

Abstract
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by multiple genetic risk factors, high levels of interferon alpha (IFN-α), and the production of autoantibodies against components of the cell nucleus. Interferon regulatory factor 5 (IRF5) is a transcription factor which induces the transcription of IFN-α and other cytokines, and genetic variants of IRF5 have been strongly linked to SLE pathogenesis. IRF5 functions downstream of Toll-like receptors and other microbial pattern-recognition receptors, and immune complexes made up of SLE-associated autoantibodies seem to function as a chronic endogenous stimulus to this pathway. In this paper, we discuss the physiologic role of IRF5 in immune defense and the ways in which IRF5 variants may contribute to the pathogenesis of human SLE. Recent data regarding the role of IRF5 in both serologic autoimmunity and the overproduction of IFN-α in human SLE are summarized. These data support a model in which SLE-risk variants of IRF5 participate in a “feed-forward” mechanism, predisposing to SLE-associated autoantibody formation, and subsequently facilitating IFN-α production downstream of Toll-like receptors stimulated by immune complexes composed of these autoantibodies.
1. Introduction
Systemic lupus erythematosus (SLE) is a complex and heterogeneous disease characterized by a strong genetic contribution and activation of a number of immune system pathways [1–3]. Recent advances in human genetics and gene expression studies have increased our understanding of the immunopathogenesis of the disorder [4]. Interferon (IFN)-α is a pleiotropic type I IFN with the potential to break self-tolerance by inducing dendritic cell differentiation, which can lead to the activation of autoreactive T and B cells [5, 6]. Serum IFN-α levels are often elevated in lupus patients [7–9] and the “IFN-α signature” of gene expression in peripheral blood mononuclear cells is present in more than 50% of SLE patients [10–14]. High IFN-α levels are associated with more severe disease and presence of particular autoantibodies [9, 14, 15]. Additionally, high levels of IFN-α are common in unaffected SLE family members, suggesting that IFN-α is a heritable risk factor [8, 16]. Moreover, some patients given recombinant human IFN-α for viral hepatitis C or malignancy have developed de novo SLE and recovered after the IFN-α was discontinued [17–19]. This body of evidence suggests that IFN-α plays a key role in etiology and pathogenesis of SLE.
Interferon regulatory factor (IRF) 5 is a transcription factor that can induce transcription of IFN-α mRNA [20]. Perhaps not surprisingly, numerous genetic studies have supported an association between SLE and various single-nucleotide polymorphisms (SNPs) and functional variants in the IRF5 gene. These genetic associations have been demonstrated across multiple ancestral backgrounds, although the exact molecular mechanisms by which these polymorphisms contribute to human disease pathogenesis are still unclear [21–33]. Other autoimmune diseases such as rheumatoid arthritis, Sjogren's syndrome, systemic sclerosis, multiple sclerosis, and inflammatory bowel disease have also been associated with IRF5 polymorphisms, suggesting a role of IRF5 in common autoimmune disease pathways [34].
Like other IRF family members, IRF5 has a prototypical helix-loop-helix and a conserved tryptophan repeat in its aminoterminal DNA-binding domain. IRF5 induces gene expression by binding to promoters containing the IFN-stimulated response element (ISRE), whose consensus sequence is GAAANN [35] and AANNNGAA [36]. IRF5 has been called the “master regulator of proinflammatory cytokines” [37] because of its role in upregulating expression of IL-6, IL-12b, IL-17, IL-23, TNF-α, IFN-β-IP-10, MCP1, and RANTES [38, 39] in addition to type 1 IFN [40]. Because IRF5 is an IFN-induced gene, its expression can potentially be enhanced via a positive feedback loop, where IFN-α production could lead to increased IRF5 expression and subsequently additional IFN-α transcription [41]. In addition, IRFs play an important role in the regulation of cell growth and apoptosis as evidenced by IRF5 playing a role in the induction of apoptosis in cancer cells [42]. While IRF5 functions in cell cycle processes and apoptosis, for the purpose of this paper we will focus on how IRF5 relates to IFN-α, and how IRF5 variants may influence the pathogenesis of SLE.
2. IRF5 and Infection
Early and accurate detection of microbial pathogens is a critical part of the immune response against pathogens. This is accomplished through the recognition of common microbial molecules called pathogen-associated molecular patterns (PAMPs) [43]. Pattern recognition receptors (PRRs) are cell surface proteins on innate immune cells that detect these PAMPs, bind them, and subsequently set off signaling cascades to initiate the immune response. PRRs include Toll-like receptors (TLRs), C-type lectin receptors, retinoic acid-inducible gene (RIG)-I-like receptors, and nucleotide-binding oligomerization domain-(NOD-) like receptors. Many downstream targets of PRRs are members of the IRF family. Type I IFN and pro-inflammatory cytokines produced downstream of PRR ligation coordinate the recruitment of other innate and adaptive immune cells, which enable the attenuation and eventual eradication of the infection.
Studies by multiple investigators show that IRF5 in particular can be induced in response to specific viral infections such as Newcastle disease virus (NDV), vesicular stomatitis virus, and herpes simplex virus type 1 [20, 41, 44]. IRF5 expression is mainly restricted to dendritic cells, B cells, macrophages, and monocytes [39, 41], a pattern which is unique from other IRF family members. Although IRF5 expression may be constitutive, its activity must be induced via several posttranslational modifications at multiple amino acid residues (discussed below).
3. IRF5 Is a Downstream Target of TLR7 and TLR9
Rather than being on the cell surface, TLR7, 8, and 9 are localized in the endosomal compartment, along with TLR3. TLR7 and TLR8 recognize single-stranded RNA viruses, while TLR9 recognizes double-stranded DNA (dsDNA) viruses or CpG motifs on bacteria. As shown in Figure 1, IRF5 is activated following engagement of TLR7 or 9, and perhaps TLR8. Of note, early studies in the characterization of TLR7 and TLR9 were performed in mutant mice [45, 46], but there is no mouse ortholog of TLR8. Therefore, less is known about the regulation and downstream signaling of TLR8, which is expressed only in humans.
Figure 1.
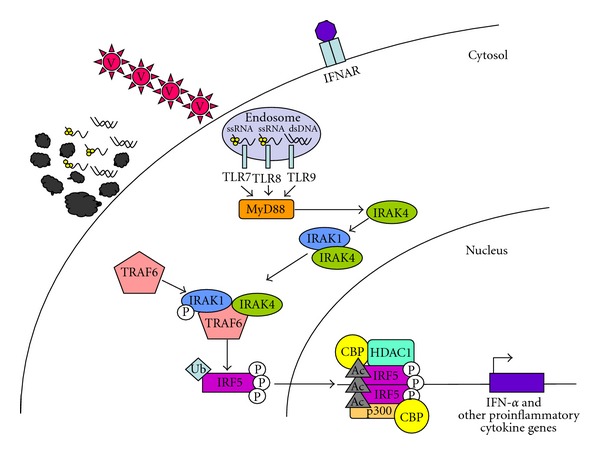
Schematic model forIRF5 activation. Cells use TLRs as sensors to detect the presence of viruses (V) via TLR7, -8, and -9. Alternatively, apoptotic debris (shown here as membrane blebs, ssRNA, and dsDNA) can also be a source of nuclear proteins and nucleic acids. Nuclear material is brought to the endosome, triggering TLR7, -8, and -9 signaling. Binding of cognate ligands to these TLRs recruits MyD88, a main signaling intermediate involved in TLR7, -8, and -9 signaling. MyD88 recruits interleukin-1 receptor associated kinase (IRAK)-4. IRAK-4 binds and phosphorylates IRAK-1, which in turn recruits Tumor necrosis factor (TNF) receptor associated factor (TRAF) 6 [46–48]. TRAF6 is an E3 ubiquitin (Ub) ligase that adds K63-Ub chains to IRF5 [49]. IRF5 is then shuttled to the nucleus and is acetylated by CBP and p300 [50]. Together, these events set the stage for the transcription of IFN-α and other pro-inflammatory cytokine genes.
