

Abstract
Frizzled (Fz) 2 and Fz7, together with Fz1, form a distinct subfamily within the Frizzled family of Wnt receptors. Using targeted gene deletion, we show that: Fz7−/− mice exhibit tail truncation and kinking with 100% penetrance and ventricular septal defects (VSDs) with ~15% penetrance; Fz2+/−;Fz7−/− mice exhibit VSDs with ~50% penetrance and cleft palate with less than 10% penetrance; and Fz2−/−;Fz7−/− mice exhibit convergent extension defects and mid-gestational lethality with 100% penetrance. When Fz2 and/or Fz7 mutations are combined with mutations in Vangl2, Dvl3, Wnt3a, Wnt5a or Wnt11, an increased frequency of VSDs is observed with Dvl3, Wnt3a and Wnt11; an increased frequency of palate closure defects is observed with Vangl2; and early lethality and enhanced tail shortening are observed with Wnt5a. To assess the signaling pathways that underlie these and other Frizzled-mediated genetic interactions, we used transfected mammalian cells to analyze (1) canonical Wnt signaling induced by all pairwise combinations of the ten mouse Frizzleds and the 19 mouse Wnts and (2) localization of each Frizzled at cell-cell junctional complexes formed by mouse Celsr1, a likely indicator of competence for planar cell polarity signaling. These in vitro experiments indicate that Fz2 and Fz7 are competent to signal via the canonical pathway. Taken together, the data suggest that genetic interactions between Fz2, Fz7 and Vangl2, Dvl3 and Wnt genes reflect interactions among different signaling pathways in developmental processes that are highly sensitive to perturbations in Frizzled signaling.
Keywords: Fz2, Fz7, Convergent extension, Planar cell polarity, Wnt signaling
INTRODUCTION
Frizzled receptors are found throughout the animal kingdom, where they play central roles in controlling cell proliferation, movement and polarity. Frizzleds function as the principal receptors for the Wnt family of ligands and they couple to at least three distinct intracellular signaling pathways: the canonical Wnt pathway, the planar cell/tissue polarity (PCP) pathway, and the calcium pathway (van Amerongen and Nusse, 2009). The first two pathways are highly conserved between vertebrates and insects; the third has been studied principally in vertebrates. Canonical Wnt signaling features a co-receptor (Lrp5 or Lrp6 in mammals) that suppresses the constitutive proteolysis of β-catenin, which then migrates to the nucleus and, in conjunction with LEF/TCF family members, controls transcription of target genes. PCP signaling has been most extensively studied in epithelia, where it controls cytoskeletal organization via a set of asymmetrically positioned plasma membrane protein complexes that mediate vectorial communication between neighboring cells. These complexes are composed of Frizzled together with several other conserved PCP membrane proteins, including Stan/Fmi/Celsr and Vang/Vangl family members (Goodrich and Strutt, 2011; Gray et al., 2011).
Targeted loss-of-function mutations in the mouse have revealed developmental phenotypes for most of the ten Frizzled receptor genes. In many cases, partial redundancy is observed, especially among Frizzleds that are closely related in sequence. Redundant pairs include frizzled (Fz) 1 and Fz2, Fz3 and Fz6, Fz4 and Fz8, and Fz5 and Fz8 (Wang et al., 2006b; Yu et al., 2010; Ye et al., 2011; Liu et al., 2012). Fz3−/− and Fz6−/− mice exhibit phenotypes that appear the most PCP-like, as judged by their similarities to PCP phenotypes in Drosophila or to phenotypes caused by mutations in other core PCP genes in mice (e.g. Celsr1-3 and Vangl2). These include defects in CNS axon guidance (Fz3) and hair follicle orientation (Fz6) (Wang et al., 2002; Wang et al., 2006a; Lyuksyutova et al., 2003; Guo et al., 2004). Fz3−/−;Fz6−/− embryos additionally exhibit a fully open neural tube (craniorrhachischisis) and defects in inner ear sensory hair cell orientation (Wang et al., 2006b), as do Vangl2Lp/Lp embryos and Vangl1+/−;Vangl2Lp/+ embryos (Strong and Hollander, 1949; Torban et al., 2008). By contrast, Fz4 appears to act predominantly or exclusively via canonical Wnt signaling to control retinal vascularization (Xu et al., 2004). Fz4 also functions redundantly with Fz8 in kidney development, with Fz4−/−;Fz8−/− embryos exhibiting renal hypoplasia (Ye et al., 2009; Ye et al., 2011). Interestingly, in cell culture, Fz4 can activate both the canonical Wnt signaling pathway and the non-canonical Rho pathway. For most mammalian Frizzled genes, the question of which signaling pathway accounts for which developmental function remains open.
The present study focuses on Fz2 and Fz7, which, together with Fz1, form a distinct subfamily among mammalian Frizzled genes. As noted above, Fz1 and Fz2 are partially redundant. Fz1−/−;Fz2−/− embryos exhibit fully penetrant defects in palate closure and incomplete, but highly penetrant, ventricular septal defects (VSDs) (Yu et al., 2010). On a Vangl2Lp/+ background, loss of Fz1 and/or Fz2 greatly increases the frequency of neural tube defects and loss of Fz2 enhances the misorientation of inner ear sensory hair cells. These data suggest an interaction between PCP signaling and Fz1/Fz2 signaling – either a direct role of Fz1 and Fz2 in PCP signaling or an additive effect of compromised PCP signaling mediated by Vangl2 and non-PCP signaling mediated by Fz1 and Fz2. In the present study, we show that Fz7 is highly redundant with Fz2, we characterize the Fz2−/−;Fz7−/− embryonic lethal phenotype, we explore genetic interactions between Fz2, Fz7 and a series of canonical and non-canonical signaling genes, and we systematically compare the canonical and PCP signaling competence of all ten Frizzled family members in cell culture assays to address the potential of each Frizzled to mediate signaling in one or both pathways.
MATERIALS AND METHODS
Construction of Fz7−/− mice
The Fz7 gene targeting vector replaced the intronless Fz7 coding region with DNA coding for a nuclear-localized lacZ followed by a LoxP-PGK-neo-LoxP cassette (supplementary material Fig. S1). ES cell targeting, Southern blot screening, blastocyst injections and excision of the LoxP-PGK-neo-LoxP cassette were performed as described (Yu et al., 2010). PCR primers for the endogenous Fz7 gene are 5′-ATGGCGTCGCTTCTACCACAGACTCAGCCACAGC-3′ and 5′-ACAACCACTTGCCTTGCAGCAGGGGATTGGACTC-3′. Primers for the knock-in Fz7lacZ allele are 5′-GCCTCCCTGTATCCAAGCCTCTCCCCAGCG-3′ and 5′-CATCAACATTAAATGTGAGCGAGTAACAACCCG-3′.
Mouse lines and husbandry
Fz2−/− mice were described by Yu et al. (Yu et al., 2010). The following lines were obtained from the Jackson Laboratories: Vangl2Lp (Strong and Hollander, 1949); Wnt3a+/− (Takada et al., 1994); Wnt5a+/− (Yamaguchi et al., 1999); and Dvl3+/− (Etheridge et al., 2008). The Vangl2Lp allele was typed by PCR and sequencing as described (Yu et al., 2010). Mice were maintained on a mixed C57BL/6 × Sv129 background.
Wnt and Frizzled plasmids
Wnt and Frizzled open reading frames for expression in mammalian cells were purchased from Open BioSystems, identified by screening cDNA libraries, or obtained by PCR amplification of genomic DNA (for Frizzled coding regions lacking introns), and inserted into vectors with a cytomegalovirus immediate-early enhancer/promoter. An optimal translation initiation context (CCACCATG) was added by PCR to each Wnt and Frizzled coding region. For the Celsr1 colocalization experiments, a monoclonal antibody (mAb) 1D4 epitope [the C-terminus of bovine rhodopsin (Molday and MacKenzie, 1983)] was added to the C-terminus of each Frizzled coding region. All coding regions were fully sequenced to rule out spurious mutations.
