

Complicated natural products, with their structures and properties evolved over millions of years, frequently display specific biological modes of action which can often be traced back to their highly preorganized three dimensional structures that perfectly complement the shape and functional group presentation of their target protein pockets.[1] A recent study analyzed the protein binding properties of compounds from natural as well as synthetic sources and found that protein binding selectivity correlated with shape complexity (defined as the relative content of sp3-hybridized carbons) and stereochemical complexity (defined as the relative content of stereogenic carbons).[2] Octahedral metal complexes may offer an attractive alternative strategy to sophisticated globular and rigid structural templates. They are constructed from a powerful single metal stereocenter with chelating ligands limiting the degree of conformational flexibility, thus achieving “natural-product-like” structural complexities and strinkingly high target specificities as demonstrated by us in several previous studies.[3–6]
An important aspect regarding the design of such metal-templated protein binders –which has not been articulated in the past– is the globular space requirement of an octahedral center. The latter significantly increases the demand for a proper design, mainly because the metal must be located at a specific position within the active site in order to be useful. For example, if the metal is located too far within the active site or too close to the protein backbone there will not be enough space available to accommodate the space-demanding octahedral coordination sphere, whereas if the metal is located too far towards the solvent, the metal center cannot easily impact on binding affinity and selectivity. A clear indicator for an advantageous metal position within the protein pocket is a strong influence of the metal coordination sphere on binding affinity and selectivity. We have identified such a priviledged position of the metal within the ATP-binding site of protein kinases by using the now well established staurosporine-inspired metallo-pyridocarbazole scaffold.