

ABSTRACT
The Toxoplasma gondii SRS gene superfamily is structurally related to SRS29B (formerly SAG1), a surface adhesin that binds host cells and stimulates host immunity. Comparative genomic analyses of three Toxoplasma strains identified 182 SRS genes distributed across 14 chromosomes at 57 genomic loci. Eight distinct SRS subfamilies were resolved. A core 69 functional gene orthologs were identified, and strain-specific expansions and pseudogenization were common. Gene expression profiling demonstrated differential expression of SRS genes in a developmental-stage- and strain-specific fashion and identified nine SRS genes as priority targets for gene deletion among the tissue-encysting coccidia. A Δsag1 ∆sag2A mutant was significantly attenuated in murine acute virulence and showed upregulated SRS29C (formerly SRS2) expression. Transgenic overexpression of SRS29C in the virulent RH parent was similarly attenuated. Together, these findings reveal SRS29C to be an important regulator of acute virulence in mice and demonstrate the power of integrated genomic analysis to guide experimental investigations.
IMPORTANCE
Parasitic species employ large gene families to subvert host immunity to enable pathogen colonization and cause disease. Toxoplasma gondii contains a large surface coat gene superfamily that encodes adhesins and virulence factors that facilitate infection in susceptible hosts. We generated an integrated bioinformatic resource to predict which genes from within this 182-gene superfamily of adhesin-encoding genes play an essential role in the host-pathogen interaction. Targeted gene deletion experiments with predicted candidate surface antigens identified SRS29C as an important negative regulator of acute virulence in murine models of Toxoplasma infection. Our integrated computational and experimental approach provides a comprehensive framework, or road map, for the assembly and discovery of additional key pathogenesis genes contained within other large surface coat gene superfamilies from a broad array of eukaryotic pathogens.
Introduction
Toxoplasma gondii, a highly successful protozoan parasite, infects nearly one-third of the world’s population. It is a member of the subclass Coccidia, which includes Eimeria, Neospora, and Sarcocystis, all genera of medical and veterinary importance. T. gondii has the ability to infect any warm-blooded animal, while other coccidians apparently possess a more restricted host range. The coccidia are part of the phylum Apicomplexa, which includes Plasmodium, Theileria, and Cryptosporidium. Their shared mode of infection relies on physical interactions between the parasite and a host cell; consequently, many apicomplexans have evolved an arsenal of surface protein virulence genes that facilitate parasite attachment and invasion of host cells and evasion or modulation of host immunity (reviewed in reference 1).
The genome of T. gondii contains several distinct, coccidian-specific multicopy gene families (2), including those that encode the SRS, ROPK, and SUSA proteins (3–5). The SRS genes encode virulence determinants that induce lethal ileitis in a murine model (SRS29B, formerly SAG1), mediate attachment to host cells (SRS57; formerly SAG3), and establish transmissible latent infections (SRS16B; formerly SRS9) (6–8). T. gondii presumably expresses and regulates this multigene family to successfully establish chronic infections in most warm-blooded vertebrates, where it persists within latent cysts for the life of the intermediate host. Unlike the serial expression of a single var or VSG gene in Plasmodium falciparum and Trypanosoma brucei, respectively, the SRS proteins are expressed in a developmentally regulated manner as distinct, largely nonoverlapping sets of SRS antigens (3). An SRS gene typically contains one or two domains, each with four to six cysteines (4-Cys or 6-Cys) that participate in disulfide bonds and a glycosylphosphatidylinositol (GPI) anchor for attachment to the parasite cell surface (9). Some SRS proteins are very closely related, while others share less than 30% sequence identity. The exact biological role of the SRS gene superfamily is poorly understood. It is known that SRS29B and SRS34A (formerly SAG2A) are highly immunogenic during infection, whereas SRS29B and SRS57 function as adhesins (6, 10). An interaction with cellular ligands is supported by structural evidence that SRS29B forms a homodimer with a deep, positively charged groove capable of docking sulfated proteoglycans (11, 12).
With the release of the type II Me49 draft genome in 2004, 161 unique SRS DNA sequences were identified (3). A combination of targeted studies and searches of expressed sequence tag (EST) data has revealed SRS orthologs in other coccidian genera, including Neospora and Sarcocystis (13, 14), and an SRS-like fold in the Plasmodium 6-Cys family of parasite adhesins (15).
Exploiting the recent availability of genome sequence data for three strains of T. gondii, here we describe a comprehensive investigation exploring the evolution and expression of SRS virulence genes. Systematic sequence searches identified eight subfamilies of SRS domains which display distinct and nonrandom patterns of co-occurrence, expansions, and losses across the three T. gondii strains. Crystal structure analyses revealed a core SRS structural scaffold with sequence divergence that presumably reflects the diversity of host ligands targeted. Integration of transcriptomic data sets further revealed lineage- and life cycle stage-specific programs of SRS gene expression that correlate with altered patterns of virulence by different parasite strains. Targeted disruption of two highly abundant, pancoccidial SRS proteins (SRS29B and SRS34A) resulted in a dramatic upregulation of the levels of SRS29C (formerly SRS2), a protein that is differentially expressed by mouse-virulent and -avirulent strains of T. gondii. Transgenic overexpression of SRS29C in a type I virulent strain significantly attenuated virulence, and the majority of the mice infected survived. Our results highlight the utility of integrating bioinformatic with targeted genetic approaches to identify a major and unexpected role for SRS29C in Toxoplasma acute virulence in mice.
RESULTS
There are eight subfamilies of SRS domains.
To identify the full complement of SRS domains in T. gondii, previously published SRS sequences were used to seed a hidden Markov model (HMM). New sequences were curated and added iteratively to expand the diversity of the HMM. A total of 763 SRS domains were found, of which 459 had annotated ToxoDB gene models, a predicted signal peptide, four to six invariant cysteine residues, and a predicted GPI anchor sequence consistent with an SRS protein (Table 1). SRS domains not found within a gene model were considered pseudogenes. The low sequence identity (~15%) prevented the use of standard methods for the construction of a robust phylogeny. Therefore, all-against-all pairwise alignments were used to generate a similarity score matrix, which was clustered hierarchically. The domains segregated into two families and eight distinct domain subfamilies (Fig. 1A), subfamilies 1 to 6 (which includes SRS34A) and subfamilies 7 and 8 (which includes SRS29B). The intrasubfamily pairwise identity percentages ranged from 23% to 45%, with the majority of the conservation mapping to the proposed β-strands in the structures (see Fig. 2, system object model [SOM] file 1).
TABLE 1.
SRS genes and pseudogenes by strain and number of domains
| Parameter | T. gondii Me49 | T. gondii GT1 | T. gondii VEG |
|---|---|---|---|
| No. of SRS protein-coding genes | 109 | 90 | 91 |
| No. (%) of SRS protein-coding genes with: | |||
| 1 domain | 44 (40) | 25 (27) | 39 (40) |
| 2 domains | 58 (53) | 60 (67) | 46 (50) |
| 3 domains | 4 (4) | 3 (3) | 3 (3) |
| ≥4 domains | 3 (3) | 2 (2) | 3 (3) |
| No. of SRS protein domains | 194 | 162 | 162 |
| No. (%) of SRS protein domains that pass filters | 172 (89) | 142 (88) | 145 (90) |
| No. of SRS pseudogenes | 35 | 57 | 67 |
| No. of domains | 48 | 87 | 110 |
| No. (%) of domains that pass filters | 29 (60) | 68 (78) | 86 (78) |
| No. of gene loci | 52 | 52 | 56 |
| No. (%) of gene loci with ≥2 genes | 26 (50) | 26 (50) | 25 (45) |
FIG 1.