Canonical Wnt signaling assay
A stable HEK293 cell line expressing a firefly luciferase reporter under the control of a minimal promoter and seven tandem LEF/TCF binding sites [Super Top Flash (STF) cells (Xu et al., 2004)] was transiently transfected in triplicate using FuGENE 6 (Roche) with expression plasmids encoding Wnt (50 ng), Frizzled (50 ng) and Lrp5 (5 ng), together with 1 ng Renilla luciferase expression plasmid and empty vector to bring the total DNA to 150 ng per well in a 24-well tray. Control transfections were performed as above, but with either the Wnt or Frizzled plasmids omitted. Luciferase activity was determined 48 hours post-transfection, and these values were corrected for any well-to-well variations in transfection efficiency and/or cell number by dividing each by the internal control Renilla luciferase activity. A second calculation normalized the canonical Wnt signaling activity from cells transfected with Wnt+Fz+Lrp5 relative to the canonical Wnt signaling activity from cells transfected with Wnt+Lrp5 and Fz+Lrp5. This value was calculated for each Wnt and Frizzled combination as follows: (Wnt+Fz+Lrp5)/[(Wnt+Lrp5)+(Fz+Lrp5)]. For example, if a particular Wnt+Fz+Lrp5 combination produced 1000 luciferase units and the individual Wnt+Lrp5 and Fz+Lrp5 samples produced 20 and 80 luciferase units, respectively, then the normalized value would be 1000/(20+80)=10.
For hierarchical clustering of Wnts and Frizzleds based on their mutual interactions, calculations were performed with both normalized and non-normalized data sets (as defined above). Using the log2 data sets, the mean Wnt+Fz+Lrp5 luciferase activity levels for each of the 19 Wnts was calculated and subtracted from the ten individual Wnt+Fz+Lrp5 luciferase activity values for that particular Wnt. The mean subtracted values were used for unsupervised hierarchical clustering using Pearson's correlation (Spotfire, Tibco).
Histology
Paraffin embedding and sectioning, anti-PECAM staining of embryo whole-mounts and X-Gal staining followed standard protocols (Ye et al., 2009; Yu et al., 2010). For plastic sections, yolk sacs were fixed with paraformaldehyde (PFA) and glutaraldehyde, osmicated, and embedded in Spurr's resin.
Transfection and immunostaining of Madin-Darby canine kidney (MDCK) cells
The plasmid coding for the mouse Celsr1-GFP fusion was a kind gift of Dr Elaine Fuchs (Rockefeller University, New York, USA), and mouse Celsr2-HA was a kind gift of Dr Tadashi Uemura (Kyoto University, Kyoto, Japan). MDCK cells were transfected with FuGENE 6 and imaged 2 days later using a Zeiss LSM700.
Immunostaining for cell surface protein
HEK293 cells (the parent cell line from which STF cells were derived) were seeded onto gelatin-coated coverslips and transiently transfected with plasmids coding for N-terminal 3×HA epitope-tagged Fz3 (3HA-Fz3), N-terminal 3×HA epitope-tagged Fz6 (3HA-Fz6), C-terminal rhodopsin epitope-tagged Fz3 (Fz3-1D4), or C-terminal rhodopsin epitope-tagged Fz6 (Fz6-1D4). The N-terminal tags were inserted between the end of the predicted signal peptide and the beginning of the ligand-binding cysteine-rich domain, with a duplication of four amino acids from the target Frizzled sequence flanking the site of 3×HA epitope insertion (SLFS for Fz3 and SLFT for Fz6). One day after transfection, coverslips were processed in one of two ways: (1) to visualize epitopes accessible exclusively from the extracellular space, living cells were stained with rabbit anti-3×HA antiserum or mouse mAb 1D4 in cell culture medium with 0.5% calf serum for 1 hour on ice, washed in ice-cold PBS, and then fixed with 4% PFA/PBS for 30 minutes on ice; and (2) to visualize epitopes in all locations, cells were fixed with 4% PFA/PBS for 30 minutes on ice, and immunostained with rabbit anti-3×HA antiserum or mouse mAb 1D4 in 0.3% Triton X-100/PBS/10% normal goat serum (NGS) overnight at 4°C and washed in 0.3% Triton X-100/PBS. After primary antibody binding, coverslips from both procedures were incubated in secondary antibodies and DAPI in 0.3% Triton X-100/PBS/10% NGS for 2 hours at room temperature. For all of the samples, transfections and immunostaining reactions were carried out in duplicate and in parallel. Confocal images of matched pairs of permeabilized and non-permeabilized samples were collected with identical parameter settings.
Microarray hybridization
Individual embryonic day (E) 8.5 embryos were stored at −80°C while their genotypes were determined from yolk sac DNA. For each RNA sample, two or three E8.5 embryos of identical Frizzled genotype but undetermined sex were pooled, and RNA was purified using Trizol (Invitrogen) and RNeasy (Qiagen) kits. Three biologically independent sets of Fz7−/− versus Fz2−/−;Fz7−/− embryo RNAs were hybridized to Affymetrix mouse genome 430 2.0 microarrays and the data analyzed using Spotfire. The microarray data are available at Gene Expression Omnibus with accession number GSE37221.
Statistics
Pairwise comparisons of phenotype frequencies were performed with 2×2 contingency tables and Fisher's exact test.
RESULTS
Targeted replacement of Fz7 with lacZ
To eliminate Fz7 function, we generated a mouse line carrying a targeted replacement of the Fz7 coding region with the coding region of a nuclear-localized derivative of E. coli β-galactosidase (we refer to this allele equivalently as Fz7lacZ or Fz7−; supplementary material Fig. S1). Fz7lacZ/lacZ mice are viable and fertile; their only apparent anatomical phenotype is a ~10% shorter tail with a distal kink, as discussed below. X-Gal staining of Fz7lacZ/lacZ embryos at E8.5 and E9.5 shows widespread expression of the lacZ reporter, including expression throughout the developing nervous system and somites. This pattern closely resembles the embryonic expression patterns observed for Fz1lacZ and Fz2lacZ alleles (Yu et al., 2010) (supplementary material Fig. S1).
As noted above, Fz1, Fz2 and Fz7 constitute a distinct branch of the Frizzled family of receptors (supplementary material Fig. S2), and Fz1 and Fz2 show partial redundancy (Yu et al., 2010). To explore the possibility of additional redundancies within this subfamily, we generated mice with various combinations of Fz1− and Fz7− alleles and Fz2− and Fz7− alleles. Fz1−/−;Fz7−/− mice are healthy and fertile but have a gray coat color, suggesting that these genes act redundantly in some aspect of melanocyte development, function or survival. Approximately 50% of Fz2+/−;Fz7−/− embryos have cardiac defects (described more fully below) and more than 50% of Fz2+/−;Fz7−/− embryos fail to survive postnatally; the surviving minority of Fz2+/−;Fz7−/− mice are viable and fertile. Genotyping of more than 100 progeny from an intercross among Fz2+/−;Fz7−/− mice revealed no Fz2−/−;Fz7−/− progeny at any postnatal age.
Early lethal phenotype in Fz2−/−;Fz7−/− embryos
An examination of mid-gestation embryos from Fz2+/−;Fz7−/− intercrosses showed growth retardation of Fz2−/−;Fz7−/− embryos starting at ~E8.5, with death at ~E10.5 (Fig. 1). At E8.5, when Fz2−/−;Fz7−/− embryos are only modestly smaller than control littermates, they exhibit a strikingly rotund morphology with a length:width ratio of ~2, as compared with the control ratio of ~3.5 (Fig. 1A,B,E). Over the ensuing day, Fz2−/−;Fz7−/− embryos fail to close the neural tube, develop left-right heart asymmetry, or complete tail turning (Fig. 1C,D,F-M). The abrupt growth arrest of Fz2−/−;Fz7−/− embryos might be caused, at least in part, by a failure of normal vascular development. At E9.5, the Fz2−/−;Fz7−/− embryo vasculature is underdeveloped and the yolk sac has failed to generate an organized vascular plexus, and by E10.5 the yolk sac vasculature is engorged with blood (Fig. 2). This last observation implies that hematopoiesis is not substantially affected in Fz2−/−;Fz7−/− embryos.
Fig. 1.