[3–7] For example, the octahedral organoruthenium complex Λ-FL172 was designed as a selective inhibitor for the p21-activated kinase 1 (PAK1)[8] in which the bidentate pyridocarbazole ligand of the ruthenium complex occupies the adenine pocket.[4] By interacting with the so-called hinge region it places the ruthenium center at a defined position within the ribose binding site, where the additional CO, chloride, and bidentate iminopyridine ligands can form important contacts with other parts of the active site and thereby strongly contribute to binding affinity and selectivity (Figure 1).[4,6] In order to better understand the design of metal-based enzyme inhibitors, we wondered if this design is unique or whether other scaffolds with metals located at other positions within the active site could yield similar or even better results. To address this question, we designed new scaffolds[9] and uncovered a simple ruthenium complex (R)-1 which places the metal at a distinct position within the ATP-binding site, contains a completely different set of coordinating ligands, but at the same time shows an improved affinity for PAK1 compared to the much more complicated, previously established pyridocarbazole complex Λ-FL172.
Figure 1.
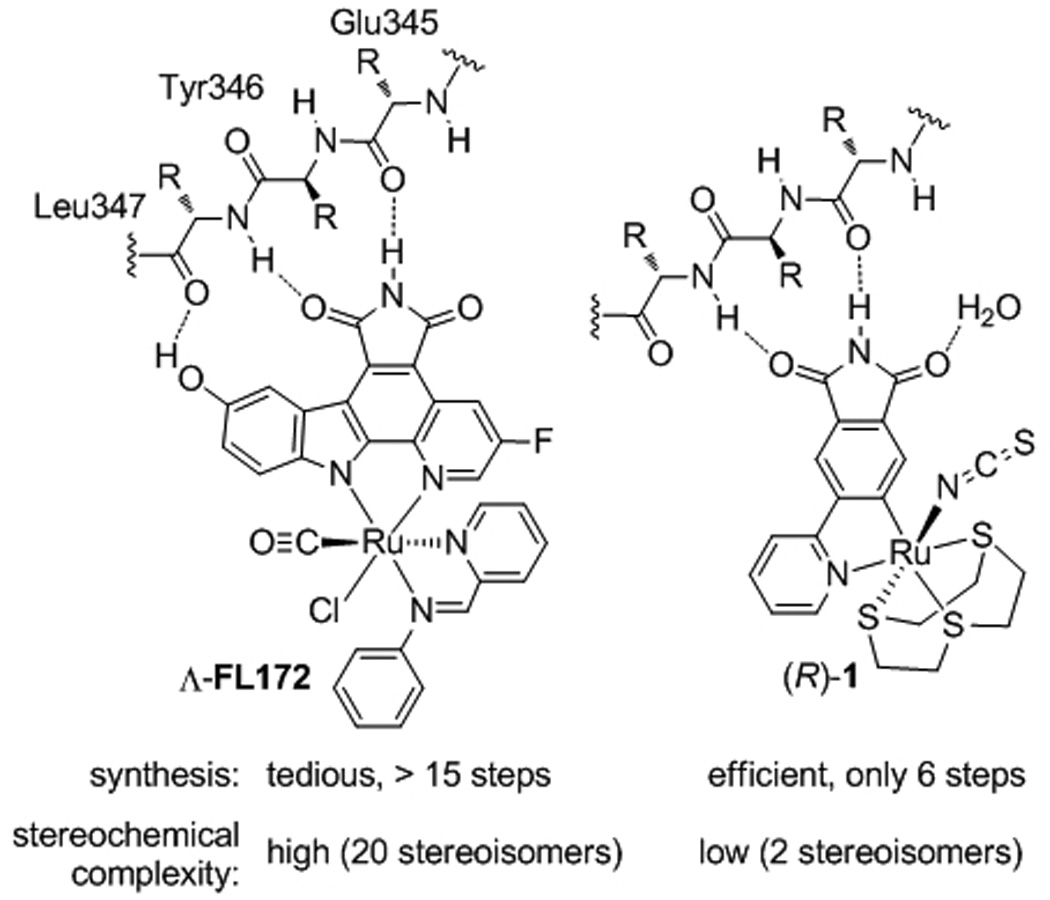
Complicated versus simple design for metal-templated inhibitors of the protein kinase PAK1.
Ruthenium complex 1 is based on a simple pyridylphthalimide scaffold, which can be synthesized in just a few steps (Scheme 1). Accordingly, a Suzuki cross-coupling of bromophthalimide 2 with 2-trimethylstannylpyridine afforded the pyridylphthalimide 3 (49%), which was subsequently reacted with the ruthenium complex [Ru(C6H12S3)(MeCN)3](CF3SO3)2 under basic conditions, followed by the addition of NaSCN to afford the racemic complex 1 (38% over two steps). A crystal structure of the N-benzylated derivative of 1 is shown in Figure 2 and demonstrates the formation of a C-Ru bond. This cyclometallation reduces the requirement for metal-coordinating heteroatoms and is thus crucial for the short and efficient synthesis.
Scheme 1.
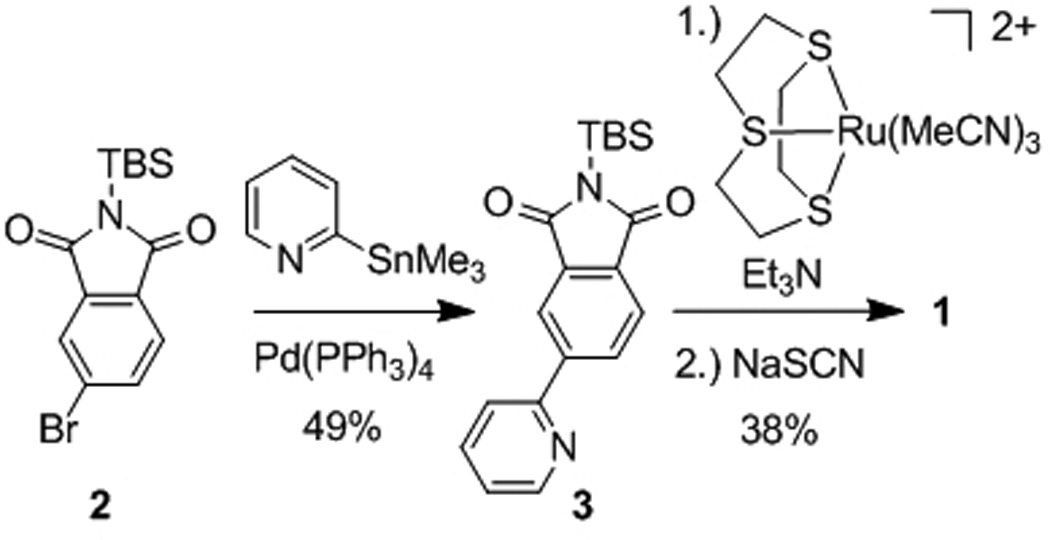
Synthesis of pyridylphthalimide ruthenium complex 1.
Figure 2.