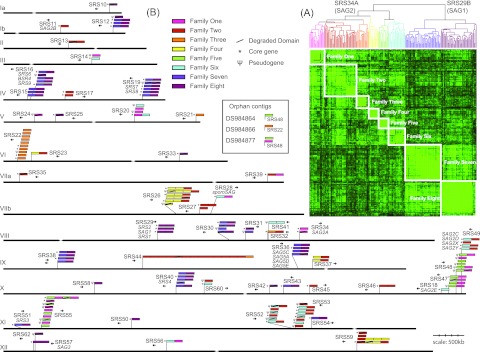
SRS domain subfamily definition and distribution. (A) Four hundred thirty-seven SRS domains were clustered by using pairwise sequence similarity. Lighter green indicates higher sequence similarity. Eight subfamilies are resolved hierarchically that bifurcate into the previously defined SRS29B and SRS34A families. (B) HMMs constructed by using previously published SRS genes identified 144 SRS genes in the T. gondii ME49 genome. Core conserved genes are indicated by asterisks. SRS domains containing fewer than four conserved cysteine residues were considered “degraded” (indicated by a slash). Pseudogenes (indicated by Ψ) are SRS domains without an associated ToxoDB gene model. Domains are colored by subfamily definitions from panel A. Four SRS genes were located on orphan contigs DS984864, DS984866, and DS984877.
A structure-guided alignment of each domain subfamily revealed that, of the six family-defining cysteines (Cys-1 to Cys-6), four are invariant (see Fig. S1 in the supplemental material). However, in subfamilies 1 and 2, a subfamily-specific cysteine upstream of Cys-5 replaces the absent Cys-3 to form the third disulfide bond (16). Members of subfamilies 3 and 6 are also missing middle cysteine residues but do not have a compensatory residue to rescue the disulfide bond. In two-domain proteins, the extent to which SRS domains vary depends upon whether the domain is solvent exposed (D1) or proximal to the membrane (D2) (SOM file 1). The membrane-distal end of D1 is poorly conserved and likely to be involved in mediating ligand interactions, whereas the membrane-proximal end of the D2 subunit is highly conserved to orient and tether the protein to the parasite surface (Fig. 2). The interface between the two domains exhibits similar patterns of conservation, reflecting the complementary surfaces required to stabilize the interface.
FIG 2.

The SRS domain is stabilized by four invariant cysteines, and sequence diversity is greatest in ligand binding regions. (A) A conservation score for individual residues was calculated for all Me49 two-domain SRS proteins and mapped onto the SRS29B/SAG1 dimer in a secondary-structure format in orthogonal orientations with the domain used for surface mapping highlighted in gray. The areas labeled I, II, and III are membrane-distal, interface, and membrane-proximal regions, respectively. The color scheme uses calculated conservation scores, with red reflecting the highest level of conservation (>0.5) and blue representing the lowest (<−0.5). (B) Conservation at the apical surface and the interdomain interface. (C) Dashed lines represent the 2-fold axis of the SRS29B dimer. Secondary structural elements (colored green) represent the symmetry-related monomer. Analysis of the D1 domain reveals that Lys33 and Lys94 are highly conserved while most of the D1 dimer interface is poorly conserved. (D) Spatial representation of family-defining residues mapped to the D1 domain of SRS29B. Top-down and side views of the SRS29B dimer reveal the localization of the family-defining residues shown as color-coded sticks that cluster near the D1 domain tip, distal from the dimer interface, but adjacent to the D2 domain. See File S1 in the supplemental material for additional details and Fig. S1 in the supplemental material for sequence number correlation.
SRS sequences form multigene loci that are distributed throughout the genome.
Genes encoding SRS proteins are found on all T. gondii chromosomes (Fig. 1B). SRS genes are organized into 57 discrete genomic sites across the three strains sequenced (each locus possesses either a single gene or a group of paralogous genes that group together in a cluster). Syntenic genes and pseudogenes common to the three strains are described as “orthologous groups.” In total, 182 SRS gene loci were identified but only 122 (67%) existed as orthologous groups common to all three strains (Fig. 3A). When pseudogenes were excluded, a stable core of only 69 orthologous groups was identified (Fig. 3A).
FIG 3.

SRS orthology/paralogy among three T. gondii strains. (A) Venn diagrams showing the distribution of orthologous SRS genes across three Toxoplasma strains: (i) orthologous groups including pseudogenes, (ii) orthologous groups excluding pseudogenes. (B) Syntenic relationships of the SRS genes represented by at least one gene model in ToxoDB. Each block represents an individual chromosome colored by strain. Links between blocks represent syntenic genes (see inset color key). (C and D) Detailed views of syntenic relationships within SRS36 and SRS40, respectively. (E) The SRS55 locus is split and found in two locations in strain GT1, as either a lineage-specific translocation or an example of misassembly.
The reference Me49 genome has 144 SRS gene sequences, 109 with annotated gene models and 35 pseudogenes (Table 1; Fig. 1B and 3B). The SRS genes are organized into 52 genomic loci, half existing as multigene clusters containing more than one related SRS gene (designated A, B, C, etc.), accounting for 118 (81%) of the 144 genes. GT1 and VEG strains had fewer SRS gene models but an increased number of pseudogenes, possibly reflecting lower levels of sequence coverage or inherent conflicts produced via scaffolding of the GT1 and VEG assemblies to the Me49 reference genome.
We found that 60/144 (42%) Me49 SRS genes are located in subtelomeric sites and all domain subfamilies are represented (Fig. 1B). This is in contrast to the var genes in P. falciparum, where strict partitioning of the A and B subfamilies to subtelomeric expression sites versus partitioning of the C subfamily to central locations along chromosomes has occurred (17). The 69 core SRS genes were distributed somewhat equally across 44 syntenic loci, and the majority of the singleton genes were represented (19/26; 73%). The remaining 49 genes were organized into 23 loci, and only SRS17, SRS29, SRS34, and SRS49 existed as multigene orthologous groups that were invariant across all three strains (Fig. 1B).
SRS proteins demonstrate only a limited repertoire of potential domain combinations.
Most SRS gene sequences consist of two SRS domains (Table 1). Only 5 to 6% of the genes contain three or more domains with a single 14-domain gene model (SRS44) found in Me49 and VEG. Investigations of domain combinations revealed nonrandom groupings. Strong associations between subfamilies 1 and 5, 2 and 4, and 2 and 6 exist, and domains from subfamily 7 are found only in genes with domains from subfamily 8 (Table 2). Domain pairing between subfamilies 7 and 8 makes up approximately 30% of the T. gondii SRS genes and half of the multidomain genes. No subfamily 7 or 4 domains were present as single-domain genes, and domain subfamily 3 is largely restricted to single-domain genes.