In human plasmacytoid dendritic cells (pDCs), recognition of cognate TLR7 and TLR9 ligands leads to the activation of IRF5 [51], via the signaling intermediate MyD88. As an adaptor protein that has a Toll/interleukin (IL)-1 domain, MyD88 recruits interleukin-1 receptor associated kinase (IRAK)-4. IRAK-4 binds and phosphorylates IRAK-1, which in turn recruits tumor necrosis factor (TNF) receptor associated factor (TRAF) 6 [46–48]. TRAF6 is an E3 ubiquitin (Ub) ligase that adds K63-Ub chains to IRF5 [49]. Together, these events set the stage for the translocation of IRF5 into the nucleus.
4. Activation and Regulation of IRF5
Regulation of IRF5 activation is still not well understood. The C-terminal end of IRF5 has been shown to be autoinhibitory in an IFN-α reporter assay [41, 52, 53]. Upon stimulation, IRF5 is modified posttranslationally by phosphorylation and ubiquitination. Multiple phosphorylated residues have recently been identified (alignment positions based on IRF5 variant (v)5: T10, S158, S309, S317, S451, and S462) [54]. An additional putative phosphorylation site has been proposed at S430 on IRF5v4 (equivalent to S456 on IRF5v5) [55]. However, the importance of each phosphorylation event on IRF5 function is not clear. Chen et al. hypothesized that phosphorylation at these positions facilitated the unfolding of the auto-inhibitory structure of IRF5 monomers, promoting self-dimerization, and exposing a surface for CREB-binding protein (CBP)/p300 binding (see Figure 1 below) [55]. While there is no doubt that IRF5 is phosphorylated following stimulation through TLR7 or 9 [41], which downstream kinases and at what sites remains an area of active investigation. It is possible that pathway-specific IRF5 activation is achieved through the use of different kinases, each of which would presumably phosphorylate distinct amino acid residues. As described below, progress has been made addressing this issue in the context of RIG-I and NOD pathway regulation.
Evidence from viral stimulation and overexpression systems has shown that RIG-I pathway kinases IκB kinase (IKK)-ε and TANK-binding kinase (TBK) 1 can phosphorylate IRF5 [41, 44, 45, 56], but this phosphorylation is not sufficient for IRF5 nuclear translocation [56]. A recent study used mass spectrometry to identify residues S158 and S462 on IRF5v5 as targets of TBK1, a kinase involved in the RIG-I pathway [54]. These events induced IL-6 transcription, but did not transactivate IFN-α promoter activity [44, 56]. Studies using viral stimulation have been less clear. Barnes et al. showed that NDV induced phosphorylation of IRF5 in 2fTGH cells transfected with IRF5 [41] as well as translocation into the nucleus and transactivation of an IFN-α reporter construct [20]. Cheng et al. demonstrated that NDV infection did not lead to phosphorylation of IRF5 in a HEC-1B/GFP-IRF5 system [44]. This discrepancy can perhaps be explained by the differences in cell type and/or cell tropism of the viruses. Interestingly, contrary to NDV infection, Sendai virus infection in 2fTGH cells led to activation of IRF3 and IRF7, but not IRF5 [20]. Moreover, IRF5 and 7 seem to have overlapping binding partners and functions, making it difficult to distinguish the dependence of either IRF on IFN-α transactivation [57]. To better understand the requirement of IRF5 on IFN-α regulation, biochemical studies need to be done in the context of IRF7−/− cells.
As with other IRF family members, IRF5 can form homodimers upon phosphorylation. This was demonstrated in a study in which GFP- and T7-tagged IRF5 were cotransfected with IKKε into HEC-1B cells. Pull-down assays with anti-T7 antibodies showed the presence of GFP-tagged IRF5 [44]. In support of this concept, crystallographic analysis of the C-terminal fragment of IRF5(v4) S430D showed the formation of stable homodimers [55]. In addition, like IRF3, IRF5 interacts with CBP/p300 [44, 55]. Size exclusion chromatography studies have shown two molecules of IRF5 S430D binding to two molecules of CBP, forming an IRF52CBP2 complex [55]. IRF5 can also form dimers with IRF1, IRF3, and IRF7 [41, 57]. This interaction was enhanced upon stimulation with virus. Whereas binding of IRF3 with IRF5 synergistically augmented IFN-α reporter activity [41], IRF5/IRF7 heterodimers blocked each other's DNA-binding domains and prevented the ability of either to bind cognate DNA sequences, resulting in the repression of IFN-α promoter activity [57].
In addition to phosphorylation, ubiquitylation represents another important means of regulating protein expression and activity. Two types of poly-ubiquitin (Ub) chains dictate the fate of proteins: K48-Ub and K63-Ub, where the number refers to the position of the lysine (K) residue upon which the chains of Ub are built. E3 Ub ligases are responsible for adding Ub chains to either proteins destined for degradation (K48-Ub) or for activating signal transducing proteins (K63-Ub) [58]. The E3 Ub ligase TRAF6 is activated by TLR7 and 9 signaling via MyD88 and IRAK-1. The addition of K63-Ub on IRF5 by TRAF6 is necessary for nuclear translocation and IFN-α transactivation. Lysines 410 and 411 are putative targets of K63-Ub since mutagenesis of these lysines to arginines abolished nuclear translocation and IFN-α promoter activity [48].
K63-Ub-IRF5 could potentially be subjected to negative regulation by deubiquitinating enzymes such as TNF-α-induced protein 3 (TNFAIP3, also known as A20) [59]. With regard to type 1 IFN-induced gene activity, it is unknown whether TNFAIP3 can influence TLR7 and TLR9-mediated signaling via IRF5. IRF5 activity in an IL-12p40 luciferase reporter assay system was reduced with increased expression of TNFAIP3 [54]. This system utilized receptor interactive protein kinase 2 (RIP2), a kinase involved in the NOD signaling pathway.
Trafficking of molecules in and out of the nucleus is a tightly controlled process coordinated by importins and exportins on the nuclear membrane. These proteins recognize and bind to nuclear localization sequences (NLS) and nuclear export sequences (NES) encoded in the amino acid sequence. IRF5 has one NES (IRF5 v5 aa150-LQRMLPSLSLT-160 [44, 56] and two NLS's (IRF5 v4 aa12-PRRVRLK-18 and aa398-PREKKLI-404) [41, 55, 56, 60]. A specific inhibitor of the nuclear export protein CRM1, leptomycin B (LMB), has been used to monitor IRF5 nuclear trafficking. Treatment with LMB results in nuclear retention of IRF5 [56], indicating that IRF5 is continuously exported out of the nucleus.