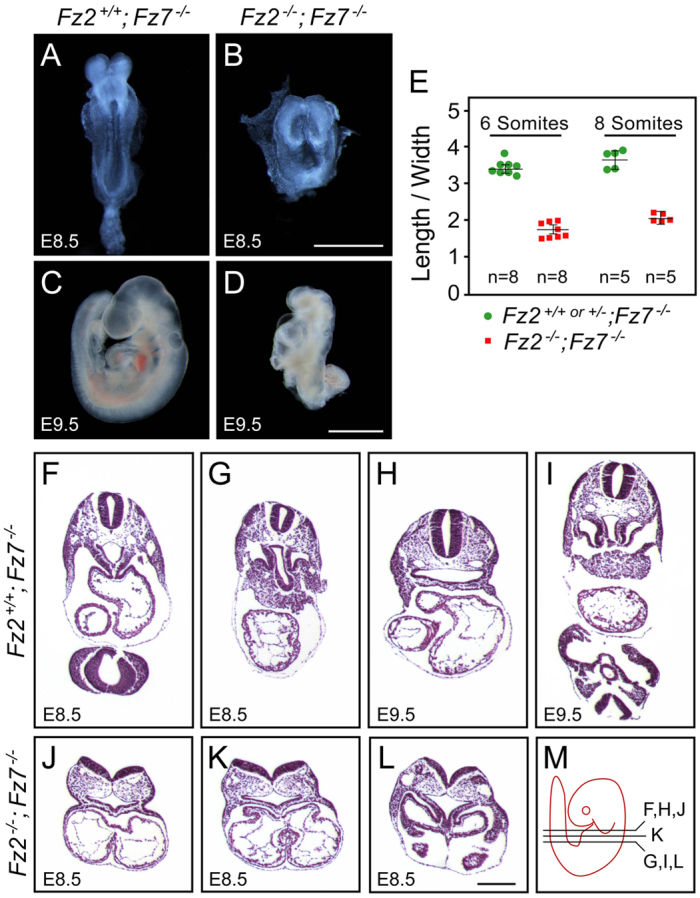
Fz2−/−;Fz7−/− embryos exhibit a severe defect in convergent extension and die at ~E10.5. (A-D) Fz2+/+;Fz7−/− mouse embryos are indistinguishable from wild type at all stages. Fz2−/−;Fz7−/− embryos arrest growth at ~E8.5; at E9.5 they fail to turn but are still alive as judged by the presence of a heartbeat. Unstained specimens are shown. (E) Quantification of convergent extension, as determined by the embryo length:width ratio, among Fz2−/−;Fz7−/− and littermate control (Fz2+/− ;Fz7−/− or Fz2+/+;Fz7−/−) embryos at ~E8.5. Error bars indicate s.e.m. (F-M) H&E-stained transverse sections (as indicated in M) of the thorax from control Fz2+/+;Fz7−/− embryos at E8.5 and E9.5 and Fz2−/−;Fz7−/− embryos at E8.5. At E8.5, the Fz2−/−;Fz7−/− embryo has an open neural tube and a symmetric heart, whereas the control has a closed neural tube and exhibits left-right heart asymmetry. Scale bars: 1 mm in A-D; 200 μm in F-L.
Fig. 2.
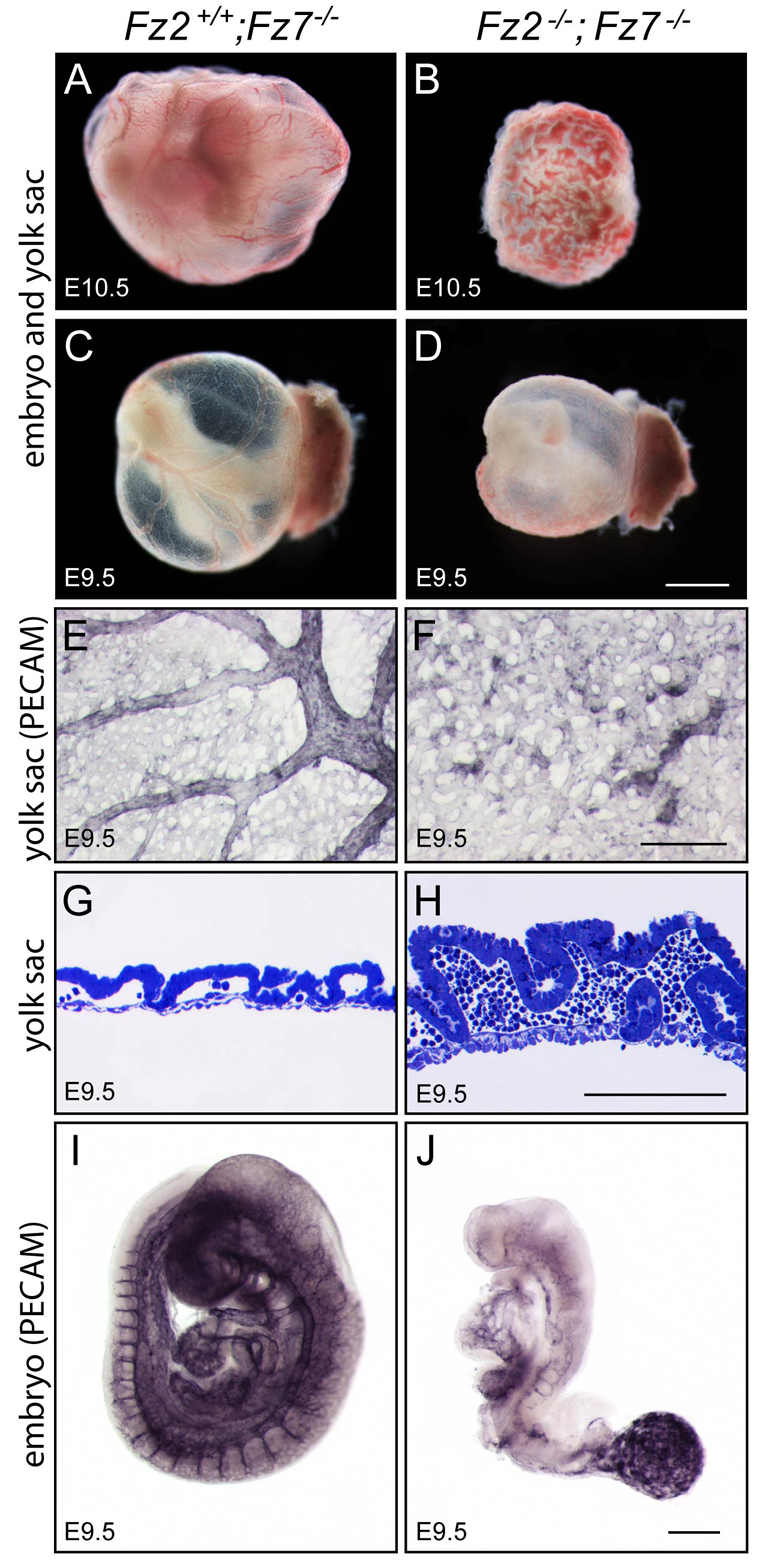
Vascular defects in Fz2−/−;Fz7−/− embryos. (A-D) Fz2−/−;Fz7−/− mouse embryos are still alive at E10.5 as judged by the presence of a heartbeat, but the yolk sac shows extensive pooling of blood. Unstained specimens are shown. (E-H) Fz2−/−;Fz7−/− yolk sac vasculature fails to differentiate into well-organized larger and smaller vessels (compare E and F; Pecam 1 immunostaining). Pooling of red blood cells in the dense capillary network is seen in transverse section (compare G and H; Toluidine Blue staining). (I,J) The Fz2−/−;Fz7−/− embryo vasculature remains rudimentary at E9.5. Scale bars: 1 mm in A-D; 500 μm in E,F,I,J; 100 μm in G,H.