TABLE 2.
Domain family architecture across three Toxoplasma strains
| Domain architecture(s)a | No. of genes in: |
||||||
|---|---|---|---|---|---|---|---|
| Coreb | Me49 |
GT1 |
VEG |
||||
| Total | Minus pseudogenes | Total | Minus pseudogenes | Total | Minus pseudogenes | ||
| fam7.fam8 | 18 | 34 | 33 | 35 | 28 | 36 | 20 |
| fam2 | 8 | 15 | 9 | 11 | 6 | 16 | 8 |
| fam3 | 5 | 12 | 11 | 14 | 5 | 15 | 11 |
| fam6.fam2 | 7 | 8 | 7 | 11 | 11 | 9 | 8 |
| fam8 | 6 | 11 | 7 | 9 | 5 | 9 | 6 |
| fam6 | 5 | 8 | 5 | 5 | 3 | 6 | 4 |
| fam8.fam8 | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
| fam1 | 3 | 13 | 7 | 9 | 6 | 13 | 8 |
| fam4{2}.fam2 | 3 | 4 | 4 | 9 | 3 | 7 | 3 |
| fam6.fam1 | 3 | 3 | 3 | 4 | 4 | 5 | 5 |
| fam2.fam2 | 2 | 3 | 3 | 3 | 3 | 2 | 2 |
| fam5.fam1 | 1 | 7 | 3 | 15 | 8 | 10 | 2 |
| fam2.fam1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| fam3.fam2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| (fam5.fam1){2} | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| fam2.fam4{2}.fam2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| fam5 | 0 | 8 | 5 | 5 | 0 | 7 | 2 |
| fam4.fam2 | 0 | 3 | 3 | 3 | 0 | 4 | 3 |
| fam4{3}.fam2 | 0 | 2 | 0 | 0 | 0 | 2 | 0 |
| fam7 | 0 | 2 | 0 | 4 | 0 | 3 | 0 |
| fam4 | 0 | 1 | 0 | 1 | 0 | 2 | 0 |
| fam6.fam6 | 0 | 1 | 0 | 1 | 0 | 1 | 0 |
| fam2{13}.fam3 | 0 | 1 | 1 | 0 | 0 | 1 | 1 |
| fam4.fam4 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| fam1.fam2 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| All | 69 | 144 | 109 | 147 | 90 | 158 | 91 |
In famX{N}, X is the number of the SRS domain subfamily and N is the number of consecutive instances of that domain subfamily in the architecture.
The core genes are the syntenic gene models, excluding pseudogenes, found in all three strains.
To determine the mechanism of SRS gene expansion genome wide, a phylogenetic investigation of subfamily 7 and 8 architecture was performed. The consensus reconstruction placed four of the multigene loci into homogeneous monophyletic clades (clades B to E) and a large heterogeneous clade (clade A), which was also monophyletic (Fig. 4). The topology of the reconstruction suggested that duplications of single genes and entire loci have occurred, consistent with gene conversion (SOM file 1).
FIG 4.

Phylogenetic relationship of the most common SRS domain architectures. The alignment and tree reconstructions were performed as described in Materials and Methods. To achieve a robust phylogeny, three tree-building methods were used, i.e., ME with DayHoff and JTT probability models and ML with the JTT model. The three methods returned highly similar topologies. The node supports shown are from 1,000 bootstrap replicates. Nodes are considered resolved if they gained >50% support in two of the three tree-building methods used. Only proteins containing nondegraded domains were included in the analysis
Interstrain comparisons reveal lineage-specific patterns of expansions and losses.
Among the three T. gondii strains, 37 strain-specific SRS gene expansions were present, 26 in multigene loci versus 9 in bona fide genes (Fig. 3A). However, the relative abundance of pseudogenes should be considered preliminary, as five of the multigene loci sit in regions of potential genome misassembly (J. D. Wasmuth et al., unpublished data). When considering gene loss or conversion of a bona fide gene into a pseudogene, large arrays of pseudogenes are found most frequently within loci composed of four or more tandemly arrayed SRS genes (Fig. 1B). Sixty gene loci were composed exclusively of pseudogenes across the three genomes analyzed and presumably represent either a mutation event in an ancestral T. gondii strain or an assembly error. Of the remaining 122 gene loci, 31 had pseudogenes that are orthologous with bona fide genes. Assuming no reactivation mutations, all of the changes are likely to be relatively recent (Fig. 3). When focusing on the SRS36 (formerly SAG5) locus, a variegated pattern of pseudogenization is observed, with only SRS36C bona fide across all three strains (Fig. 3C). Similar patchworks of genes and pseudogenes are present at the SRS40 (Fig. 3D) and SRS53 loci (Fig. 3E).
The SRS gene family is polymorphic and differentially regulated in a strain- and developmental-stage-specific manner.
Single-nucleotide polymorphisms in the 69 core SRS genes were assessed (see Table S1 in the supplemental material). In 58% of the genes, intertypic variation was biallelic (interstrain polymorphisms, >0.4%), consistent with the tenet that lineage types I, II, and III are recombinant progeny from a limited number of crosses between two distinct ancestries (18, 19). Nineteen (28%) of the core SRS genes were monoallelic, whereas 10 (14%) of the genes were triallelic. Manual inspection identified problematic gene models for three triallelic genes (SRS12B, SRS18, and SRS26A). The remaining seven were essentially biallelic but had elevated mutation rates.
To identify the evolutionary forces acting on individual SRS genes, we estimated ω (dN/dS ratio) (Fig. 5; see Table S1 in the supplemental material). In general, SRS genes are not under strong selection pressure, with 38 (55%) of 69 core Me49 SRS genes displaying an ω value of >0.5 and <1. Six SRS genes are under diversifying selection, whereas 25 SRS genes were highly conserved and under purifying selection, where synonymous mutations are significantly enriched (ω, <0.5). Indeed, 14 of these genes were 100% identical across the three strains. No skewing of subfamilies toward purifying or diversifying selection was observed. To unequivocally show that selection is operating at these loci will, however, require more extensive population level sampling.
FIG 5.

SRS gene expression patterns. The SRS gene name is used, and where appropriate, the original gene designation is included. Core genes are denoted by asterisks. Twenty core genes are not listed, as there were no expression data available for them. Column A shows the EST data for N. caninum, S. neurona, and different T. gondii life cycle stages. Column B shows stage specificity calculated from microarray data. Column C depicts microarray-derived relative gene expression differences between different T. gondii types. Column D shows evolutionary selection in core genes. Column E provides the subfamily domain architecture from Fig. 1.
By interrogating EST and microarray data, transcript expression for 95 SRS genes was confirmed (see Table S2 in the supplemental material). The EST analyses, composed of a mixture of cDNAs derived from different developmental stages of T. gondii, identified developmental-stage-specific expression patterns for those SRS genes that had large numbers of ESTs. Eleven genes were expressed primarily in tachyzoites, whereas four were apparently restricted to bradyzoites and only SRS28 was enriched in sporozoites. Strict partitioning of SRS gene expression to a particular parasite stage appeared to be the rule, with one exception: SRS25 was abundantly expressed in both tachyzoites and sporozoites (Fig. 5). Analysis of VEG strain oocyst ESTs has previously identified SRS19 as sporozoite specific (20).
