. Acceleration of the folding of hen lysozyme by trifluoroethanol. J Mol Biol 265, 112–117 (1997). [DOI] [PubMed] [Google Scholar]
- Buck M., Radford S. E. & Dobson C. M. A partially folded state of hen egg white lysozyme in trifluoroethanol: structural characterization and implications for protein folding. Biochemistry 32, 669–678 (1993). [DOI] [PubMed] [Google Scholar]
- Fitzkee N. C. & Rose G. D. Reassessing random-coil statistics in unfolded proteins. Proc Natl Acad Sci U S A 101, 12497–12502 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernado P. & Svergun D. I. Structural analysis of intrinsically disordered proteins by small-angle X-ray scattering. Mol Biosyst 8, 151–167 (2012). [DOI] [PubMed] [Google Scholar]
- Rambo R. P. & Tainer J. A. Characterizing flexible and intrinsically unstructured biological macromolecules by SAS using the Porod-Debye law. Biopolymers 95, 559–571 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svergun D. I., Petoukhov M. V. & Koch M. H. Determination of domain structure of proteins from X-ray solution scattering. Biophys J 80, 2946–2953 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernado P., Mylonas E., Petoukhov M. V., Blackledge M. & Svergun D. I. Structural characterization of flexible proteins using small-angle X-ray scattering. J Am Chem Soc 129, 5656–5664 (2007). [DOI] [PubMed] [Google Scholar]
- Chalovich J. M. & Schroeter M. M. Synaptopodin family of natively unfolded, actin binding proteins: physical properties and potential biological functions. Biophysical Reviews 2, 181–189 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapp H. et al. MARCKS is a natively unfolded protein with an inaccessible actin-binding site: evidence for long-range intramolecular interactions. J Biol Chem 280, 9946–9956 (2005). [DOI] [PubMed] [Google Scholar]
- Li H., Chen G., Zhou B. & Duan S. Actin filament assembly by myristoylated alanine-rich C kinase substrate-phosphatidylinositol-4,5-diphosphate signaling is critical for dendrite branching. Mol Biol Cell 19, 4804–4813 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Jiang Z. G., Khan A. A., Chishti A. H. & McKnight C. J. Dematin exhibits a natively unfolded core domain and an independently folded headpiece domain. Protein Sci 18, 629–636 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohseni M. & Chishti A. H. Regulatory models of RhoA suppression by dematin, a cytoskeletal adaptor protein. Cell Adh Migr 3, 191–194 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler H. & Peti W. Structure-function analysis of the filamentous actin binding domain of the neuronal scaffolding protein spinophilin. FEBS J 275, 59–68 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shvetsov A., Berkane E., Chereau D., Dominguez R. & Reisler E. The actin-binding domain of cortactin is dynamic and unstructured and affects lateral and longitudinal contacts in F-actin. Cell Motil Cytoskeleton 66, 90–98 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turunen O., Wahlstrom T. & Vaheri A. Ezrin has a COOH-terminal actin-binding site that is conserved in the ezrin protein family. J Cell Biol 126, 1445–1453 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q. et al. Self-masking in an intact ERM-merlin protein: an active role for the central alpha-helical domain. J Mol Biol 365, 1446–1459 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson M. A., Reczek D., Bretscher A. & Karplus P. A. Structure of the ERM protein moesin reveals the FERM domain fold masked by an extended actin binding tail domain. Cell 101, 259–270 (2000). [DOI] [PubMed] [Google Scholar]
- Gary R. & Bretscher A. Ezrin self-association involves binding of an N-terminal domain to a normally masked C-terminal domain that includes the F-actin binding site. Mol Biol Cell 6, 1061–1075 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretscher A., Edwards K. & Fehon R. G. ERM proteins and merlin: integrators at the cell cortex. Nat Rev Mol Cell Biol 3, 586–599 (2002). [DOI] [PubMed] [Google Scholar]