Structure of the N-benzylated derivative of complex 1 (1Bn). Disordered solvent and a disordered position of S101 are not shown. ORTEP drawing with 50% probability thermal ellipsoids. Selected bond distances (Å): C15-Ru1 = 2.033(4), N1-Ru1 = 2.098(3), N100-Ru1 = 2.084(3), S1-Ru1 = 2.2770(9), S2-Ru1 = 2.3972(9), S3-Ru1 = 2.2923(9).
The racemic complex 1 displays an IC50 value of 83 ± 20 nM (1 µM ATP) against PAK1, which is slightly more potent than the previously reported Λ-FL172 (IC50 = 130 nM, 1 µM ATP)[4] (Figure 3). Interestingly, the pyridylphthalimide ligand itself is not an inhibitor for PAK1 at all (IC50 > 100 µM), thus demonstrating the importance of the entire coordination sphere for protein kinase binding.
Figure 3.

IC50 curves with PAK1 of ligand 4, complex 1, and some derivative complexes 5–7. ATP concentration was 1 µM.
PAK1 harbors a very atypical, open ATP-binding site which is probably responsible for the difficulties to develop high affinity inhibitors of this kinase.[4,8,10] A cocrystal structure at the resolution of 2.0 Å reveals the binding of the (R)-enantiomer of 1 to the ATP binding site of PAK1 (amino acids 249–545 with mutation Lys299Arg)[11] (Figures 4 and 5). The van der Waals surface representation shown in Figure 4 demonstrates how nicely the ATP-binding site is filled with (R)-1 in a shape complementary fashion with the phthalimide moiety additionally forming two hydrogen bonds with the hinge region (Glu345 and Leu347) and one water-mediated contact to Thr406 (Figure 5). Interestingly, superimposition of this structure with the recently disclosed PAK1/Λ-FL172[4] cocrystal structure demonstrates that even though both compounds are ATP-competitive binders, they significantly differ in there binding modes (Figure 6). While both form the canonical hydrogen bonds between the maleimide moieties of the inhibitor and the hinge region of the kinase, the aromatic heterocycles are rotated towards each other by approximately 30° demonstrating the range that is possible for the directionality of hydrogen bonds. Furthermore, the metals of the two inhibitor scaffolds are 3.0 Å apart from each other and point the monodentate ligands into different areas of the active site. Whereas the CO of FL172 is located right below the center of the glycine-rich loop (P-loop), the NCS ligand of 1 points towards the interface of the glycine-rich loop (connecting strands β1 and β2) and the methylene groups of Arg299 of the β-sheet strand β3 and thereby perfectly filling a small hydrophobic pocket as visualized in the van der Waals representation of Figure 4. The importance of the NCS ligand is manifested by the loss of the binding affinity of related complexes, which carry instead a SeCN (IC50 = 0.625 ± 0.06 µM, 7.5-fold weaker inhibitor), NCO (IC50 = 15.7 ± 2 µM, 193-fold weaker inhibitor), or CO (IC50 = 12.7 ± 2 µM, 153-fold weaker inhibitor) ligand (Figure 3). This intriguing example demonstrates how a different position of the metal within the active site, even if it is just shifted by 3.0 Å, requires a completely different coordination sphere to fill the protein pocket in a comparable fashion.
Figure 4.

Cocrystal structure of PAK1 (amino acids 249–545 with mutation Lys299Arg) and (R)-1 at 2.0 Å with displayed van der Waals surfaces. Coordinates of the structure have been deposited in the Protein Data Bank (PDB ID: 4DAW).
Figure 5.

Hydrogen bonding of (R)-1 within the ATP-binding site of PAK1.
Figure 6.

Relative binding position of (R)-1 and Λ-FL172 (PDB ID: 3FXZ) within the ATP-binding site of PAK1. Superimposed with the PyMOL Molecular Graphics System, Version 1.3, Schrödinger, LLC.
In conclusion, the here presented metal-based enzyme inhibitor design together with the analysis of an X-ray cocrystal structure reveals the scope of fitting octahedral metal complexes within an enzyme active site. Despite the shortness of the synthesis (overall only 6 steps) and simplicity of the structure (only two stereoisomers possible), complex 1 displays an IC50 value of 83 nM (1 µM ATP) which is superior to FL172, a complex that is much more tedious to synthesize (>15 steps) and contains a much higher stereochemical complexity (20 possible stereoisomers). To date, the ruthenium phthalimide complex 1 described here belongs to the most potent ATP-competitive inhibitors known for the protein kinase PAK1,[12] demonstrating the advantages of filling large or open pockets with globular octahedral metal complexes.