The morphology of Fz2−/−;Fz7−/− embryos at E8.5 suggests a severe defect in convergent extension, a process that has been associated with PCP signaling in Xenopus, zebrafish and mice (Goodrich and Strutt, 2011; Gray et al., 2011). Interestingly, morpholino oligonucleotide knockdowns of zebrafish fz2 and Xenopus Fz7 are also associated with convergent extension defects (Sumanas and Ekker, 2001; Sumanas et al., 2001). Current evidence suggests that PCP signaling controls the cytoskeleton with little or no effect on gene expression (Goodrich and Strutt, 2011; Gray et al., 2011). Microarray hybridization of RNA from Fz7−/− control versus Fz2−/−;Fz7−/− embryos at E8.5 shows fewer than ten transcripts with abundance changes greater than 2-fold and P<0.05 (supplementary material Fig. S3). Moreover, a separate analysis of 25 genes known to be induced by canonical Wnt signaling in mammals, of which 19 have been characterized as direct targets of LEF/TCF/β-catenin, showed no evidence for a decrease in canonical Wnt signaling in Fz2−/−;Fz7−/− embryos (supplementary material Fig. S4 and Table S1). The changes in transcript abundance cluster near to zero on the log2 plot, and, to the extent that those values deviate from zero, there is an accompanying increase in the P-value to a level that is statistically insignificant. Stated equivalently, there is no statistically significant evidence for the hypothesis of a change in transcript abundance compared with the null hypothesis of no change in abundance. These data are consistent with the idea that the morphological defect in E8.5 Fz2−/−;Fz7−/− embryos reflects a disruption of PCP signaling, which may involve few or no changes in gene expression.
Genetic interactions between Fz2, Fz7 and Vangl2, Dvl3 and Wnt genes
To broadly explore the possibility that Fz2 and Fz7 interact genetically with PCP and/or canonical Wnt signaling components, we determined the frequencies of anatomical anomalies in crosses with each of the following: VanglLp [Lp is a semi-dominant allele (Strong and Hollander, 1949; Song, H. et al., 2010; Yin et al., 2012)], a null allele of dishevelled 3 [Dvl3; one of three highly homologous and partially redundant cytosolic signaling proteins involved in both canonical and PCP signaling (Etheridge et al., 2008)], and null alleles of Wnt3a, Wnt5a and Wnt11. Wnt3a was chosen for these experiments because there is strong in vivo evidence that it acts via the canonical pathway in the early embryo (Galceran et al., 2001; Nakaya et al., 2005). Wnt5a was chosen because it has been implicated in PCP signaling in the inner ear and is required for cardiac outflow tract development, trunk elongation and tail development (Yamaguchi et al., 1999; Qian et al., 2007; Schleiffarth et al., 2007). In cell culture, Wnt5a has been reported to act via Fz2 as well as ROR receptors and to either activate or inhibit canonical signaling depending on the receptors present (Oishi et al., 2003; Mikels and Nusse, 2006; Sato et al., 2010; Ho et al., 2012). Wnt11 was chosen because it appears to act via a non-canonical pathway in collaboration with Fz7 during convergent extension in zebrafish and Xenopus and during cardiac development in Xenopus (Heisenberg et al., 2000; Pandur et al., 2002; Kim et al., 2008).
Among the many combinations of genotypes examined with one or more null alleles of Fz2 and/or Fz7 in combination with mutant alleles of Vangl2, Dvl3, Wnt3a, Wnt5a or Wnt11, the most informative were the combinations with Fz2+/−;Fz7−/− (Fig. 3). Fz2+/−;Fz7−/− embryos appear to be at threshold for cardiac defects – in particular VSD, or less commonly VSD in combination with double-outlet right ventricle (DORV). These embryos also appear to be near threshold for palate closure defects. Cardiac defects were analyzed from Hematoxylin and Eosin (H&E) stained transverse paraffin sections obtained at late gestation (E15-18; Fig. 3A). The presence of a single Vangl2Lp allele had little or no effect on the frequency of cardiac defects on a Fz2+/−;Fz7−/− background: 37% of Fz2+/−;Fz7−/−;Vangl2Lp embryos (n=18) were affected compared with 50% of Fz2+/−;Fz7−/− embryos (n=22; P=0.537). A single Vangl2Lp allele produced a modestly elevated frequency of cardiac defects on a Fz7−/− background: 35% of Fz7−/−;Vangl2Lp embryos were affected (n=20) compared with 14% for Fz7−/− (n=14; P=0.250), and, more significantly, a 50% frequency of cardiac defects was observed for Fz2+/−;Vangl2Lp embryos (n=6) compared with 0% for Vangl2Lp (n=8; P=0.055) and 0% for Fz2−/− [n=11; reported in Yu et al. (Yu et al., 2010); P=0.029] embryos.
Fig. 3.

Tests for genetic interactions between Fz2, Fz7 and Vangl2, Dvl3, Wnt3a and Wnt11. (A) (Top) H&E-stained transverse paraffin sections showing cardiac defects in E15-18 mouse embryos. The vast majority were ventricular septal defects (VSDs), as seen upon comparing the normal cardiac anatomy of a Fz2+/−;Fz7−/− heart (left) with a pair of sections from a Fz2+/−;Fz7−/−;Dvl3+/− heart (right). PA, pulmonary artery; LV, left ventricle; RV, right ventricle. (Bottom) Summary of the numbers of embryos with the indicated genotypes and cardiac phenotypes. Each set of crosses is represented as a separate cluster of red and blue bars, with blue numbers and bars indicating embryos with no cardiac defect, and red numbers and bars indicating embryos with cardiac defects. Bar heights are normalized to the total number of embryos of the indicated genotype. (B) (Top) Palate closure defects in E18.5 embryos, as seen upon comparing the normal palate anatomy at anterior (A) and posterior (P) sites in an E18.5 Fz2+/−;Fz7−/− head (left pair of panels) with the cleft palate (blue arrows) of an E18.5 Fz2+/−;Fz7−/−;Vangl2Lp/+ head (right pair of panels). Most palates were scored intact after removing the lower jaw. Bottom, summary of genotypes and palate phenotypes as described for A. Scale bars: 1 mm.
Loss of one allele of Dvl3 increased the frequency of cardiac defects from 14% for Fz7−/− embryos (n=14) to 50% for Fz7−/−;Dvl3+/− embryos (n=14; P=0.103) and from 50% for Fz2+/−;Fz7−/− embryos (n=22) to 83% for Fz2+/−;Fz7−/−;Dvl3+/− embryos (n=12; P=0.075). Dvl3+/− embryos did not exhibit cardiac defects (n=5). Consistent with the high penetrance of cardiac defects observed by Etheridge et al. (Etheridge et al., 2008) in Dvl3−/− embryos, we observed three out of three Fz7−/−;Dvl3−/− embryos with cardiac defects.
Loss of a single allele of Wnt3a or Wnt11 similarly increased the frequency of cardiac defects. For Wnt3a, the increase was from 14% for Fz7−/− embryos (n=14) to 37% for Fz7−/−;Wnt3a+/− embryos (n=16; P=0.226), and from 50% for Fz2+/−;Fz7−/− embryos (n=22) to 92% for Fz2+/−;Fz7−/−;Wnt3a+/− embryos (n=12; P=0.024). With loss of one Wnt11 allele, no cardiac defects were seen on Fz7+/− or Fz7−/− backgrounds, but 100% of Fz2+/−;Fz7−/−;Wnt11+/− embryos had cardiac defects (n=10) compared with 50% of Fz2+/−;Fz7−/− embryos (n=22; P=0.006).
In a large number of crosses between Fz2+/−;Fz7−/− parents, we did not see palate closure defects in Fz7−/− or Fz2+/−;Fz7−/− embryos (n>50 each; Fig. 3B). Similarly, the presence of one or two Vangl2Lp alleles on a wild-type background did not produce palate closure defects (n=20 for Vangl2Lp/+ and n=10 for Vangl2Lp/Lp embryos). However, the presence of a single Vangl2Lp allele produced a higher frequency of cleft palate in combination with Fz2+/−;Fz7−/− (53% of Fz2+/−;Fz7−/−;Vangl2Lp/+ embryos affected; n=17) compared with Fz2+/−;Fz7−/− controls from the same set of crosses (15% affected; n=13; P=0.057). The occurrence of palate closure defects among Fz2+/−;Fz7−/− littermate controls in this last set of crosses suggests that the Fz2+/−;Fz7−/− genotype is very close to threshold for palate closure defects. This last observation suggests that non-genetic variation and/or differences in genetic background account, at least in part, for the palate closure defects seen in 4% of Fz7−/−;Vangl2Lp/+ embryos (n=25), 14% of Fz2+/−;Fz7−/−;Wnt3a+/− embryos (n=14) and 11% of Fz2+/−;Fz7−/−;Wnt11+/− embryos (n=9).