In the microarray experiments, parasites were induced to differentiate from tachyzoites into bradyzoites. Sufficient transcript data existed for 70 genes. Fourteen genes were more abundantly expressed in tachyzoites, including the SRS29 gene cluster; 31 genes possessed more transcripts after bradyzoite induction, including 10 genes whose relative expression was greater than 20-fold (Fig. 5B). Genes within the same cluster possessed similar expression profiles. Given the imperfect nature of the artificial induction of the life cycle switch, additional experimental validation is required in order to assign a relative stage expression pattern.
Comparing SRS gene expression between strains revealed 44 differentially expressed SRS genes; 8 genes were downregulated in type I, 2 genes were downregulated and 25 genes were upregulated in type II, and 7 genes were downregulated and 8 genes were upregulated in type III (Fig. 5C). Dramatic differences in relative expression among tachyzoite-specific SRS genes were detected for SRS20C (down 12-fold in VEG), SRS42 (up 9-fold in VEG), and SRS54 (up 15-fold in Me49).
Comparative and cross-species analyses prioritize genes for targeted deletion.
SRS domains have also been identified in Neospora caninum, N. hughesi, Sarcocystis muris, S. neurona, and Hammondia hammondii. Putative orthologs of 29 genes were found in the EST data from N. caninum and S. neurona (Fig. 5A). Despite the use of sensitive HMMs for each subfamily, the taxonomic distribution of the SRS superfamily was not extended beyond the Sarcocystidae, tissue cyst-forming coccidian parasites that have broad intermediate host ranges.
Integration of the bioinformatic and comparative analyses identified nine SRS genes that were pursued for targeted gene deletion studies. SRS17A and SRS17B are nonpolymorphic, are abundantly expressed in N. caninum, and likewise show differential expression among strains of T. gondii. SRS25 possesses a degraded SRS domain containing only three cysteines, is expressed in both Neospora and Toxoplasma, and is not strictly stage specific. SRS33 is the only gene with evidence of strong expression across all coccidian parasites, SRS35 (SAG4) is the most abundant bradyzoite-specific SRS gene that is under positive selection and is differentially expressed across the archetypal strains of Toxoplasma, and SRS54 is highly polymorphic and differentially expressed among Toxoplasma strains. SRS29C is differentially expressed among Toxoplasma strains (21) but not at the level of transcription (Fig. 5C), and its ortholog in Neospora is one of the most abundant proteins expressed (13). Finally, SRS29B and SRS34A are highly expressed in all T. gondii strains and are known to induce strong immunity during infection (22).
Targeted disruption of SRS29B and SRS34A alters expression of SRS29C.
In this section, and throughout this paper, we have adopted the Toxoplasma genome annotation for SAG1 and SAG2, respectively, SRS29B and SRS34A. Targeted gene deletion constructs were engineered to disrupt SRS17A, SRS17B, SRS25, SRS29B, SRS29C, SRS33, SRS34A, and SRS35 to test their relevance in virulence and pathogenesis during in vivo infection in the natural murine host. Herein we present data on a dramatic virulence phenotype uncovered after the targeted deletion of SRS29B and SRS34A by double-crossover homologous recombination in the mouse-virulent type I RH strain of Toxoplasma.
Western blot analyses and flow cytometry with monoclonal antibodies (MAbs) specific for SRS29B and SRS34A confirmed the absence of expression of SRS29B and/or SRS34A in the mutant parasite lines (Fig. 6). Western blotting for the cross-reacting determinant (CRD), which is exposed after a GPI-anchored protein is cleaved by GPI-phospholipase C (PLC) (22), identified a lack of SRS29B, SRS34A, or both proteins in the Δsrs29B, Δsrs34A, and Δsrs29B ∆srs34A double-knockout (DKO) mutant lines, respectively (Fig. 6A), confirming the targeted disruption of the genes in question. Anti-CRD immunoblotting identified no real differences in the relative expression of other tachyzoite-expressed SRS proteins in the single-knockout lines but did identify an apparent increased expression of two bands running above 35 kDa, consistent with those of SRS29C (3). To confirm that SRS29C protein levels were increased in the DKO, the surface expression of SRS29B, SRS34A, SRS57, SRS29A, and SRS29C was tested by flow cytometry. With fluorescence-activated cell sorter (FACS) staining, no significant differences in relative SRS29B, SRS34A, SRS57, and SRS29A surface expression, apart from the obvious lack of the SRS29B and SRS34A proteins in the respective mutant parasite lines, was detected (Fig. 6D). However, a substantial increase in SRS29C protein expression was detected in the DKO mutant, confirming the Western blotting data. TaqMan quantitative reverse transcription (qRT)-PCR results for the 18 most abundant SRS genes confirmed the bioinformatic analyses reported in Fig. 5; the relative order and hierarchy of SRS genes from most to least abundant were reproducibly similar (Fig. 6C). Importantly, RNA levels were only moderately increased for SRS29C, SRS51, and SRS52A in the Δsrs29B and DKO lines, indicating that the increased SRS29C protein expression in the DKO mutant line was likely regulated posttranscriptionally, as has been previously shown for SRS16C (BSR4) (23).
FIG 6.

Targeted disruption of SRS29B and SRS34A alters SRS29C expression and acute virulence in mice. (A) Anti-CRD Western blot profile of Srs29B−, Srs34A−, and DKO parasite strains to identify relative expression levels of SRS antigens expressed on knockout and WT tachyzoites. A total of 107 tachyzoites were solubilized in 1% NP-40, incubated in the presence of GPI-PLC purified from T. brucei for 1 h at 37°C, separated by 12% SDS-PAGE, and transferred to nitrocellulose. SRS proteins were detected by Western blotting with a rabbit anti-CRD polyclonal antibody, followed by a horseradish peroxidase-conjugated anti-rabbit secondary antibody, and enhanced chemiluminescence. (B) Death kinetics of seropositive CD-1 mice infected with engineered strains of T. gondii. The numbers of seropositive mice infected with the WT, Srs29B−, and Srs34A− strains were five, five, and four, respectively. The total infectious dose was 25 parasites. The numbers of seropositive mice infected with the DKO strain at infectious doses of 25, 250, and 2,500 parasites were 9, 10, and 10, respectively. Mortality data are a composite of three independent experiments with independently derived knockout parasite clones. (C) Gene expression was measured by TaqMan qRT-PCR. The experiment shown is for one representative data set from two independent mRNA-cDNA extractions. Data are clustered from most abundant (SRS29B) to least abundant (SRS17A) transcripts. Transcript levels are represented as 2−∆∆CT to show absolute levels of transcripts relative to every SRS gene examined and were normalized against 18S rRNA genes transcripts. (D) Relative levels of surface-expressed SRS proteins on WT, Srs29B−, Srs34A−, and DKO parasites were determined by flow cytometry by using DG52, 5A6, 4F12, SUS1, and CL15/4 to detect SRS29B, SRS34A, SRS57, SRS29A, and SRS29B, respectively.