Recently, investigators have presented evidence demonstrating the regulation of transcription factor activity by acetylation/deacetylation [61]. IRF1, 2, and 7 have been shown to be acetylated by histone acetylases [62, 63]. In a study by Feng et al., IRF5 appears to be one transcription factor subject to this form of regulation as well [50]. When 2fTGH cells expressing human IRF5 and either an ISRE- or an IFNA1-dependent luciferase reporter construct were stimulated with virus in the presence of trichostatin A (histone deacetylase (HDAC) inhibitor), luciferase activity was ablated. Furthermore, they showed that under uninfected conditions, IRF5 forms a multicomponent complex with the corepressors HDAC1, silencing mediator of retinoic acid and thyroid hormone receptor (SMRT), and Sin3a to inhibit the luciferase reporter activity. Upon infection with NDV, IRF5 binds to histone acetylase (HAT) proteins p300, CBP, and PCAF while SMRT is exported out of the nucleus. It appears that IRF5 may be acetylated at several lysine residues since an antibody against acetylated lysine, which was used to immunoprecipitate overexpressed IRF5 fragments, pulled out both N- and C-terminal IRF5 fragments. Taken together, IRF5 activity is highly regulated post-translationally. Multiple phosphorylation, ubiquitylation, and acetylation events must all be coordinated to induce IRF5 transactivation.
Not only is IRF5 activation regulated by different enzymes, but also IRF5 gene expression is complex. There are up to eleven distinct isoforms of IRF5 resulting from alternative splicing [22, 60]. Four different IRF5 transcripts result from alternative usage of the first, noncoding exon (as shown in Figure 2(b)). In the study by Mancl et al., IRF5 isoforms were differentially expressed in various purified immune cell subpopulations, though more than one isoform could be expressed in the same subpopulation [60]. For example, pDCs constitutively expressed IRF5 variants 1–4 [60]. Moreover, different IRF5 isoforms activated the IFN-α and IFN-β promoters to varying degrees, where isoform 3/4 induced the highest levels of activity [60]. In summary, many points of IRF5 regulation are possible, and greater IRF5 activity could generate an IFN-α-rich environment which could lead to SLE disease susceptibility.
Figure 2.
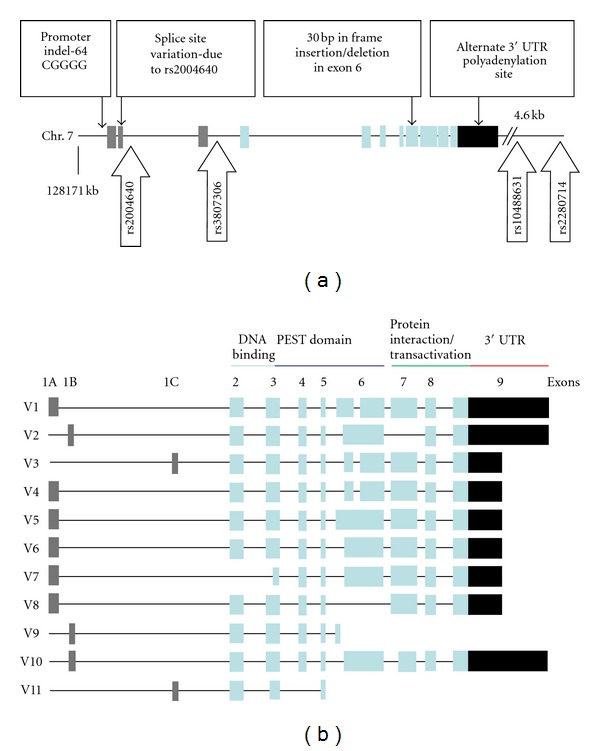
(a) IRF5 gene marked with previously reported functional variants along with studied SNPs [64]. The first three grey boxes represent differentially spliced first exons (1A, 1B, and 1C), the next light blue boxes represent the exons 2–9, and the last black box indicates the 3′ UTR. SNPs rs2280714 and rs10488631 were used as proxies for rs10954213 in the 3′ UTR due to high LD. (b) IRF5 mRNA isoforms [22]. There are eleven different variants. PEST, proline-, glutamic acid-, serine-, and threonine-rich.
5. Genetic Variants in IRF5 Are Associated with Systemic Lupus Erythematosus
The IRF5 locus was first implicated in SLE through a candidate gene analysis involving patients of Nordic ancestry. The SNP rs2004640 which was associated in this study introduced a new donor splice site, suggesting alternate exon 1 splicing may occur in the context of this variant [21]. A subsequent study by Graham et al. strongly replicated the association of rs2004640 with SLE in multiple independent case-control cohorts, including cases and controls from Europe, North Americans of European ancestry, and a cohort from Argentina [22]. This study also confirmed that the risk allele allowed for alternate splicing of the first exon [22]. This study described three different alternate first exons (1A, 1B, and 1C) and showed that mRNAs containing 1B could only be made when the rs2004640 risk allele was present (Figure 2(b)). The first exon is not translated, so despite this clear impact upon splicing, the functional significance of exon 1B transcripts is not clear. Even when exon 1B transcripts are produced in the setting of the splice variant, they are present at levels which are 100 times lower than those derived from other exon 1 transcripts, such as exon 1A [22, 65].
A second SNP in the 3′ region of the IRF5 locus was associated with increased IRF5 expression [22], and an SLE-risk haplotype was described that was composed of the high expression variant of this SNP along with the alternate splice variant of rs2004640. The high expression allele was not dependent upon the splice variant in this study, suggesting that there were multiple functional elements in IRF5. The high expression allele was correlated with a SNP in the 3′UTR region which introduces an alternate poly-adenylation (poly-A) site and provides a potential explanation for higher IRF5 mRNA abundance in the presence of this allele [23, 65, 66]. The SLE-risk allele of this SNP results in the production of a shorter poly-A tail, which is more stable and resistant to degradation, leading to a longer IRF5 mRNA half-life and greater mRNA abundance (Figure 2) [23, 65].
6. Insertion/Deletion Polymorphisms in IRF5
In addition to the SNP variants detailed above, common insertion/deletion (indel) polymorphisms in IRF5 have been reported, including a 30-base pair (bp) in-frame indel in exon 6, and a promoter indel [23, 28, 67]. The exon 6 insertion is present on both risk and nonrisk haplotypes. While this would suggest that it does not independently contribute to SLE-risk related to IRF5, the insertion is present on the risk haplotype and a cooperative role in pathogenesis cannot be ruled out. The exon 6 insertion is located in a proline-, glutamic acid-, serine-, and threonine-rich domain which can affect protein stability and function of IRF5 (Figure 2(b)) [8, 23, 67]. Moreover, a promoter indel has been described, which is 5-base long (CGGGG/−), and this insertion polymorphism in the promoter is also present on the SLE-risk haplotype. This promoter variant confers risk of SLE independently from the risk haplotype presented by Graham et al. [23, 28], as shown in Table 1. The promoter indel is in high linkage disequilibrium (LD) with the exon 1 splice site variation, and it is possible that this variant could explain the risk signal from the 5′ region of the gene (Figure 2(a)). The SLE-associated insertion creates an additional SP1 transcription factor binding site and leads to increased IRF5 expression [28]. Whether the promoter indel or the 3′ UTR variant is more important for IRF5 mRNA abundance is not currently understood, and SLE-associated haplotypes carry both of these polymorphisms, suggesting that both may be required to result in risk of SLE. IRF5 polymorphisms found to be associated with SLE in seminal candidate gene case-control studies are summarized in Table 1. Subsequent candidate gene and genome-wide association studies have strongly replicated these findings [24–27, 29–33].