Wnt5a+/− mice have no apparent phenotype, but Wnt5a+/−;Fz7−/− mice show an accentuated tail truncation phenotype relative to Fz7−/− mice (Fig. 4A,B). With the additional loss of one copy of Fz2, no embryos survive beyond E11. Fz2+/−;Fz7−/−;Wnt5a+/− embryos arrest growth starting at ~E9.5 (Fig. 4C); the mechanism of the arrest has not been investigated.
Fig. 4.
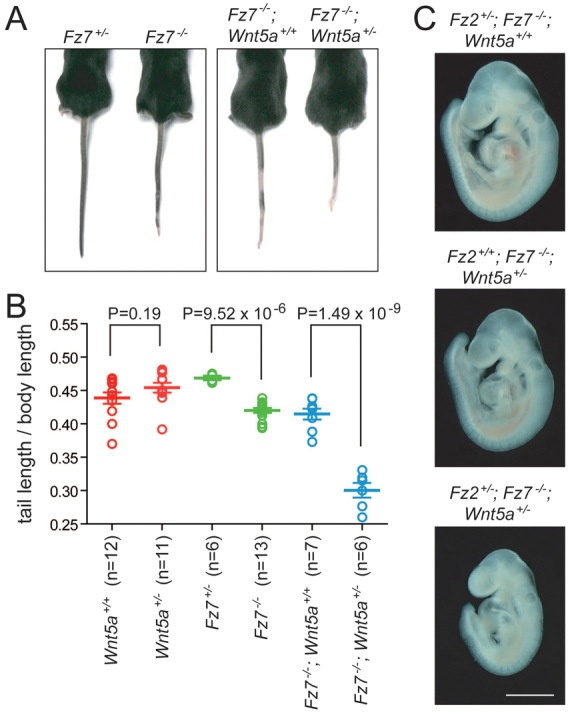
Wnt5a+/− in combination with Fz7−/− enhances the short tail phenotype and in combination with Fz2+/−;Fz7−/− causes mid-gestational lethality. (A,B) The only anatomical phenotype evident in Fz7−/− mice is a ~10% decrease in tail length and a small kink at the end of the tail. The scatter plot shows tail length divided by body length, measured from the nose to the base of the tail, at 4 weeks of age. Wnt5a+/− mice show no tail phenotype, but Wn5a+/− enhances the Fz7−/− tail shortening phenotype. Error bars indicate s.e.m. (C) E9.5 littermates illustrate the lethal growth retardation in Fz2+/−;Fz7−/−;Wnt5a+/− embryos. Fz2+/−;Fz7−/−;Wnt5a+/+ and Fz2+/+;Fz7−/−;Wnt5a+/+ embryos are unaffected at this age. Unstained specimens are shown. Scale bar: 1 mm.
In summary, Fz7−/− mice exhibit tail truncation and kinking with full penetrance and cardiac defects with low penetrance; Fz2−/− mice exhibit cleft palate with ~50% penetrance; Fz2+/−;Fz7−/− mice exhibit cardiac defects with ~50% penetrance and cleft palate with low penetrance; and Fz2−/−;Fz7−/− mice exhibit convergent extension defects and mid-gestational lethality with 100% penetrance. When Fz2 and/or Fz7 mutations are combined with mutations in one or both copies of Vangl2, Dvl3, Wnt3a, Wnt5a or Wnt11, increased frequencies of cardiac phenotypes are observed with Dvl3, Wnt3a and Wnt11; an increased frequency of palate closure defects is observed with Vangl2; and enhanced tail shortening and early lethality are observed with Wnt5a.
Cell culture assay of Frizzled signaling potential: canonical signaling
The genetic interactions described above that link Fz2 and Fz7 to Vangl2, Dvl3, Wnt3a, Wnt5a and Wnt11 imply a convergence in the actions of these signaling components, but the data do not define how direct or indirect that convergence is. At one extreme, a reduction in Wnt ligand level could synergize with a reduction in Frizzled receptor level because the two components act directly as a ligand-receptor pair. However, there are many possibilities that are less direct. For example, the ligand and receptor could act through the same signaling pathway but via distinct cell surface complexes, or through different signaling pathways that later converge via their effects on some general behavior such as cell motility. It is also possible that the ligand and receptor could act on different cells and/or at different times but converge on the same biological process. For example, one component might act in one group of cells to facilitate their migration, whereas the other component might act in a second group of cells that serve as guideposts or substrates for that migration. The same ambiguity regarding the mechanism also applies to genetic interaction between receptors (Frizzled) and non-ligand signaling components (Vangl2 and Dvl3).
As one approach to constraining the mechanisms that might explain the Fz2 and Fz7 phenotypes and their genetic interactions, as well as to more generally assess the signaling capacities of Frizzled receptors, we have tested the activities of each mouse Frizzled protein in two assays that currently represent the best cell culture correlates of canonical and PCP signaling capacity, as described below.
For canonical signaling, we transfected cDNAs encoding each of the ten mouse Frizzleds and each of the 19 mouse Wnts in all pairwise combinations into a HEK293 cell line that responds to β-catenin stabilization and LEF/TCF activation by expression of a firefly luciferase reporter [Super Top-Flash (STF) cells (Xu et al., 2004)]. Fig. 5 shows the results of three averaged experiments for each of the 190 Wnt-Frizzled combinations, as well as individual transfections of the 19 Wnts and ten Frizzleds. All transfections included the canonical Wnt signaling co-receptor Lrp5, and the resulting firefly luciferase values were normalized to a cotransfected Renilla luciferase control.
Fig. 5.

Canonical Wnt signaling in STF cells induced by each of the 19 Wnts in pairwise combination with each of the ten Frizzleds. (A) Luciferase activity was determined following transient transfection of STF cells with: (1) each of the 19 mouse Wnts together with Lrp5 (vertical columns), (2) each of the ten mouse Frizzleds together with Lrp5 (horizontal rows), and (3) the 10×19=190 pairwise combinations of mouse Wnt and mouse Frizzled together with Lrp5 (central rectangles). Four representations of the data are shown, and all values are averages of three transfections. (Left) Not normalized, signifying that the firefly luciferase values were corrected only with respect to the Renilla luciferase internal control. (Right) Normalized, signifying that the firefly luciferase values were additionally normalized by calculating (Wnt+Fz+Lrp5)/[(Wnt+Lrp5)+(Fz+Lrp5)] to reveal the fold increase in luciferase activity referable specifically to the interaction between Frizzled and Wnt. Upper pair of panels, linear values; lower pair of panels, log2 values. The numerical ranges for the yellow-blue calibration bar are as follows (lowest value is pure yellow; highest value is pure blue): Wnt+Lrp5 (linear), 0.14 to 140.80; Wnt+Lrp5 (log2), −2.84 to 7.14; Fz+Lrp5 (linear), 0.26 to 16.30; Fz+Lrp5 (log2), −1.94 to 4.03; Wnt+Fz+Lrp5 (linear, not normalized), 0.19 to 170.79; Wnt+Fz+Lrp5 (log2, not normalized), −2.40 to 7.42; Wnt+Fz+Lrp5 (linear, normalized), 0.28 to 51.27; Wnt+Fz+Lrp5 (log2, normalized), −1.84 to 5.68. For each of the individual data sets – Wnt+Lrp5, Fz+Lrp5 and Wnt+Fz+Lrp5 (normalized and not normalized) – the yellow-blue calibration has been set so that the full color range corresponds to the full numerical range. Numerical values are listed in supplementary material Table S2. The luciferase activity for cells transfected with Lrp5 alone is not statistically distinguishable from the lowest values of Wnt+Lrp or Fz+Lrp combinations. (B) Hierarchical clustering of Wnts and Frizzleds based on luciferase activity for the not normalized and normalized data sets as defined in A (see Materials and methods for details). For the blue-white-red spectrum, the range of values for the not normalized and normalized data sets are, respectively, −4.338 to 4.2225 and −3.548 to 3.831.