The Δsrs29B ∆srs34A DKO mutant is less virulent in mice.
In vitro replication of the Δsrs29B, Δsrs34A, and DKO strains, as well as their ability to adhere to fibroblasts, was comparable to that of wild-type (WT) strain RH, with the Δsrs29B strain exhibiting a greater invasion rate than the parental WT strain, as previously described (8). To investigate the virulence phenotype of the Δsrs29B, Δsrs34A, and DKO mutant parasites, groups of five outbred CD-1 mice were injected intraperitoneally with ~25 tachyzoites and the resulting disease was monitored. Results of seropositive mice are shown in Fig. 6B. Mice infected with the Srs29B− and Srs34A− mutant singly deficient parasites exhibited delayed kinetics to death (by several days), with one survivor, relative to WT-infected mice, which all died between days 11 to 12. The DKO parasite line, however, was significantly attenuated in virulence, the majority of mice survived an inoculum of 25 tachyzoites, and mice also survived infection at doses of 250 and 2,500 tachyzoites (Fig. 6B).
Overexpression of SRS29C attenuates virulence.
To test whether overexpression of either SRS29C allele in the RH type I WT strain is sufficient to alter virulence in mice, we engineered a series of SRS29C transgenic strains (designated SRS29C-I and SRS29C-II, respectively) that express either the type I or the type II SRS29C allele at levels equivalent to those found on the surface of avirulent type II strains (Fig. 7A). The type II SRS29C allele possesses five amino acid polymorphisms relative to the type I allele. No difference in growth, attachment, or invasion of cell lines in vitro was detected among the WT, a parallel green fluorescent protein (GFP) mutant (as a heterologous overexpression control), and the SRS29C transgenic overexpression clones (data not shown). CD-1 outbred mice were infected with WT, GFP overexpression control, SRS29C-I, and SRS29C-II parasite clones. All mice infected with WT parasites died (Fig. 7B). Remarkably, SRS29C overexpression attenuated the virulence of the WT strain in mice by 60 to 70% in five independent experiments. Bioluminescent imaging (BLI) to monitor in vivo parasite burdens showed that the parasites in WT-infected mice increased exponentially up to day 10, when the mice died. In contrast, the parasites in mice infected with SRS29C-I (Fig. 7C) increased exponentially for the first 5 days, peaked by day 8, and then progressively decreased on days 10 to 15, coincident with resolution of the acute infection phase. BLI of day 10 SRS29C-I-infected mice showed dramatically lower photonic signals than that of WT-infected mice in the ex vivo spleen, gastrointestinal tract, and thymus, indicating significant differences in the parasite loads in their organs (Fig. 7D). Hence, WT parasites achieve substantially greater parasite burdens and/or greater parasite dissemination to peripheral organs than do mice infected with SRS29C-I or -II parasites. The similar phenotypes observed with the two alleles argues that the level of SRS29C expression is more important to the virulence phenotype than the type-specific allele is.
FIG 7.

Transoverexpression of SRS29C is sufficient to attenuate acute virulence in mice. (A) Flow cytometric analysis of SRS29C surface expression in transgenic clones overexpressing type II (SRS29C-II) and type I (SRS29C-I) alleles. Endogenous SRS29C expression levels for type I (RH) and type II (Me49) are shown for comparison. Unstained control refers to staining without a primary antibody. (B) Effect of SRS29C overexpression on acute virulence in mice. Infection with SRS29C-I or SRS29C-II parasites resulted in significantly lower mortality than that caused by the WT. The number of seropositive mice infected with WT strain 22 was 17 for SRS29C-I and 14 for SRS29C-II. The total infectious dose for all experiments was 50 parasites. Mortality data are a composite of six independent experiments by using independently derived transgenic clones (two for SRS29C-I, one for SRS29C-II) and the WT parent. (C) Bioluminescent detection of parasite burdens and dissemination in vivo. CD-1 mice infected with 50 luciferase-positive WT or SRS29C-I parasites and imaged daily. Representative images with photon output intensity in photons/second/cm2/surface radiance (sr) are shown for days 3, 5, 8, and 11. The minimum number of photons was set at 3,932, and the maximum was set at 1.3 × 106. To control for background luminescence on days 8 and 11, the minimum was set at 2 × 105. (D) BLI of luciferase-positive parasites in ex vivo organs. Spleens, gastrointestinal tracts (GIT), and thymuses from mice sacrificed on day 10 were imaged ex vivo to quantify parasite burdens.
DISCUSSION
Here we present a comprehensive data set used for a systematic investigation of the genetic organization, evolution, regulation, and expansion of the coccidian SRS gene superfamily. The proteins they encode are the major constituents expressed on the surface coats of prevalent coccidian parasites, including T. gondii, N. caninum, and S. neurona. Comparative genomic analyses identified 182 SRS genes that were distributed throughout the genome, which is in contrast to surface antigens described in other protozoan species, where their subtelomeric location is thought to drive antigenic variation or sequential expression of a single gene on the parasite surface coat (i.e., VSG genes in T. brucei) or promote rapid sequence variation via increased rates of ectopic recombination (var genes in P. falciparum) (24, 25). In contrast, distinct, largely nonoverlapping sets of SRS mRNA transcripts were found to be codominantly expressed in a strain-dependent and developmental life cycle stage-dependent fashion. Although no data currently exist for merozoites, gametes, and developing oocysts that represent the sexual stages of T. gondii development, it is likely that their surface coats will likewise be populated by a mutually exclusive set of SRS proteins. Hence, the genetic mechanism(s) that regulates SRS gene expression is clearly different from that of the vsg or var genes, whereby only a single locus is expressed at any one time. Analysis of the expression data is a first step toward understanding how SRS genes are regulated as a system. Stage specificity data will become more robust as new RNA sequence data sets emerge for bradyzoites sampled from cysts in vivo, for merozoites growing in feline enterocytes, and for oocysts undergoing sporulation. By moving away from single-gene studies, future work will be able to look for factors that regulate the coordinated coexpression of these life cycle stage-specific SRS genes, whether it be by shared transcription factor binding sites and promoters or via epigenetic factors.
The size of the SRS superfamily, its sequence diversity, and its limited taxonomic distribution point to a relatively recent and rapid expansion coupled to a high mutation rate. The tolerance of these events is likely due to the SRS structural fold serving as a scaffold for the design of new biological properties, similar to the proposed evolution of the T. brucei transferrin receptor (TfR) from the VSG gene (26). Given the plasticity of this gene family, the assignment of SRS domains was nontrivial. The subfamily HMMs identified related domains, which were curated by match length and the presence of at least four cysteine residues. Domains that failed either of the latter two criteria were labeled as “degraded.” This led to the assignment of an SRS domain to SRS35, the SAG4 gene. It is noteworthy that SAG4.2 does not meet the HMM cutoff and contains too few cysteines; however, it is likely that this gene was duplicated from an SRS gene and evolved to fulfill a new functional niche. It is possible that a similar event led to the establishment of the SUSA gene family (5). Structural analysis and cross-species comparisons will help reveal the role of such SRS-related sequences.