Table 1.
Summary of genetic variants found in early seminal studies.
| Ancestry | Samples | Study type | Genetic variants | OR, P values |
Functions | |
|---|---|---|---|---|---|---|
| Sigurdsson et al., 2005 [21] | Swedish, Finnish |
589 cases 377 controls |
FB and CC association | rs2004640 |
OR = 1.59 P = 7.1 × 10−7 |
Altered exon 1 spicing |
|
| ||||||
| Graham et al., 2006 [22]* | Argentina, Spain, Sweden, USA |
1661 cases 2508 controls |
CC association | rs2004640 | OR = 1.45 P = 4.4 × 10−16 |
Altered exon 1 splicing |
|
| ||||||
| 555 trio pedigrees, |
Risk haplotype | OR = 1.78 P = 1.4 × 10−19 |
Altered exon 1 splicing, exon 6 in, short poly-A | |||
| Graham et al., 2007 [23]** | USA, UK, Sweden |
2188 cases | FB and CC association | Protective haplotype 1 | OR = 0.76 P = 5.0 × 10−8 |
Nonaltered exon 1 splicing, exon 6 in, long poly-A |
| 3596 controls | Protective haplotype 2 | OR = 0.76 P = 2.8 × 10−5 |
Nonaltered exon 1 splicing, exon 6 del, short poly-A | |||
|
| ||||||
| Sigurdsson et al., 2008 [28]*** | Sweden | 485 cases 563 controls |
CC association | CGGGG/− | OR = 1.69 P = 4.6 × 10−9 |
Promoter indel |
| rs10488631 | OR = 2.07 P = 9.4 × 10−10 |
Altered exon 1 splicing, exon 6 in, short poly-A | ||||
*The populations were mostly of European ancestry.
**Only the haplotype analysis is shown here. SNP rs2070197 was found to be a proxy for the risk haplotype.
***SNP rs10488631 is in high LD with rs2070197 and was used as a proxy for the risk haplotype. OR and P values are obtained from nonconditional analysis.
FB: family based, CC: case-control, OR: odds ratio, P: P value, poly-A: poly-adenylation, in: insertion, del: deletion, indel: insertion/deletion, LD: linkage disequilibrium.
7. Genetic Similarities and Differences by Ancestry
The risk alleles described above were initially found in European ancestry subjects, and while an association between IRF5 and SLE has been subsequently confirmed in other ancestral backgrounds, the particular associated polymorphisms differ somewhat [24–27, 29]. For example, intron 1 SNPs (rs6953165 and rs41298401) but not exon 6 indel or 3′ UTR poly-A polymorphisms were found to be associated with SLE in Japanese population, and they were related to differential expression of several IFN pathway genes although not IRF5 itself [26]. On the other hand, the European risk haplotype and its homozygosity appear more frequently in Mexican SLE patients compared to European patients [25], and in this ancestral background the European haplotype is a strong risk factor. In African Americans, a novel SNP rs3807306 was associated with SLE, although a functional role has not been defined [27]. We have performed follow-up work in African American and African populations which suggests that the European SLE-risk haplotype is present in African Americans due to European admixture and is associated with risk of SLE, but this haplotype was not present in African populations, and an African-derived SLE-risk haplotype was not observed in this study [23].
8. Autoantibodies, IFN-α and IRF5 Variants
Further studies are needed to clarify how different combinations of the genetic elements of IRF5 lead to SLE susceptibility, and what roles they play in the molecular pathogenesis of the disease. We have shown that the European risk haplotype is associated with increased serum IFN-α in SLE patients [68], and subsequent studies have supported this concept by showing that SLE-associated IRF5 variants are associated with increased activation of the IFN-α pathway [69, 70]. However, the association between the risk haplotype and increased serum IFN-α in SLE patients was only observed in those patients who had anti-dsDNA or anti-RNA-binding protein (RBP) autoantibodies [68]. We expanded these findings in a study involving 1034 and 555 SLE patients with European and African ancestries, respectively [64]. The functional variants and SNPs studied are depicted in Figure 2(a). As shown in Table 2, the previously reported SLE-risk haplotype TACA [23] was associated with anti-dsDNA and anti-Ro antibodies, whereas the TATA haplotype which has previously been reported as a neutral haplotype [23] was associated with anti-dsDNA antibodies in case-case analysis. Similar patterns were detected in case-control analysis where the TACA and TATA haplotypes were associated with anti-dsDNA positive patients versus controls (Odds Ratio (OR) = 2.79, P = 2.9 × 10−20) and the TACA haplotype with anti-Ro positive patients versus controls (OR = 2.57, P = 1.8 × 10−14). The TACA haplotype is characterized by the presence of all four functional variants, the insertions in the IRF5 promoter and exon 6, the spice variant, and the poly-A variant, whereas the TATA haplotype has all but the exon 6 insertion [64]. The fact that these two haplotypes which differ only at the exon 6 insertion are associated with different autoantibody profiles suggests a functional relevance of the exon 6 insertion. Functional studies of the exon 6 insertion to date support a role for exon 6 variants in altering its nuclear translocation, impacting apoptosis and cytokine production [67]. Moreover, our study showed that the haplotypes associated with particular auto-antibodies resulted in increased levels of serum IFN-α only in the presence of that particular associated autoantibody. The above data support a pathogenic model in which these auto-antibodies chronically stimulate the endosomal TLR system, and specific IRF5 variants in conjunction with particular autoantibodies dysregulate IFN-α production, resulting in increased risk of SLE (Figure 3) [64].
Table 2.
European ancestry case-case analysis showing IRF5 haplotypes with associated functional elements and serological associations [64].
| Tag SNP haplotype | Promoter indel | Splice variant | Exon 6 indel | Poly-A variant | Serologic association |
|---|---|---|---|---|---|
| (1) TACA | In | Present | In | Present | Anti-Ro: OR = 1.50, P = 2.0 × 10−3
Anti-dsDNA: OR = 1.51, P = 7.4 × 10−3 |
| (2) TATA | In | Present | Del | Present | Anti-dsDNA: OR = 1.68, P = 4.9 × 10−5 |
| (3) TCTA | Del | Present | In | Absent | Anti-La: OR = 3.51, P = 7.5 × 10−3 |
| (4) GCTA | Del | Absent | Del | Present | — |
| (5) GCTG | Del | Absent | In | Absent | — |
The haplotypes are shown as each of the four alleles in order from 5′ to 3′ (rs2004640, rs3807306, rs10488631, rs2280714).
SNP: single nucleotide polymorphism, indel: insertion/deletion, Poly-A: poly-adenylation, In: insertion, Del: deletion, OR: odds ratio, P: P value.
Figure 3.