The heat maps in Fig. 5A show four representations of the same luciferase data sets (supplementary material Table S2). Each set of panels shows the individual Wnt+Lrp5 luciferase levels (left column), the individual Fz+Lrp5 luciferase levels (top row), and the 190 pairwise combinations of Wnt+Fz+Lrp5 (10×19 rectangle). Each of these three data sets have been independently color-coded with minimal and maximal values represented by fully saturated yellow and blue, respectively. Strikingly, transfection of Wnt1+Lrp5 produces a high level of luciferase activity, suggesting that STF cells might express a receptor for Wnt1. Similarly, transfection of Fz9+Lrp5 produces a high level of luciferase activity, suggesting that Fz9 can signal in a ligand-independent manner or that an Fz9 ligand might be either produced by STF cells or present in bovine serum. For the two sets of panels on the right of Fig. 5A, the individual Wnt+Fz+Lrp5 values have been corrected to show the fold change relative to the level of canonical signaling conferred by the individual Wnt+Lrp5 and Fz+Lrp5 samples. Fig. 5A also displays both linear (top) and log2 transformed (bottom) versions of the data sets; the log2 version reveals more subtle differences in signal strength. Fig. 5B shows an unsupervised hierarchical clustering of Wnts and Frizzleds based on signaling specificity and amplitude.
Several patterns emerge from these data. First, among the Frizzleds, Fz3 and Fz6 show the weakest signaling in combination with nearly all Wnts, the single exception being the combination of Wnt5b and Fz6. The low level of canonical signaling exhibited by Fz3 and Fz6 is consistent with the PCP-like phenotype of Fz6−/− (aberrant hair follicle orientation) and Fz3−/−;Fz6−/− (aberrant inner ear sensory hair orientation) mice and the high sequence homology between Fz3 and Fz6 (supplementary material Fig. S2). Second, several Wnts (e.g. Wnt2, Wnt3, Wnt3a and Wnt7b) activate multiple Frizzleds, and several Frizzleds (e.g. Fz1, Fz2, Fz4, Fz5, Fz7 and Fz8) are activated by multiple Wnts. Third, Frizzleds with a high degree of amino acid sequence identity show similar patterns of Wnt activation. Thus, Fz1, Fz2 and Fz7 form one subgroup, Fz5 and Fz8 form a second subgroup, and Fz3 and Fz6 form a third subgroup.
In extrapolating these luciferase data to the in vivo situation, we note that there are a number of uncertainties. First, the efficiencies with which different Wnts and Frizzleds are correctly folded, processed and transported in STF cells are unknown. In this regard, we note that C-terminal epitope-tagged versions of all ten Frizzleds accumulate in transfected HEK293 cells (the STF parent line) to levels that are readily detected by immunostaining, and that an examination of the cell surface localization of the two Frizzleds that exhibit the weakest canonical signaling (Fz3 and Fz6) by immunostaining of living cells for an extracellular epitope tag reveals readily detectable signals (supplementary material Fig. S5). Second, differential effects of co-receptors (Lrp5 versus Lrp6) or chaperones (such as Tspan12) have not been tested. Third, differential effects of serum-derived components have not been tested. Fourth, the STF assay does not take into account the in vivo levels of ligands, receptors or intra- and extracellular modulators of canonical Wnt signaling. Finally, the biological significance of a particular level of canonical signaling is difficult to predict – a low level of signaling, could, in the appropriate context, have a large biological effect. Despite these caveats, these data represent the first comprehensive analysis of Wnt-Frizzled signaling and they are a useful starting point for assessing ligand-receptor relationships in this system.
Cell culture assay of Frizzled signaling potential: PCP signaling
At present, there is no cell culture system that reproduces PCP signaling. However, in the Drosophila wing disc and in the mammalian inner ear and skin, Frizzleds are arrayed on one side of each epithelial cell, Vang/Vangl proteins are arrayed on the opposite side, and Fmi/Stan/Celsr proteins are present on both sides where they appear to mediate homophilic adhesion between neighboring cells via their cadherin repeats (Goodrich and Strutt, 2011; Gray et al., 2011). This distinctive localization of a subset of core PCP proteins in epithelial cells suggests that the assembly of asymmetric cell surface complexes could serve as an assay to predict which Frizzleds have the capacity to participate in PCP signaling. Importantly, a rudimentary version of these asymmetric complexes has recently been observed in mouse keratinocytes transfected with Celsr1, Fz6 and Vangl2 (Devenport and Fuchs, 2008).
To assess the competence of each Frizzled to co-assemble with Celsr1 in surface complexes, we transfected MDCK epithelial cells with plasmids coding for: (1) Celsr1-GFP; or (2) each of the ten mouse Frizzleds (with a C-terminal epitope tag corresponding to the C-terminal ten amino acids of bovine rhodopsin, referred to as 1D4); or (3) Celsr1-GFP together with each 1D4-tagged Frizzled. Two days after transfection, we analyzed neighboring cell pairs (‘doublets’). As the overall transfection efficiency was relatively low (<5%), we presume that most doublets arose from a cell division that occurred after DNA uptake. Doublets expressing Celsr1-GFP alone or in combination with a Frizzled-1D4 typically display strong accumulation of Celsr1-GFP at the junction between the adjacent cells (Fig. 6). When Frizzled-1D4 proteins are expressed without Celsr1-GFP, there is variable accumulation of Frizzled-1D4 at the plasma membrane, with Fz9-1D4 showing the most efficient accumulation. Under these conditions, most doublets do not show preferential accumulation of Frizzled-1D4 at the cell interface; two representative doublets are shown for each Frizzled-1D4 in Fig. 6. However, when Frizzled-1D4 and Celsr1-GFP are co-expressed, Fz3-1D4, Fz5-1D4 and Fz6-1D4 exhibit efficient colocalization with Celsr1-GFP at the cell interface (five representative doublets are shown for each Frizzled-1D4 + Celsr1-GFP set in Fig. 6). Fz4-1D4 occasionally (<50%) and Fz2-1D4 rarely (~10%) showed colocalization with Celsr1-GFP at the cell interface. The other Frizzled-1D4 proteins did not colocalize with Celsr1-GFP complexes (n>20 doublets). Interestingly, close inspection of MDCK cells co-expressing Fz10-1D4 and Celsr1-GFP suggests that Fz10-1D4 is actively excluded from the cell interface where Celsr1-GFP accumulates. Fz3-1D4 and Fz6-1D4 occasionally exhibited enrichment at the cell interface in the absence of Celsr1-GFP co-expression, perhaps mediated by expression of endogenous Celsr proteins in MDCK cells.
Fig. 6.

Test of colocalization of each of the ten Frizzleds with Celsr1 in MDCK cells. Each of the ten mouse Frizzled-1D4 cDNAs was transiently transfected either alone or with mouse Celsr1-GFP cDNA into MDCK cells. Two days later, the subcellular distributions of the two proteins were visualized in adjacent pairs of transfected cells. For each Frizzled-1D4, two pairs of cells are shown from the transfection lacking Celsr1-GFP (first two panels in each row) followed by five pairs of cells from the transfection with Celsr1-GFP (green, top rows). Celsr1-GFP accumulates at the junction between adjacent cells, but the subcellular localization of Frizzled (red, middle rows) varies depending on the identity of the Frizzled protein. White arrowheads indicate Frizzled-1D4 co-enrichment with Celsr1-GFP at the junction between adjacent transfected cells. Bottom panel shows the merged images, with DAPI staining in blue. Scale bar: 20 μm.
These data are consistent with the idea that Fz3 and Fz6 are largely dedicated to PCP signaling, and that Fz4 and Fz5 may be capable of PCP signaling. Although these data imply that other Frizzleds might be dedicated predominantly or exclusively to non-PCP signaling, there are two uncertainties regarding this assay that limit its interpretation. First, Celsr2 and Celsr3 have not been tested for their ability to colocalize with Frizzleds. In the case of Celsr2, we do not see an accumulation of transfected Celsr2 (with a C-terminal HA tag) at cell-cell junctions, precluding a test of Celsr2-Frizzled colocalization. Second, to associate with Celsr, some Frizzleds might require additional proteins that are not present in MDCK cells or, if present, are too divergent for the canine versions to functionally interact with the expressed mouse proteins. Finally, the colocalization assay does not measure the activity of Frizzled receptors in the non-canonical regulation of Jun kinase and Rho.