The eight domain subfamilies present an excellent opportunity to study the SRS superfamily. An important caveat is that intrasubfamily relationships must remain ambiguous. Within each multigene locus, member genes have similar architectures of domain families and similar gene structures (number of introns) and, with the exception of the SRS38 locus, are transcribed in the same direction. These strong associations between domain families and the small number of observed domain architectures (only 9 of a potential 36 nondirectional two-domain combinations) imply that exon switching is not a significant mechanism for increasing protein divergence. Taken together, these data suggest that these loci arose by tandem gene duplication. Whether the expansion is due to duplications of single genes or whole loci is unclear. Considering a subset of two-domain proteins (families 7 and 8), it appears that a mixture of the two is most likely (Fig. 4). The situation is further complicated by convergent evolution. A recent analysis of the expanded SRS gene family in N. caninum has revealed intralocus genetic exchange as an important mechanism homogenizing SRS genes (27).
The spectrum of biological functions for known SRS proteins has led to the hypothesis that the SRS family has evolved distinct subsets that either function as adhesins (e.g., SRS57) or immune decoys (e.g., SRS34A) or possess some form of dual activity (e.g., SRS29B). In fact, the tachyzoite-specific SRS29B, SRS34A, and SRS29C proteins elicit high-titer antibodies during the acute infection phase (28). Such immune dominance has been postulated to focus immunity against the disseminating tachyzoite stage to limit their proliferation and away from the transmissible tissue-encysting bradyzoite form (which no longer expresses SRS29B, SRS34A, and SRS29C). Alternatively, immunity to these dominant antigens might function, at least in part, to provide a differentiation signal to switch tachyzoites into bradyzoites. This could represent a novel type of surface coat variation that is coupled to the developmental program of the parasite. These two mechanisms are not necessarily mutually exclusive, and the latter possibility is further supported by previous studies that implicate components of host immunity as differentiation signals (29). At least 29 orthologs of the SRS superfamily are present in other tissue-dwelling coccidians. In contrast, no SRS orthologs were identified in other apicomplexan parasites (Eimeria, Plasmodium, or Cryptosporidium) on the basis of primary sequence similarity searches. Recent studies, however, identified an SRS-like fold in the 10-member Pfs230-related 6-Cys family of gamete surface proteins, potentially extending the role of SRS domains as fertility factors in gamete-gamete recognition and attachment during fertilization (15, 30). It is unclear whether this represents the convergent evolution of unrelated proteins or is the result of divergent evolution of genes that arose from the same ancestral gene, possibly a precursor of the 6-Cys proteins that are a feature of the apicomplexan genomes (1).
Toxoplasma presumably regulates the expression of specific SRS antigens to successfully initiate infection and regulate the development of host immunity in order to establish patent, transmissible infections. Differential expression of certain alleles of the ROPK genes ROP5, ROP16, and ROP18 has previously been shown to facilitate parasite infection by actively altering immunity-related GTPases (IRGs) and cytokine signaling to promote parasite infection (4, 31–33). Integration of the SRS expression and polymorphism data identified numerous parasite lineage-specific differences and prioritized nine SRS genes for functional investigation to determine their role in parasite virulence. Targeted disruption of the SRS29B and SRS34A genes in the virulent type I RH strain did not affect parasite replication in vitro but did have a dramatic effect on mouse virulence. These DKO parasites showed a strong and specific upregulation in their SRS29C surface expression to a level found on the surface of mouse avirulent type II and III parasites. Upregulation was not at the level of transcription (Fig. 6C) but more likely to be the result of increased mRNA stability or to be regulated posttranscriptionally, since similar levels of SRS29C message were detected in type I strains, which express low levels of SRS29C protein, and the DKO parasites and type II and III strains, which are high expressers. Importantly, the SRS29C-I and SRS29C-II strains that transgenically overexpress the two SRS29C alleles in the virulent RH strain possessed similar lethal dose kinetics in mice, suggesting that the SRS29C expression level, rather than polymorphism, is the more relevant factor modulating the acute-virulence phenotype. The decreased virulence seen is most likely the function of an altered host immune response because no intrinsic growth defect was observed in the transgenic parasites and they were not impaired in the ability to invade host cells. Our preliminary results measuring systemic gamma interferon (IFN-γ) levels at the peak of infection showed that parasites expressing high levels of SRS29C protein had substantially reduced levels of IFN-γ during in vivo infection (V. Pszenny et al., unpublished data). Hence, the absolute levels of IFN-γ produced in mice infected with the SRS29C-I or SRS29C-II parasites relative to the WT RH parent strain are controlled throughout infection. Murine infections with type I strains typically produce dysregulated levels of interleukin-12 and IFN-γ and induce severe immunopathology and death (34, 35). It is possible that antigens such as SRS29C have evolved to fine-tune and/or negatively regulate the proinflammatory capacity of other immunodominant antigens (SRS29B) or polymorphic effector molecules that directly alter cytokine induction and/or signaling (ROP5, ROP16, ROP18, ROP38, GRA15) in a parasite strain- and host genetics-dependent fashion, resulting in dysregulated immunity and gross pathology (4, 8, 31, 32, 36). Ultimately, these two conceivably opposing mechanisms might ensure the persistence of the bradyzoite form of the parasite by actively subverting and/or fine-tuning the potent proinflammatory immune response that tachyzoite-specific antigens such as SRS29B, ROP16, ROP18, and GRA15 induce. This notion is strongly reinforced by a recent study implicating SRS genes that regulate the persistence of parasite cysts (7). This incredibly successful Toxoplasma parasite might thus have evolved such an intricate mechanism to allow for early parasite proliferation and dissemination to sites of chronic infection in the host (e.g., brain, heart) before a potent host response is mounted against the tachyzoite form to limit the parasite burden and the pathology of infection.
It is conceivable that Toxoplasma’s complex virulence traits are multifactorial, with a variety of different positive and/or negative modulatory factors that cooperatively function to confer various degrees of virulence among parasite strains. Ultimately, this would allow the efficient expansion of the range of hosts and ecological niches this successful parasite occupies. Attempts to alter the expression levels and/or engineer a targeted deletion of SRS29C in the avirulent type II Prugniaud strain are ongoing. Genetic disruption of the endogenous gene and/or subsequent transgenic manipulation to allow allele-specific and regulated expression of SRS29C will be required to elucidate the mechanistic basis governing the role of SRS29C in the acute-virulence phenotype across a variety of parasite genotypes.
In conclusion, this report highlights the importance of integrating comparative and functional genomic data sets to generate testable scientific hypotheses that are assumptionless in their approach. The identification of SRS29C as an important regulator of virulence capable of altering parasite pathogenesis and the development of protective immunity underpins the critical role apicomplexan parasite surface-expressed antigens play among eukaryotic pathogens that interact with and subvert host immune responses in order to establish patent, transmissible infections.
MATERIALS AND METHODS
Sequences, SRS domain family searches, phylogeny, and selection.