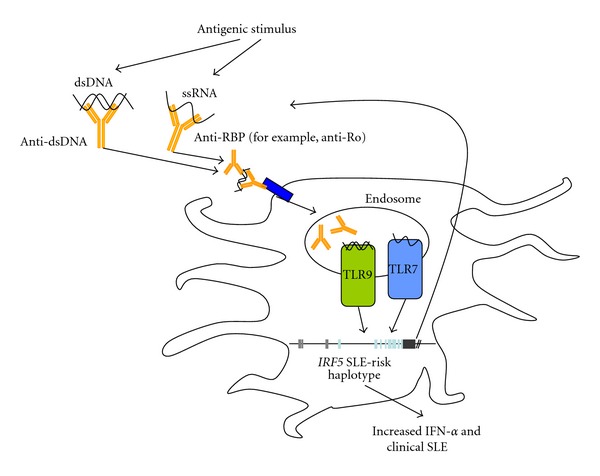
Diagram showing relationships between SLE-associated autoantibodies, IRF5 genotype and IFN-α involved in the pathogenesis of SLE [64]. This suggests a “feed-forward” model in which specific auto-antibodies interact with particular IRF5 risk variants which also predispose to the same antibody formation.
The data presented above support a “gene + autoantibody = high IFN-α and risk of SLE” model, and presumably the associations between IRF5 genotype and autoantibodies may be due to this interaction. Based upon these data, we cannot rule out the possibility that IRF5 risk genotype could directly predispose to the formation of SLE-associated autoantibodies. In fact, IRF5 knockouts of murine SLE models have decreased levels of SLE-associated auto-antibodies [71, 72]. This may be due to the role of IRF5 in regulating transcription of Prdm1 which encodes Blimp-1, an essential regulator of plasma cell differentiation [73]. To answer this question in humans, we studied IRF5 genotype in a unique cohort of anti-Ro autoantibody positive European subjects who carried a variety of diagnoses, including many who were asymptomatic and generally did not have high levels of circulating IFN-α [74]. We found that the IRF5 SLE-risk haplotype was enriched even in these asymptomatic subjects with positive anti-Ro antibody, and that this enrichment was even greater (OR ~ 5) in those initially asymptomatic Ro-positive individuals who later developed SLE [75]. Taken together, these data support a “feed-forward” hypothesis in which the risk haplotype predisposes to the formation of autoantibodies, and these autoantibodies subsequently lead to increased production of IFN-α in conjunction with the same IRF5 variant (Figure 3) [75].
9. Conclusions
In this paper, we examined how IRF5 is regulated and activated, and how its genetic variants can influence the risk of SLE by differentially activating the IFN-α pathway along with affecting the production of SLE-associated autoantibodies. The above data support an interesting novel model of SLE pathogenesis, in which genetic variations lead to serologic autoimmunity, subsequently creating a microenvironment which stimulates PRRs and results in high IFN-α [76].
A number of other SLE-associated genetic variants in the IFN-α and PRR pathways result in increased IFN-pathway activation [77–82], further supporting the concept that gain-of-function polymorphisms in the IFN-α and PRR pathways contribute to SLE susceptibility. While the exact initial trigger of autoimmunity in SLE remains unclear, possible antigenic sources include ultraviolet light, viruses, and demethylating drugs [83]. Recently, several studies point toward neutrophils as a factor in lupus pathogenesis [84, 85]. It has been hypothesized that chronic activation of neutrophils by immune complexes via Fc receptors induces them to release neutrophil extracellular traps (NETs) in a suicidal process called NETosis. NETs contain genomic DNA, providing a source of antigenic self-DNA. These would in turn stimulate TLRs on pDCs, putting in motion a vicious cycle of increased IFN-α and eventual autoimmune disease.
It is clear that IRF5 is a major pathogenic factor in human lupus, which will impact upon aspects of SLE diagnosis, prognosis, and management. Predictive models which include autoantibodies, IFN-α and other molecular measurements, and genetic variants may prove useful in diagnosis or prognosis. It seems unlikely that a purely genetic model will be sufficiently predictive, but the work summarized here demonstrates how other molecular phenotypes can greatly enhance the predictive capacity of genetic data. Additionally, the pathway in which IRF5 functions is currently being targeted by therapeutics directed at the endosomal TLRs and IFN-α [86, 87], and it is possible that IRF5 genotype may help to define responder/nonresponder groups with respect to these therapies. The complexity demonstrated by this one disease-associated locus is staggering and suggests that we still have much work to do in understanding the genetic basis of human autoimmune disease.
Disclosure
The authors report no financial conflict of interests.
Conflict of Interests
The authors declare that they have no conflict of interests.
Authors' Contribution
C. M. Cham and K. Ko contributed equally.
Acknowledgments
This work is supported by K Ko-Arthritis Foundation Clinical to Research Transition Award: TB Niewold-NIH R01 AR060861, K08 AI083790, P30 DK42086, NIAID Clinical Research Loan Repayment AI071651, NIH CTSA Core Subsidy Grant, and CTSA Pilot Grants from UL1 RR024999, Lupus Research Institute Novel Research Grant, and an Alliance for Lupus Research Target Identification in Lupus Grant.
References
- 1.Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis and Rheumatism. 1997;40(9):p. 1725. doi: 10.1002/art.1780400928. [DOI] [PubMed] [Google Scholar]
- 2.Alarcón-Segovia D, Alarcón-Riquelme ME, Cardiel MH, et al. Familial aggregation of systemic lupus erythematosus, rheumatoid arthritis, and other autoimmune diseases in 1,177 lupus patients from the GLADEL cohort. Arthritis and Rheumatism. 2005;52(4):1138–1147. doi: 10.1002/art.20999. [DOI] [PubMed] [Google Scholar]
- 3.Rhodes B, Vyse TJ. The genetics of SLE: an update in the light of genome-wide association studies. Rheumatology. 2008;47(11):1603–1611. doi: 10.1093/rheumatology/ken247. [DOI] [PubMed] [Google Scholar]
- 4.Tsokos GC. Systemic lupus erythematosus. The New England Journal of Medicine. 2011;365(22):2110–2121. doi: 10.1056/NEJMra1100359. [DOI] [PubMed] [Google Scholar]
- 5.Blanco P, Palucka AK, Gill M, Pascual V, Banchereau J. Induction of dendritic cell differentiation by IFN-α in systemic lupus erythematosus. Science. 2001;294(5546):1540–1543. doi: 10.1126/science.1064890. [DOI] [PubMed] [Google Scholar]
- 6.Niewold TB, Clark DN, Salloum R, Poole BD. Interferon alpha in systemic lupus erythematosus. Journal of Biomedicine and Biotechnology. 2010;2010:8 pages. doi: 10.1155/2010/948364.948364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim T, Kanayama Y, Negoro N, Okamura M, Takeda T, Inoue T. Serum levels of interferons in patients with systemic lupus erythematosus. Clinical and Experimental Immunology. 1987;70(3):562–569. [PMC free article] [PubMed] [Google Scholar]
- 8.Niewold TB, Hua J, Lehman TJA, Harley JB, Crow MK. High serum IFN-α activity is a heritable risk factor for systemic lupus erythematosus. Genes and Immunity. 2007;8(6):492–502. doi: 10.1038/sj.gene.6364408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weckerle CE, Franek BS, Kelly JA, et al. Network analysis of associations between serum interferon-α activity, autoantibodies, and clinical features in systemic lupus erythematosus. Arthritis and Rheumatism. 2011;63(4):1044–1053. doi: 10.1002/art.30187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baechler EC, Batliwalla FM, Karypis G, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(5):2610–2615. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bennett L, Palucka AK, Arce E, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. Journal of Experimental Medicine. 2003;197(6):711–723. doi: 10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Han GM, Chen SL, Shen N, Ye S, Bao CD, Gu YY. Analysis of gene expression profiles in human systemic lupus erythematosus using oligonucleotide microarray. Genes and Immunity. 2003;4(3):177–186. doi: 10.1038/sj.gene.6363966. [DOI] [PubMed] [Google Scholar]