DISCUSSION
The results presented here define a crucial role for Fz2 and Fz7 in convergent extension and cardiovascular development. They also reveal an extended web of genetic interactions between this pair of Frizzleds and a diverse set of signaling proteins involved in canonical and non-canonical signaling. In the broader context of the signaling capacities of Frizzled family members, this study also presents the first comprehensive survey of canonical signaling across the entire Wnt and Frizzled families and the first survey of PCP-associated protein localization activity across the entire Frizzled family.
Fz1, Fz2 and Fz7 are closely related receptors that define one branch of the Frizzled family and exhibit partial redundancy. In cell culture assays, these Frizzleds can activate canonical signaling when cotransfected with a variety of Wnts, but unlike Fz3 and Fz6 they show little or no colocalization with Celsr1, suggesting that they might have a limited capacity for PCP signaling. How can one reconcile the apparently conflicting evidence that Fz2 and Fz7 mediate canonical Wnt signaling based on cell culture assays with the in vivo evidence for their role as mediators of non-canonical signaling based on the convergent extension phenotype and an apparent absence of transcript changes in Fz2−/−;Fz7−/− E8.5 embryos? A number of explanations could potentially reconcile this apparent contradiction, as listed below.
First, the Celsr1 colocalization assay should be interpreted with caution as the ability of this assay to predict PCP signaling competence is not established. This caution applies most strongly in the case of a negative outcome, i.e. a failure to observe colocalization. By contrast, the activity of Fz2 and Fz7 in the STF assay can be taken as strong evidence for competence in canonical signaling. Thus, the data can be reconciled if, like Drosophila Fz, mammalian Fz2 and Fz7 can mediate both canonical and PCP signaling in vivo, with the latter required for convergent extension. Second, convergent extension might require canonical Wnt signaling, an idea supported by the observation of spina bifida in Lrp6−/− embryos (Pinson et al., 2000; Zhou et al., 2010). In this case, the data can be reconciled if Fz2 and Fz7 mediate canonical signaling in a minor population of cells, such that transcriptome changes are too small to detect against a large background of whole-embryo RNA. Alternately, the time window during which the relevant canonical signaling occurs might substantially predate the appearance of the convergent extension phenotype, in which case the microarray analysis at E8.5 might not reveal the relevant transcriptome changes. Third, Frizzled-mediated signaling pathways other than the canonical Wnt and PCP pathways could be relevant. We note that Fz7 signaling via the Wnt/calcium pathway has been implicated in tissue movements during Xenopus gastrulation (Winklbauer et al., 2001); an analogous role in mice could explain the convergent extension defect. Moreover, a new Frizzled signaling pathway – the Frizzled nuclear import (FNI) pathway – which involves proteolytic cleavage and nuclear translocation of the C-terminus of Frizzled receptors, has recently been described in Drosophila (Mathew et al., 2005; Speese et al., 2012). Whether this pathway operates in mammals is at present unknown. Finally, it is possible that, although the immediate cause of the Fz2−/−;Fz7−/− convergent extension phenotype is a decrease in PCP signaling, this could occur indirectly if loss of Fz2 and Fz7 perturbs canonical or other signaling pathways that are linked to PCP by cross-regulatory interactions, such as those mediated by inversin (Lienkamp et al., 2012).
Genetic network analysis of Fz2 and Fz7 interactions
Genetic interactions are typically identified when alterations in one gene enhance or suppress the phenotype(s) caused by mutations in a second gene. When applied in an unbiased manner, the network of interactions thus defined is useful for identifying proteins that interact directly, act in the same pathway, or act in different pathways to affect the same biological process. Genetic interaction screens have been conducted on a genome-wide scale in prokaryotes, S. cerevisiae, C. elegans and Drosophila, either with the aid of mutagens or, more recently, with RNAi libraries (Dixon et al., 2009; Gunsalus and Rhrissorrakrai, 2011). In mice, genome-wide genetic interaction screens are prohibitively expensive, but targeted analyses of genetic interactions are useful for testing specific hypotheses.
In the present study, we observed genetic interactions between Fz2 and Fz7 and five canonical and/or non-canonical signaling molecules: mutations in Dvl3, Wnt3a or Wnt11 enhanced the cardiac phenotype; mutation of Vangl2 enhanced the palate closure phenotype; and mutation of Wnt5a enhanced the tail shortening phenotype and produced a synthetic mid-gestational lethality. If interpreted as evidence for a direct interaction between the proteins encoded by the interacting loci, this result would imply that Fz2 and Fz7 interact with three different Wnts and also participate in PCP signaling. We favor a more nuanced interpretation: developmental processes such as directed cell migration during palate and cardiac development (in the latter case, both long-range migration of neural crest cells and local morphogenetic movements) depend on a delicate balance of signal strength in multiple pathways, and many of these pathways are sensitive to changes of 2-fold or less in the levels of their constituent proteins. Importantly, some phenotypes with severe functional consequences, such as VSDs and palate defects, arise from a nearly correct execution of the appropriate tissue movements (Fig. 3). Apparently, small perturbations in any of a variety of signaling pathways can move the embryo from a position just beyond a critical threshold on the phenotypic landscape to a position just short of that threshold.
Redundancy among Frizzleds and other Wnt/PCP signaling components
The present work provides additional evidence that redundancy among homologous genes is widespread in vertebrate Wnt-Frizzled signaling, a phenomenon that might relate to the evolutionary expansion in the size of the gene families that encode proteins involved in these signaling pathways. For the Wnt, Frizzled, Dsh/Dvl, Vang/Vangl, Fmi/Stan/Celsr, and Arrow/Lrp families, the number of Drosophila and mammalian members are, respectively: seven and 19, four and ten, one and three, one and two, one and three, and one and two (supplementary material Table S1). Among mouse Frizzled genes, the functional redundancies reported thus far occur principally between closely related pairs, the one exception being Fz4 and Fz8 (Ye et al., 2011) (supplementary material Fig. S2). Additionally, the degree of genetic redundancy varies among pairs of Frizzled genes. The partial redundancy of Fz3 and Fz6 represents one end of this spectrum: defects in axon guidance are seen in Fz3−/− but not Fz6−/− mice, and defects in hair follicle orientation are seen in Fz6−/− but not Fz3−/− mice, with additional defects in neural tube closure, inner ear sensory hair cell orientation, and eyelid closure appearing in Fz3−/−;Fz6−/− embryos (Wang et al., 2002; Wang et al., 2006b; Lyuksyutova et al., 2003; Guo et al., 2004). At the other end of the spectrum, Fz1−/− and Fz8−/− mice show no apparent phenotype, and the effects of gene loss are only revealed in double mutant combinations: Fz1−/−;Fz2−/− embryos have fully penetrant palate closure defects compared with 50% penetrance for Fz2−/− embryos (Yu et al., 2010), Fz4−/−;Fz8−/− embryos have severe renal hypoplasia compared with mild hypoplasia in Fz4−/− mice (Ye et al., 2011), and Fz5−/−;Fz8−/−embryos (with Fz5 function maintained in extra-embryonic tissue) die at mid-gestation compared with the adult viability of Fz5−/− mice (Liu et al., 2012). As described here, Fz7−/− represents an intermediate case, as Fz7−/− mice exhibit a mild tail phenotype and a low-penetrance VSD phenotype, whereas Fz2−/−;Fz7−/− mice show a severe defect in convergent extension. A pattern of partial redundancy similarly characterizes the Dvl1/Dvl2/Dvl3 (Wang et al., 2006c; Etheridge et al., 2008), Vangl1/Vangl2 (Torban et al., 2008; Song, H. et al., 2010) and Lrp5/Lrp6 (Kelly et al., 2004; Joeng et al., 2011) gene families.
It is also striking that many combinations of mutations involving homologous genes implicated in Frizzled signaling show dosage effects, such that in comparing the progressive loss of two, three or four alleles, the phenotypes become more penetrant, more severe and/or novel. Three examples illustrate this pattern: (1) the increase in severity of renal hypoplasia in Fz4−/− versus Fz4−/−;Fz8+/− versus Fz4−/−;Fz8−/− embryos (Ye et al., 2011); (2) the earlier embryonic dysmorphology phenotypes in Lrp6−/− versus Lrp6−/−;Lrp5+/− versus Lrp6−/−;Lrp5−/− embryos (Kelly et al., 2004); and (3) the increase in severity of cardiac, inner ear and neural tube defects associated with loss of increasing numbers of Dvl family members (Wang et al., 2006c; Etheridge et al., 2008). In the present work, the comparison is between a ~15% penetrant cardiac phenotype in Fz7−/− embryos versus a ~50% penetrant cardiac phenotype with a dramatic decrease in viability in Fz2+/−;Fz7−/− embryos versus a 100% penetrant convergent extension defect with mid-gestational lethality in Fz2−/−;Fz7−/− embryos.