The genome sequences, protein sequences, and coding sequences were downloaded from ToxoDB (version 5.3 [Tg]), and the ESTs were downloaded from dbEST. HMMer (v3.0.3b) was used to build HMMs to perform domain searches against Toxoplasma protein models, as well as six-frame translations (transeq) of the genome (http://hmmer.janelia.org/). Pairwise distances were used to cluster the domains (metric, Kendall’s tau; method, complete linkage). The seed model for Pfam domain PF04092 (v. 23) was searched against the T. gondii protein sequences. The gathering score (GA) cutoff was used. The CD-Hit program was used to build a representative set of domain sequences (95% identity). All-against-all global alignments were produced by using the Needleman-Wunsch algorithm, and the bit score was used to hierarchically cluster the domains (metric, Kendall’s tau; method, complete linkage). The eight SRS subfamilies were aligned by using PROMALS. The alignment was seeded with SRS29B (1KZQ), SRS28 (2X28), and SRS16C (2JKS) structures. Three rules were used in domain identification; a sequence was accepted as an SRS domain if it aligned with one of the domain models with an E value of <e−5, was at least 90 amino acids in length, and contained at least four cysteine residues. Aligned regions with fewer than four cysteines were designated “degraded” domains. Assignment of orthologous groups was done manually and guided by MultiParanoid. The phylogenetic trees were generated by using the minimum-evolution (ME) and maximum-likelihood (ML) methods. To estimate ω (dN/dS ratio), the Slr program was used.
Pseudogenes were defined as SRS domains that met the search criteria but were not part of a ToxoDB gene model. Two domains separated by less than 1,000 bp were considered multidomain pseudogenes separated by an intron. Stop codons in SRS domains were present in 11% of the pseudogenes; for the remaining 89%, the absence of a gene model was likely due to a stop codon elsewhere in the sequence but outside the SRS domain.
Expression profiling.
ESTs were mapped onto SRS gene models by using BLASTX. Microarray data were downloaded from ToxoDB. To detect stage-specific expression, an arbitrary cutoff of ±2-fold divergence was used.
Polymorphism analyses.
Strain type-specific single-nucleotide polymorphisms were downloaded for each SRS gene model, and the ratio of synonymous-to-nonsynonymous changes was calculated. Protein-based multiple-sequence alignments were generated for orthologous groups found in all three strains. Coding sequences were mapped onto the alignment, and pairwise distances were calculated by using DNADIST. To estimate ω (dN/dS ratio), the Slr program was used; an ω of >1 is positive selection, and an ω of <0.5 is purifying.
Plasmid constructs and parasite strains.
Single-knockout parasites were engineered by replacing the SRS17A/B, SRS25, SRS29B/C, SRS33, SRS34A, and SRS35 genes with the drug-selectable hxgprt marker in the RH Δhxgprt “WT” strain. To construct the SRS29B/SRS34A DKO, the hxgprt gene inserted in the SRS29B locus was negatively selected by using 6-thioxanthine to remove the hxgprt gene and yield RH Δsrs29B ∆hxgprt. SRS34A was then disrupted by using the SRS34A single-knockout construct. A 3.2-kb fragment encompassing the SRS29C gene was amplified from type I RH and type II ME49 and cloned into pTOPO. Heterologous overexpression of SRS29C in the parental RH (WT) strain was selected by FACS using MAb CL15/4, which recognizes SRS29C.
Flow cytometry.
Parasites were fixed and stained with anti-SRS29B MAb DG52, anti-SRS34A MAb 5A6, anti-SRS57 MAb 4F12, anti-SRS29A polyclonal antibody SUS1, and anti-SRS29C MAb CL15/4. Stained parasites were analyzed by flow cytometry (FACS) with a FACSCalibur (BD Biosciences).
Mouse infection.
Seven- to 10-week-old female CD-1 outbred mice were infected by intraperitoneal injection with a total of 25, 50, 250, or 2,500 tachyzoite parasites diluted in 400 µl of phosphate-buffered saline. Mice were imaged daily to detect firefly luciferase activity by using an IVIS BLI system from Xenogen to monitor parasite burdens. Mice were injected with 200 µl (3 mg) of d-luciferin substrate and imaged for 5 min to detect the photons emitted.
qRT-PCR.
Total RNA (2 µg) isolated from tachyzoites with an RNeasy minikit (Qiagen) was reverse transcribed by using random primers and SuperScript II (Invitrogen). Gene expression was measured by TaqMan qRT-PCR by using an Applied Biosystems 7900HT real-time PCR system. For the primer and probe sets used, see Table S3 in the supplemental material. The cycling program included 2 min at 50°C and 10 min of incubation at 95°C, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. The Toxoplasma 18S rRNA gene was used as a reference to normalize the quantity of transcripts. Transcript levels were represented as 2−∆∆CT to show absolute levels of transcripts relative to every SRS gene examined.
SUPPLEMENTAL MATERIAL
Detailed Materials and Methods. Download File S1, DOCX file, 0.1 MB.
Structure-guided alignment of each domain subfamily. Download Figure S1, PDF file, 0.3 MB.
Single-nucleotide polymorphisms in 69 core SRS genes and ω values.
Evaluation of EST and microarray data confirming transcript expression for 95 SRS genes.
Primer and probe sets used for qRT-PCR.
ACKNOWLEDGMENTS
This study was financially supported by the CIHR (MOP no. 84556 to J.P. and M.E.G.), the Intramural Research Program of the NIH and NIAID (M.E.G.), and the Ontario MRI (J.P.). M.J.B. was supported by the CIHR (MOP no. 82915).
We acknowledge EuPathDB (http://eupathdb.org/) for providing a publicly available repository for all Toxoplasma genomic data sources and annotations. M.E.G. is a scholar of the Canadian Institute for Advanced Research Integrated Microbial Biodiversity Program. We thank Jonathan Wastling, Adam Reid, and Arnab Pain for critical reading of the manuscript.
Footnotes
Citation Wasmuth JD, et al. 2012. Integrated bioinformatic and targeted deletion analyses of the SRS gene superfamily identify SRS29C as a negative regulator of Toxoplasma virulence. mBio 3(6):e00321-12. doi:10.1128/mBio.00321-12.