- 13.Ishii T, Onda H, Tanigawa A, et al. Isolation and expression profiling of genes upregulated in the peripheral blood cells of systemic lupus erythematosus patients. DNA Research. 2005;12(6):429–439. doi: 10.1093/dnares/dsi020. [DOI] [PubMed] [Google Scholar]
- 14.Kirou KA, Lee C, George S, et al. Coordinate overexpression of interferon-α-induced genes in systemic lupus erythematosus. Arthritis and Rheumatism. 2004;50(12):3958–3967. doi: 10.1002/art.20798. [DOI] [PubMed] [Google Scholar]
- 15.Ko K, Franek BS, Marion M, Kaufman KM, Langefeld CD, Harley JB. Genetic ancestry, serum interferon-alpha activity, and autoantibodies in systemic lupus erythematosus. Journal of Rheumatology. 2012;39(6):1238–1240. doi: 10.3899/jrheum.111467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Niewold TB, Adler JE, Glenn SB, Lehman TJA, Harley JB, Crow MK. Age- and sex-related patterns of serum interferon-α activity in lupus families. Arthritis and Rheumatism. 2008;58(7):2113–2119. doi: 10.1002/art.23619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ronnblom LE, Oberg KE, Alm GV. Possible induction of systemic lupus erythematosus by interferon α-treatment in a patient with a malignant carcinoid tumour. Journal of Internal Medicine. 1990;227(3):207–210. doi: 10.1111/j.1365-2796.1990.tb00144.x. [DOI] [PubMed] [Google Scholar]
- 18.Niewold TB, Swedler WI. Systemic lupus erythematosus arising during interferon-alpha therapy for cryoglobulinemic vasculitis associated with hepatitis C. Clinical Rheumatology. 2005;24(2):178–181. doi: 10.1007/s10067-004-1024-2. [DOI] [PubMed] [Google Scholar]
- 19.Niewold TB. Interferon alpha-induced lupus: proof of principle. Journal of Clinical Rheumatology. 2008;14(3):131–132. doi: 10.1097/RHU.0b013e318177627d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barnes BJ, Moore PA, Pitha PM. Virus-specific activation of a novel interferon regulatory factor, IRF-5, results in the induction of distinct interferon α genes. Journal of Biological Chemistry. 2001;276(26):23382–23390. doi: 10.1074/jbc.M101216200. [DOI] [PubMed] [Google Scholar]
- 21.Sigurdsson S, Nordmark G, Göring HHH, et al. Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lupus erythematosus. American Journal of Human Genetics. 2005;76(3):528–537. doi: 10.1086/428480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Graham RR, Kozyrev SV, Baechler EC, et al. A common haplotype of interferon regulatory factor 5 (IRF5) regulates splicing and expression and is associated with increased risk of systemic lupus erythematosus. Nature Genetics. 2006;38(5):550–555. doi: 10.1038/ng1782. [DOI] [PubMed] [Google Scholar]
- 23.Graham RR, Kyogoku C, Sigurdsson S, et al. Three functional variants of IFN regulatory factor 5 (IRF5) define risk and protective haplotypes for human lupus. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(16):6758–6763. doi: 10.1073/pnas.0701266104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shin HD, Sung YK, Choi CB, Lee SO, Lee HW, Bae SC. Replication of the genetic effects of IFN regulatory factor 5 (IRF5) on systemic lupus erythematosus in a Korean population. Arthritis Research and Therapy. 2007;9, article R32 doi: 10.1186/ar2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reddy MVPL, Velázquez-Cruz R, Baca V, et al. Genetic association of IRF5 with SLE in Mexicans: higher frequency of the risk haplotype and its homozygozity than Europeans. Human Genetics. 2007;121(6):721–727. doi: 10.1007/s00439-007-0367-6. [DOI] [PubMed] [Google Scholar]
- 26.Kawasaki A, Kyogoku C, Ohashi J, et al. Association of IRF5 polymorphisms with systemic lupus erythematosus in a Japanese population: support for a crucial role of intron 1 polymorphisms. Arthritis and Rheumatism. 2008;58(3):826–834. doi: 10.1002/art.23216. [DOI] [PubMed] [Google Scholar]
- 27.Kelly JA, Kelley JM, Kaufman KM, et al. Interferon regulatory factor-5 is genetically associated with systemic lupus erythematosus in African Americans. Genes and Immunity. 2008;9(3):187–194. doi: 10.1038/gene.2008.4. [DOI] [PubMed] [Google Scholar]
- 28.Sigurdsson S, Göring HHH, Kristjansdottir G, et al. Comprehensive evaluation of the genetic variants of interferon regulatory factor 5 (IRF5) reveals a novel 5 bp length polymorphism as strong risk factor for systemic lupus erythematosus. Human Molecular Genetics. 2008;17(6):872–881. doi: 10.1093/hmg/ddm359. [DOI] [PubMed] [Google Scholar]
- 29.Siu HO, Yang W, Lau CS, et al. Association of a haplotype of IRF5 gene with systemic lupus erythematosus in Chinese. Journal of Rheumatology. 2008;35(2):360–362. [PubMed] [Google Scholar]
- 30.Hom G, Graham RR, Modrek B, et al. Association of systemic lupus erythematosus with C8orf13-BLK and ITGAM-ITGAX. New England Journal of Medicine. 2008;358(9):900–909. doi: 10.1056/NEJMoa0707865. [DOI] [PubMed] [Google Scholar]
- 31.Gateva V, Sandling JK, Hom G, et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nature Genetics. 2009;41(11):1228–1233. doi: 10.1038/ng.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Han JW, Zheng HF, Cui Y, Sun LD, Ye DQ, Hu Z. Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nature Genetics. 2009;41(11):1234–1237. doi: 10.1038/ng.472. [DOI] [PubMed] [Google Scholar]
- 33.Jarvinen TM, Hellquist A, Zucchelli M, Koskenmies S, Panelius J, Hasan T. Replication of GWAS-identified systemic lupus erythematosus susceptibility genes affirms B-cell receptor pathway signalling and strengthens the role of IRF5 in disease susceptibility in a Northern European population. Rheumatology. 2012;51(1):87–92. doi: 10.1093/rheumatology/ker263. [DOI] [PubMed] [Google Scholar]
- 34.Nordang GB, Viken MK, Amundsen SS, Sanchez ES, Flato B, Forre OT. Interferon regulatory factor 5 gene polymorphism confers risk to several rheumatic diseases and correlates with expression of alternative thymic transcripts. Rheumatology. 2012;51(4):619–626. doi: 10.1093/rheumatology/ker364. [DOI] [PubMed] [Google Scholar]
- 35.Nelson N, Marks MS, Driggers PH, Ozato K. Interferon consensus sequence-binding protein, a member of the interferon regulatory factor family, suppresses interferon-induced gene transcription. Molecular and Cellular Biology. 1993;13(1):588–599. doi: 10.1128/mcb.13.1.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Escalante CR, Yie J, Thanos D, Aggarwal AK. Structure of IRF-1 with bound DNA reveals determinants of interferon regulation. Nature. 1998;391(6662):103–106. doi: 10.1038/34224. [DOI] [PubMed] [Google Scholar]