- Jahn O., Tenzer S. & Werner H. B. Myelin proteomics: molecular anatomy of an insulating sheath. Mol Neurobiol 40, 55–72 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia W. & Liang F. 4.1G promotes arborization and tight junction formation of oligodendrocyte cell line OLN-93. J Cell Physiol 227, 2730–2739 (2012). [DOI] [PubMed] [Google Scholar]
- Majava V. et al. Structural analysis of the complex between calmodulin and full-length myelin basic protein, an intrinsically disordered molecule. Amino Acids 39, 59–71 (2010). [DOI] [PubMed] [Google Scholar]
- Barylko B. & Dobrowolski Z. Ca2+-calmodulin-dependent regulation of F-actin-myelin basic protein interaction. Eur J Cell Biol 35, 327–335 (1984). [PubMed] [Google Scholar]
- Boggs J. M. & Rangaraj G. Interaction of lipid-bound myelin basic protein with actin filaments and calmodulin. Biochemistry 39, 7799–7806 (2000). [DOI] [PubMed] [Google Scholar]
- Hill C. M. & Harauz G. Charge effects modulate actin assembly by classic myelin basic protein isoforms. Biochem Biophys Res Commun 329, 362–369 (2005). [DOI] [PubMed] [Google Scholar]
- Bifulco M., Laezza C., Stingo S. & Wolff J. 2′,3′-Cyclic nucleotide 3′-phosphodiesterase: a membrane-bound, microtubule-associated protein and membrane anchor for tubulin. Proc Natl Acad Sci U S A 99, 1807–1812 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartels T., Choi J. G. & Selkoe D. J. alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 477, 107–110 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole C., Barber J. D. & Barton G. J. The Jpred 3 secondary structure prediction server. Nucleic Acids Res 36, W197–201 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prilusky J. et al. FoldIndex: a simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics 21, 3435–3438 (2005). [DOI] [PubMed] [Google Scholar]
- Linding R., Russell R. B., Neduva V. & Gibson T. J. GlobPlot: Exploring protein sequences for globularity and disorder. Nucleic Acids Res 31, 3701–3708 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward J. J., McGuffin L. J., Bryson K., Buxton B. F. & Jones D. T. The DISOPRED server for the prediction of protein disorder. Bioinformatics 20, 2138–2139 (2004). [DOI] [PubMed] [Google Scholar]
- Xue B., Dunbrack R. L., Williams R. W., Dunker A. K. & Uversky V. N. PONDR-FIT: a meta-predictor of intrinsically disordered amino acids. Biochim Biophys Acta 1804, 996–1010 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busso D., Delagoutte-Busso B. & Moras D. Construction of a set Gateway-based destination vectors for high-throughput cloning and expression screening in Escherichia coli. Anal Biochem 343, 313–321 (2005). [DOI] [PubMed] [Google Scholar]
- van den Berg S., Lofdahl P. A., Hard T. & Berglund H. Improved solubility of TEV protease by directed evolution. J Biotechnol 121, 291–298 (2006). [DOI] [PubMed] [Google Scholar]
- Konarev P. V., Petoukhov M. V., Volkov V. V. & Svergun D. I. ATSAS 2.1, a program package for small-angle scattering data analysis. J Appl Cryst 39, 277–286 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konarev P. V. et al. PRIMUS - a Windows-PC based system for small-angle scattering data analysis. J Appl Cryst 36, 1277–1282 (2003). [Google Scholar]
- Svergun D. I. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Cryst 25, 495–503 (1992). [Google Scholar]
- Calmettes P. et al. How random is a highly denatured protein? Biophys Chem 53, 105–113 (1994). [DOI] [PubMed] [Google Scholar]
- Ignatev A., Bhargav S. P., Vahokoski J., Kursula P. & Kursula I. The lasso segment is required for functional dimerization of the Plasmodium formin 1 FH2 domain. PLoS One 7, e33586 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardee J. D. & Spudich J. A. Purification of muscle actin. Methods Cell Biol 24, 271–289 (1982). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1