REFERENCES
- 1. Templeton TJ. 2007. Whole-genome natural histories of apicomplexan surface proteins. Trends Parasitol. 23:205–212 [DOI] [PubMed] [Google Scholar]
- 2. Wasmuth J, Daub J, Peregrín-Alvarez JM, Finney CA, Parkinson J. 2009. The origins of apicomplexan sequence innovation. Genome Res. 19:1202–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jung C, Lee CY, Grigg ME. 2004. The SRS superfamily of Toxoplasma surface proteins. Int. J. Parasitol. 34:285–296 [DOI] [PubMed] [Google Scholar]
- 4. Peixoto L, et al. 2010. Integrative genomic approaches highlight a family of parasite-specific kinases that regulate host responses. Cell Host Microbe 8:208–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pollard AM, Onatolu KN, Hiller L, Haldar K, Knoll LJ. 2008. Highly polymorphic family of glycosylphosphatidylinositol-anchored surface antigens with evidence of developmental regulation in Toxoplasma gondii. Infect. Immun. 76:103–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dzierszinski F, Mortuaire M, Cesbron-Delauw MF, Tomavo S. 2000. Targeted disruption of the glycosylphosphatidylinositol-anchored surface antigen SAG3 gene in Toxoplasma gondii decreases host cell adhesion and drastically reduces virulence in mice. Mol. Microbiol. 37:574–582 [DOI] [PubMed] [Google Scholar]
- 7. Kim SK, Karasov A, Boothroyd JC. 2007. Bradyzoite-specific surface antigen SRS9 plays a role in maintaining Toxoplasma gondii persistence in the brain and in host control of parasite replication in the intestine. Infect. Immun. 75:1626–1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rachinel N, et al. 2004. The induction of acute ileitis by a single microbial antigen of Toxoplasma gondii. J. Immunol. 173:2725–2735 [DOI] [PubMed] [Google Scholar]
- 9. Manger ID, Hehl AB, Boothroyd JC. 1998. The surface of Toxoplasma tachyzoites is dominated by a family of glycosylphosphatidylinositol-anchored antigens related to SAG1. Infect. Immun. 66:2237–2244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mineo JR, et al. 1993. Antibodies to Toxoplasma gondii major surface protein (SAG-1, P30) inhibit infection of host cells and are produced in murine intestine after peroral infection. J. Immunol. 150:3951–3964 [PubMed] [Google Scholar]
- 11. Boulanger MJ, Tonkin ML, Crawford J. 2010. Apicomplexan parasite adhesins: novel strategies for targeting host cell carbohydrates. Curr. Opin. Struct. Biol. 20:551–559 [DOI] [PubMed] [Google Scholar]
- 12. He XL, Grigg ME, Boothroyd JC, Garcia KC. 2002. Structure of the immunodominant surface antigen from the Toxoplasma gondii SRS superfamily. Nat. Struct. Biol. 9:606–611 [DOI] [PubMed] [Google Scholar]
- 13. Howe DK, Crawford AC, Lindsay D, Sibley LD. 1998. The p29 and p35 immunodominant antigens of Neospora caninum tachyzoites are homologous to the family of surface antigens of Toxoplasma gondii. Infect. Immun. 66:5322–5328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Howe DK, et al. 2008. Strains of Sarcocystis neurona exhibit differences in their surface antigens, including the absence of the major surface antigen SnSAG1. Int. J. Parasitol. 38:623–631 [DOI] [PubMed] [Google Scholar]
- 15. Gerloff DL, Creasey A, Maslau S, Carter R. 2005. Structural models for the protein family characterized by gamete surface protein Pfs230 of Plasmodium falciparum. Proc. Natl. Acad. Sci. U. S. A. 102:13598–13603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Crawford J, et al. 2010. Structural and functional characterization of SporoSAG: a SAG2-related surface antigen from Toxoplasma gondii. J. Biol. Chem. 285:12063–12070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kraemer SM, et al. 2007. Patterns of gene recombination shape var gene repertoires in Plasmodium falciparum: comparisons of geographically diverse isolates. BMC Genomics 8:45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Boyle JP, et al. 2006. Just one cross appears capable of dramatically altering the population biology of a eukaryotic pathogen like Toxoplasma gondii. Proc. Natl. Acad. Sci. U. S. A. 103:10514–10519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Grigg ME, Bonnefoy S, Hehl AB, Suzuki Y, Boothroyd JC. 2001. Success and virulence in Toxoplasma as the result of sexual recombination between two distinct ancestries. Science 294:161–165 [DOI] [PubMed] [Google Scholar]
- 20. Li L, et al. 2003. Gene discovery in the Apicomplexa as revealed by EST sequencing and assembly of a comparative gene database. Genome Res. 13:443–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Manger ID, et al. 1998. Expressed sequence tag analysis of the bradyzoite stage of Toxoplasma gondii: identification of developmentally regulated genes. Infect. Immun. 66:1632–1637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boothroyd JC, Hehl A, Knoll LJ, Manger ID. 1998. The surface of Toxoplasma: more and less. Int. J. Parasitol. 28:3–9 [DOI] [PubMed] [Google Scholar]
- 23. Knoll LJ, Boothroyd JC. 1998. Isolation of developmentally regulated genes from Toxoplasma gondii by a gene trap with the positive and negative selectable marker hypoxanthine-xanthine-guanine phosphoribosyltransferase. Mol. Cell. Biol. 18:807–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cross GA, Wirtz LE, Navarro M. 1998. Regulation of vsg expression site transcription and switching in Trypanosoma brucei. Mol. Biochem. Parasitol. 91:77–91 [DOI] [PubMed] [Google Scholar]
- 25. Scherf A, et al. 1998. Antigenic variation in malaria: in situ switching, relaxed and mutually exclusive transcription of var genes during intra-erythrocytic development in Plasmodium falciparum. EMBO J. 17:5418–5426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Salmon D, et al. 1997. Characterization of the ligand-binding site of the transferrin receptor in Trypanosoma brucei demonstrates a structural relationship with the N-terminal domain of the variant surface glycoprotein. EMBO J. 16:7272–7278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Reid AJ, et al. 2012. Comparative genomics of the apicomplexan parasites Toxoplasma gondii and Neorospora caninum: coccidia differing in host range and transmission strategy. PLoS Pathog. 8:e1002567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lekutis C, Ferguson DJ, Grigg ME, Camps M, Boothroyd JC. 2001. Surface antigens of Toxoplasma gondii: variations on a theme. Int. J. Parasitol. 31:1285–1292 [DOI] [PubMed] [Google Scholar]
- 29. Bohne W, Heesemann J, Gross U. 1993. Induction of bradyzoite-specific Toxoplasma gondii antigens in gamma interferon-treated mouse macrophages. Infect. Immun. 61:1141–1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. van Dijk MR, et al. 2010. Three members of the 6-cys protein family of Plasmodium play a role in gamete fertility. PLoS Pathog. 6:e1000853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Saeij JP, et al. 2006. Polymorphic secreted kinases are key virulence factors in toxoplasmosis. Science 314:1780–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Saeij JP, et al. 2007. Toxoplasma co-opts host gene expression by injection of a polymorphic kinase homologue. Nature 445:324–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Taylor S, et al. 2006. A secreted serine-threonine kinase determines virulence in the eukaryotic pathogen Toxoplasma gondii. Science 314:1776–1780 [DOI] [PubMed] [Google Scholar]
- 34. Gavrilescu LC, Denkers EY. 2001. IFN-gamma overproduction and high level apoptosis are associated with high but not low virulence Toxoplasma gondii infection. J. Immunol. 167:902–909 [DOI] [PubMed] [Google Scholar]
- 35. Mordue DG, Monroy F, La Regina M, Dinarello CA, Sibley LD. 2001. Acute toxoplasmosis leads to lethal overproduction of Th1 cytokines. J. Immunol. 167:4574–4584 [DOI] [PubMed] [Google Scholar]
- 36. Reese ML, et al. 2011. Polymorphic family of injected pseudokinases is paramount in Toxoplasma virulence. Proc. Natl. Acad. Sci. U. S. A. 108:10568–10573 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Detailed Materials and Methods. Download File S1, DOCX file, 0.1 MB.
Structure-guided alignment of each domain subfamily. Download Figure S1, PDF file, 0.3 MB.
Single-nucleotide polymorphisms in 69 core SRS genes and ω values.
Evaluation of EST and microarray data confirming transcript expression for 95 SRS genes.
Primer and probe sets used for qRT-PCR.