- 37.Santana-de Anda K, Gomez-Martin D, Diaz-Zamudio M, Alcocer-Varela J. Interferon regulatory factors: beyond the antiviral response and their link to the development of autoimmune pathology. Autoimmunity Reviews. 2011;11(2):98–103. doi: 10.1016/j.autrev.2011.08.006. [DOI] [PubMed] [Google Scholar]
- 38.Sweeney SE. Targeting interferon regulatory factors to inhibit activation of the type I IFN response: implications for treatment of autoimmune disorders. Cellular Immunology. 2011;271(2):342–349. doi: 10.1016/j.cellimm.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krausgruber T, Blazek K, Smallie T, et al. IRF5 promotes inflammatory macrophage polarization and T H1-TH17 responses. Nature Immunology. 2011;12(3):231–238. doi: 10.1038/ni.1990. [DOI] [PubMed] [Google Scholar]
- 40.Kozyrev SV, Alarcon-Riquelme ME. The genetics and biology of Irf5-mediated signaling in lupus. Autoimmunity. 2007;40(8):591–601. doi: 10.1080/08916930701510905. [DOI] [PubMed] [Google Scholar]
- 41.Barnes BJ, Kellum MJ, Field AE, Pitha PM. Multiple regulatory domains of IRF-5 control activation, cellular localization, and induction of chemokines that mediate recruitment of T lymphocytes. Molecular and Cellular Biology. 2002;22(16):5721–5740. doi: 10.1128/MCB.22.16.5721-5740.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Taniguchi T, Ogasawara K, Takaoka A, Tanaka N. IRF family of transcription factors as regulators of host defense. Annual Review of Immunology. 2001;19:623–655. doi: 10.1146/annurev.immunol.19.1.623. [DOI] [PubMed] [Google Scholar]
- 43.Kawasaki T, Kawai T, Akira S. Recognition of nucleic acids by pattern-recognition receptors and its relevance in autoimmunity. Immunological Reviews. 2011;243(1):61–73. doi: 10.1111/j.1600-065X.2011.01048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheng TF, Brzostek S, Ando O, Van Scoy S, Kumar KP, Reich NC. Differential activation of IFN regulatory factor (IRF)-3 and IRF-5 transcription factors during viral infection. Journal of Immunology. 2006;176(12):7462–7470. doi: 10.4049/jimmunol.176.12.7462. [DOI] [PubMed] [Google Scholar]
- 45.Schoenemeyer A, Barnes BJ, Mancl ME, et al. The interferon regulatory factor, IRF5, is a central mediator of toll-like receptor 7 signaling. Journal of Biological Chemistry. 2005;280(17):17005–17012. doi: 10.1074/jbc.M412584200. [DOI] [PubMed] [Google Scholar]
- 46.Takaoka A, Yanai H, Kondo S, et al. Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature. 2005;434(7030):243–249. doi: 10.1038/nature03308. [DOI] [PubMed] [Google Scholar]
- 47.Kawai T, Sato S, Ishii KJ, et al. Interferon-α induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nature Immunology. 2004;5(10):1061–1068. doi: 10.1038/ni1118. [DOI] [PubMed] [Google Scholar]
- 48.Balkhi MY, Fitzgerald KA, Pitha PM. Functional regulation of MyD88-activated interferon regulatory factor 5 by K63-linked polyubiquitination. Molecular and Cellular Biology. 2008;28(24):7296–7308. doi: 10.1128/MCB.00662-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Deng L, Wang C, Spencer E, et al. Activation of the Iκb kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 2000;103(2):351–361. doi: 10.1016/s0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- 50.Feng D, Sangster-Guity N, Stone R, et al. Differential requirement of histone acetylase and deacetylase activities for IRF5-mediated proinflammatory cytokine expression. Journal of Immunology. 2010;185(10):6003–6012. doi: 10.4049/jimmunol.1000482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baccala R, Hoebe K, Kono DH, Beutler B, Theofilopoulos AN. TLR-dependent and TLR-independent pathways of type I interferon induction in systemic autoimmunity. Nature Medicine. 2007;13(5):543–551. doi: 10.1038/nm1590. [DOI] [PubMed] [Google Scholar]
- 52.Au WC, Yeow WS, Pitha PM. Analysis of functional domains of interferon regulatory factor 7 and its association with IRF-3. Virology. 2001;280(2):273–282. doi: 10.1006/viro.2000.0782. [DOI] [PubMed] [Google Scholar]
- 53.Lin R, Mamane Y, Hiscott J. Structural and functional analysis of interferon regulatory factor 3: localization of the transactivation and autoinhibitory domains. Molecular and Cellular Biology. 1999;19(4):2465–2474. doi: 10.1128/mcb.19.4.2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chang Foreman HC, Van Scoy S, Cheng TF, Reich NC. Activation of interferon regulatory factor 5 by site specific phosphorylation. PloS ONE. 2012;7(3) doi: 10.1371/journal.pone.0033098.e33098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen W, Lam SS, Srinath H, et al. Insights into interferon regulatory factor activation from the crystal structure of dimeric IRF5. Nature Structural and Molecular Biology. 2008;15(11):1213–1220. doi: 10.1038/nsmb.1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lin R, Yang L, Arguello M, Penafuerte C, Hiscott J. A CRM1-dependent nuclear export pathway is involved in the regulation of IRF-5 subcellular localization. Journal of Biological Chemistry. 2005;280(4):3088–3095. doi: 10.1074/jbc.M408452200. [DOI] [PubMed] [Google Scholar]
- 57.Barnes BJ, Field AE, Pitha-Rowe PM. Virus-induced heterodimer formation between IRF-5 and IRF-7 modulates assembly of the IFNA enhanceosome in vivo and transcriptional activity of IFNA genes. Journal of Biological Chemistry. 2003;278(19):16630–16641. doi: 10.1074/jbc.M212609200. [DOI] [PubMed] [Google Scholar]
- 58.Pickart CM, Fushman D. Polyubiquitin chains: polymeric protein signals. Current Opinion in Chemical Biology. 2004;8(6):610–616. doi: 10.1016/j.cbpa.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 59.Hutti JE, Turk BE, Asara JM, Ma A, Cantley LC, Abbott DW. IκB kinase β phosphorylates the K63 deubiquitinase A20 to cause feedback inhibition of the NF-κB pathway. Molecular and Cellular Biology. 2007;27(21):7451–7461. doi: 10.1128/MCB.01101-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mancl ME, Hu G, Sangster-Guity N, et al. Two discrete promoters regulate the alternatively spliced human interferon regulatory factor-5 isoforms: multiple isoforms with distinct cell type-specific expression, localization, regulation, and function. Journal of Biological Chemistry. 2005;280(22):21078–21090. doi: 10.1074/jbc.M500543200. [DOI] [PubMed] [Google Scholar]
- 61.Nusinzon I, Horvath CM. Histone deacetylases as transcriptional activators? Role reversal in inducible gene regulation. Science’s STKE. 2005;2005(296, article re11) doi: 10.1126/stke.2962005re11. [DOI] [PubMed] [Google Scholar]
- 62.Caillaud A, Prakash A, Smith E, et al. Acetylation of interferon regulatory factor-7 by p300/CREB-binding protein (CBP)-associated factor (PCAF) impairs its DNA binding. Journal of Biological Chemistry. 2002;277(51):49417–49421. doi: 10.1074/jbc.M207484200. [DOI] [PubMed] [Google Scholar]