Supplementary Material
Acknowledgments
This work was supported by the German Research Foundation (DFG) and the US National Institutes of Health (CA114046). S.B. acknowledges a stipend from the Fonds der Chemischen Industrie.
Contributor Information
Sebastian Blanck, Fachbereich Chemie, Philipps-Universität Marburg, Hans-Meerwein-Straße, 35043 Marburg (Germany).
Jasna Maksimoska, The Wistar Institute, 3601 Spruce Street, Philadelphia, PA, 19104 (USA).
Julia Baumeister, Fachbereich Chemie, Philipps-Universität Marburg, Hans-Meerwein-Straße, 35043 Marburg (Germany).
Klaus Harms, Fachbereich Chemie, Philipps-Universität Marburg, Hans-Meerwein-Straße, 35043 Marburg (Germany).
Ronen Marmorstein, The Wistar Institute, 3601 Spruce Street, Philadelphia, PA, 19104 (USA).
Eric Meggers, Fachbereich Chemie, Philipps-Universität Marburg, Hans-Meerwein-Straße, 35043 Marburg (Germany).
References
- 1.Carlson EE. ACS Chem. Biol. 2010;5:639–653. doi: 10.1021/cb100105c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clemons PA, Bodycombe NE, Carrinski HA, Wilson JA, Shamji AF, Wagner BK, Koehler AN, Schreiber SL. Proc. Nat. Acad. Sci. USA. 2010;107:18787–18792. doi: 10.1073/pnas.1012741107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bregman H, Carroll PJ, Meggers E. J. Am. Chem. Soc. 2006;128:877–884. doi: 10.1021/ja055523r. [DOI] [PubMed] [Google Scholar]
- 4.Maksimoska J, Feng L, Harms K, Yi C, Kissil J, Marmorstein R, Meggers E. J. Am. Chem. Soc. 2008;130:15764–15765. doi: 10.1021/ja805555a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilbuer A, Vlecken DH, Schmitz DJ, Kräling K, Harms K, Bagowski CP, Meggers E. Angew. Chem. Int. Ed. 2010;49:3839–3842. doi: 10.1002/anie.201000682. [DOI] [PubMed] [Google Scholar]
- 6.Feng L, Geisselbrecht Y, Blanck S, Wilbuer A, Atilla-Gokcumen GE, Filippakopoulos P, Kräling K, Celik MA, Harms K, Maksimoska J, Marmorstein R, Frenking G, Knapp S, Essen L-O, Meggers E. J. Am. Chem. Soc. 2011;133:5976–5986. doi: 10.1021/ja1112996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meggers E, Atilla-Gokcumen GE, Bregman H, Maksimoska J, Mulcahy SP, Pagano N, Williams DS. Synlett. 2007;8:1177–1189. [Google Scholar]
- 8.The p21-activated kinases are important mediators of signaling pathways that regulate cellular behaviours and are implicated in pathological conditions including cancer. See, for example: Yi C, Maksimoska J, Marmorstein R, Kissil JL. Biochem. Pharmacol. 2010;80:683–689. doi: 10.1016/j.bcp.2010.03.012.
- 9.Other designs of metal-based kinase inhibitors: Spencer J, Mendham AP, Kotha AK, Richardson SCW, Hillard EA, Jaouen G, Male L, Hursthouse MB. Dalton Trans. 2009:918–921. doi: 10.1039/b816249b. Biersack B, Zoldakova M, Effenberger K, Schobert R. Eur. J. Med. Chem. 2010;45:1972–1975. doi: 10.1016/j.ejmech.2010.01.040. Blanck S, Cruchter T, Vultur A, Riedel R, Harms K, Herlyn M, Meggers E. Organometallics. 2011;30:4598–4606. doi: 10.1021/om200366r.
- 10.a.) Lei M, Lu W, Meng W, Parrini M-C, Eck MJ, Mayer BJ, Harrison SC. Cell. 2000;102:387–397. doi: 10.1016/s0092-8674(00)00043-x. [DOI] [PubMed] [Google Scholar]; b.) Lei M, Robinson MA, Harrison SC. Structure. 2005;13:769–778. doi: 10.1016/j.str.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 11.The lysine to arginine mutation at position 299 inactivates the enzyme but is important for the large-scale cellular expression of PAK1 for crystallization purposes. However, this mutation apparently does not lead to any significant structural changes. See ref. [10].
- 12.See Supporting Information for a kinase selectivity profile of 1.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.