- 63.Masumi A, Yamakawa Y, Fukazawa H, Ozato K, Komuro K. Interferon regulatory factor-2 regulates cell growth through its acetylation. Journal of Biological Chemistry. 2003;278(28):25401–25407. doi: 10.1074/jbc.M213037200. [DOI] [PubMed] [Google Scholar]
- 64.Niewold TB, Kelly JA, Kariuki SN, Franek BS, Kumar AA, Kaufman KM. IRF5 haplotypes demonstrate diverse serological associations which predict serum interferon alpha activity and explain the majority of the genetic association with systemic lupus erythematosus. Annals of the Rheumatic Diseases. 2012;71(3):463–468. doi: 10.1136/annrheumdis-2011-200463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kozyrev SV, Lewén S, Reddy PMVL, et al. Structural insertion/deletion variation in IRF5 is associated with a risk haplotype and defines the precise IRF5 isoforms expressed in systemic lupus erythematosus. Arthritis and Rheumatism. 2007;56(4):1234–1241. doi: 10.1002/art.22497. [DOI] [PubMed] [Google Scholar]
- 66.Graham DSC, Manku H, Wagner S, et al. Association of IRF5 in UK SLE families identifies a variant involved in polyadenylation. Human Molecular Genetics. 2007;16(6):579–591. doi: 10.1093/hmg/ddl469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wen F, Ellingson SM, Kyogoku C, Peterson EJ, Gaffney PM. Exon 6 variants carried on systemic lupus erythematosus (SLE) risk haplotypes modulate IRF5 function. Autoimmunity. 2011;44(2):82–89. doi: 10.3109/08916934.2010.491842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Niewold TB, Kelly JA, Flesch MH, Espinoza LR, Harley JB, Crow MK. Association of the IRF5 risk haplotype with high serum interferon-α activity in systemic lupus erythematosus patients. Arthritis and Rheumatism. 2008;58(8):2481–2487. doi: 10.1002/art.23613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Feng D, Stone RC, Eloranta ML, et al. Genetic variants and disease-associated factors contribute to enhanced interferon regulatory factor 5 expression in blood cells of patients with systemic lupus erythematosus. Arthritis and Rheumatism. 2010;62(2):562–573. doi: 10.1002/art.27223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rullo OJ, Woo JMP, Wu H, et al. Association of IRF5 polymorphisms with activation of the interferon α pathway. Annals of the Rheumatic Diseases. 2010;69(3):611–617. doi: 10.1136/ard.2009.118315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Richez C, Yasuda K, Bonegio RG, et al. IFN regulatory factor 5 is required for disease development in the FcγRIIB−/−Yaa and FcγRIIB−/− mouse models of systemic lupus erythematosus. Journal of Immunology. 2010;184(2):796–806. doi: 10.4049/jimmunol.0901748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Savitsky DA, Yanai H, Tamura T, Taniguchi T, Honda K. Contribution of IRF5 in B cells to the development of murine SLE-like disease through its transcriptional control of the IgG2a locus. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(22):10154–10159. doi: 10.1073/pnas.1005599107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lien C, Fang CM, Huso D, Livak F, Lu R, Pitha PM. Critical role of IRF-5 in regulation of B-cell differentiation. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(10):4664–4668. doi: 10.1073/pnas.0911193107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Niewold TB, Rivera TL, Buyon JP, Crow MK. Serum type I interferon activity is dependent on maternal diagnosis in anti-SSA/Ro-positive mothers of children with neonatal lupus. Arthritis and Rheumatism. 2008;58(2):541–546. doi: 10.1002/art.23191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cherian TS, Kariuki SN, Franek BS, Buyon JP, Clancy RM, Niewold TB. IRF5 SLE-risk haplotype is associated with asymptomatic serologic autoimmunity and progression to clinical autoimmunity in neonatal lupus mothers. Arthritis and Rheumatism. 2012;64(10):3383–3387. doi: 10.1002/art.34571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Salloum R, Niewold TB. Interferon regulatory factors in human lupus pathogenesis. Translational Research. 2011;157(6):326–331. doi: 10.1016/j.trsl.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Robinson T, Kariuki SN, Franek BS, Kumabe M, Kumar AA, Badaracco M. Autoimmune disease risk variant of IFIH1 is associated with increased sensitivity to IFN-α and serologic autoimmunity in lupus patients. Journal of Immunology. 2011;187(3):1298–1303. doi: 10.4049/jimmunol.1100857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pothlichet J, Niewold TB, Vitour D, Solhonne B, Crow MK, Si-Tahar M. A loss-of-function variant of the antiviral molecule MAVS is associated with a subset of systemic lupus patients. EMBO Molecular Medicine. 2011;3(3):142–152. doi: 10.1002/emmm.201000120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Salloum R, Franek BS, Kariuki SN, et al. Genetic variation at the IRF7/PHRF1 locus is associated with autoantibody profile and serum interferon-α activity in lupus patients. Arthritis and Rheumatism. 2010;62(2):553–561. doi: 10.1002/art.27182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Agik S, Franek BS, Kumar AA, Kumabe M, Utset TO, Mikolaitis RA. The autoimmune disease risk allele of UBE2L3 in African American patients with systemic lupus erythematosus: a recessive effect upon subphenotypes. Journal of Immunology. 2012;39(1):73–78. doi: 10.3899/jrheum.110590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Niewold TB. Interferon alpha as a primary pathogenic factor in human lupus. Journal of Interferon & Cytokine Research. 2011;31(12):887–892. doi: 10.1089/jir.2011.0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kariuki SN, Franek BS, Kumar AA, et al. Trait-stratified genome-wide association study identifies novel and diverse genetic associations with serologic and cytokine phenotypes in systemic lupus erythematosus. Arthritis Research and Therapy. 2010;12(4, article R151) doi: 10.1186/ar3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Deng Y, Tsao BP. Genetic susceptibility to systemic lupus erythematosus in the genomic era. Nature Reviews Rheumatology. 2010;6(12):683–692. doi: 10.1038/nrrheum.2010.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Garcia-Romo GS, Caielli S, Vega B, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Science Translational Medicine. 2011;3(73) doi: 10.1126/scitranslmed.3001201.73ra20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lande R, Ganguly D, Facchinetti V, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Science Translational Medicine. 2011;3(73) doi: 10.1126/scitranslmed.3001180.73ra19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hennessy EJ, Parker AE, O’Neill LAJ. Targeting Toll-like receptors: emerging therapeutics? Nature Reviews Drug Discovery. 2010;9(4):293–307. doi: 10.1038/nrd3203. [DOI] [PubMed] [Google Scholar]
- 87.Lichtman EI, Helfgott SM, Kriegel MA. Emerging therapies for systemic lupus erythematosus—focus on targeting interferon-alpha. Clinical Immunology. 2012;143(3):210–221. doi: 10.1016/j.clim.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]