The widespread redundancy among canonical and non-canonical signaling components implies that these signaling systems have achieved a robustness to single-allele loss and, in many cases, to two-allele loss by expanding and diversifying the underlying gene repertoire. However, the gene dosage experiments imply that, even in cases with redundancy, the abundances of many signaling proteins are only 2- or 3-fold higher than the minimum threshold for normal function. These observations suggest the possibility of a selective disadvantage associated with a signal strength that is greater than several fold above the minimum threshold. Thus, in many developmental contexts, there might be a relatively narrow range of optimal signal strength, with either reduced or excessive signaling leading to loss of fitness. If this general idea is correct, it predicts that during the evolutionary process of gene duplication there might be a bottleneck in Darwinian fitness caused by the 2-fold excess of signal strength that would accompany the duplication of any gene that codes for a rate-limiting component. The present pattern of partial redundancy among closely related signaling genes might reflect the combined effects of selective pressure for gene duplication/diversification and for levels of gene expression that maintain signal strength within the optimal window.
Implications for common congenital anomalies in humans
In humans, defects in closing the ventricular septum, palate and neural tube are found in ~0.4%, ~0.1% and ~0.1% of live births, respectively, making these among the most common congenital anatomical anomalies (Centers for Disease Control, 1997; Dolk et al., 2010). Each of these anomalies appears to have complex genetic and environmental influences (Dixon et al., 2011; Au et al., 2010; Wessels and Willems, 2010; Grosen et al., 2011). For rare patients with neural tube defects, a connection to PCP signaling comes from the finding of VANGL1, CELSR1, SCRIB and FUZ (the latter two are homologs of the Drosophila PCP genes scribbled and fuzzy, respectively) sequence variants (Kibar et al., 2007; Kibar et al., 2009; Kibar et al., 2011; Seo et al., 2011; Robinson et al., 2012). For human palate closure defects, there are statistically significant associations with single-nucleotide polymorphisms in the WNT3, WNT3A, WNT5A, WNT8A and WNT11 genes (Chiquet et al., 2008; Menezes et al., 2010; Yao et al., 2011; Mostowska et al., 2012). To our knowledge, no genetic studies have linked canonical or non-canonical signaling to human cardiac defects.
In mice, mutations in the following canonical and non-canonical signaling genes either alone or in combination lead to defects in ventricular septum (V), palate (P) and/or neural tube (N) closure: Fz1 [V, P, N (Yu et al., 2010)]; Fz2 [V, P, N (Yu et al., 2010) (present work)]; Fz7 [V, P, N (present work); we include here the Fz2−/−;Fz7−/− convergent extension phenotype in the neural tube category]; Lrp6 [V, P, N (Pinson et al., 2000; Song et al., 2009; Song, L. et al., 2010; Zhou et al., 2010)]; Dvl1, Dvl2 and Dvl3 [V, N (Wang et al., 2006c; Etheridge et al., 2008)]; Vangl1 [N (Torban et al., 2008)]; Vangl2 [V, P, N (Henderson et al., 2001; Kibar et al., 2001; Murdoch et al., 2001) (present work)]; Celsr1 [N (Curtin et al., 2003)]; and Wnt5a [N, P (He at al., 2008; Andersson et al., 2010)]. These mouse data, together with the human genetic studies described above, suggest that additional genetic variations in canonical and non-canonical signaling genes are likely to be identified in association with each of these common human anomalies.
Supplementary Material
Acknowledgements
We thank Amir Rattner, Connie Talbot, Yanshu Wang, John Williams and two anonymous referees for assistance and/or advice.
Footnotes
Funding
This work was supported by the Howard Hughes Medical institute. Deposited in PMC for release after 6 months.
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.083352/-/DC1
References
- Andersson E. R., Bryjova L., Biris K., Yamaguchi T. P., Arenas E., Bryja V. (2010). Genetic interaction between Lrp6 and Wnt5a during mouse development. Dev. Dyn. 239, 237-245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au K. S., Ashley-Koch A., Northrup H. (2010). Epidemiologic and genetic aspects of spina bifida and other neural tube defects. Dev. Disabil. Res. Rev. 16, 6-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control (1997). Surveillance summaries and temporal trends in the incidence of birth defects – United States. Morb. Mortal. Wkly. Rep. 46, 1171-1176 [PubMed] [Google Scholar]
- Chiquet B. T., Blanton S. H., Burt A., Ma D., Stal S., Mulliken J. B., Hecht J. T. (2008). Variation in WNT genes is associated with non-syndromic cleft lip with or without cleft palate. Hum. Mol. Genet. 17, 2212-2218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtin J. A., Quint E., Tsipouri V., Arkell R. M., Cattanach B., Copp A. J., Henderson D. J., Spurr N., Stanier P., Fisher E. M., et al. (2003). Mutation of Celsr1 disrupts planar polarity of inner ear hair cells and causes severe neural tube defects in the mouse. Curr. Biol. 13, 1129-1133 [DOI] [PubMed] [Google Scholar]
- Devenport D., Fuchs E. (2008). Planar polarization in embryonic epidermis orchestrates global asymmetric morphogenesis of hair follicles. Nat. Cell Biol. 10, 1257-1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon S. J., Costanzo M., Baryshnikova A., Andrews B., Boone C. (2009). Systematic mapping of genetic interaction networks. Annu. Rev. Genet. 43, 601-625 [DOI] [PubMed] [Google Scholar]
- Dixon M. J., Marazita M. L., Beaty T. H., Murray J. C. (2011). Cleft lip and palate: understanding genetic and environmental influences. Nat. Rev. Genet. 12, 167-178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolk H., Loane M., Garne E. (2010). The prevalence of congenital anomalies in Europe. Adv. Exp. Med. Biol. 686, 349-364 [DOI] [PubMed] [Google Scholar]
- Etheridge S. L., Ray S., Li S., Hamblet N. S., Lijam N., Tsang M., Greer J., Kardos N., Wang J., Sussman D. J., et al. (2008). Murine dishevelled 3 functions in redundant pathways with dishevelled 1 and 2 in normal cardiac outflow tract, cochlea, and neural tube development. PLoS Genet. 4, e1000259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galceran J., Hsu S. C., Grosschedl R. (2001). Rescue of a Wnt mutation by an activated form of LEF-1: regulation of maintenance but not initiation of Brachyury expression. Proc. Natl. Acad. Sci. USA 98, 8668-8673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich L. V., Strutt D. (2011). Principles of planar polarity in animal development. Development 138, 1877-1892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray R. S., Roszko I., Solnica-Krezel L. (2011). Planar cell polarity: coordinating morphogenetic cell behaviors with embryonic polarity. Dev. Cell 21, 120-133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosen D., Bille C., Petersen I., Skytthe A., Hjelmborg J., Pedersen J. K., Murray J. C., Christensen K. (2011). Risk of oral clefts in twins. Epidemiology 22, 313-319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunsalus K. C., Rhrissorrakrai K. (2011). Networks in Caenorhabditis elegans. Curr. Opin. Genet. Dev. 21, 787-798 [DOI] [PubMed] [Google Scholar]
- Guo N., Hawkins C., Nathans J. (2004). Frizzled6 controls hair patterning in mice. Proc. Natl. Acad. Sci. USA 101, 9277-9281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F., Xiong W., Yu X., Espinoza-Lewis R., Liu C., Gu S., Nishita M., Suzuki K., Yamada G., Minami Y., et al. (2008). Wnt5a regulates directional cell migration and cell proliferation via Ror2-mediated noncanonical pathway in mammalian palate development. Development 135, 3871-3879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heisenberg C. P., Tada M., Rauch G. J., Saúde L., Concha M. L., Geisler R., Stemple D. L., Smith J. C., Wilson S. W. (2000). Silberblick/Wnt11 mediates convergent extension movements during zebrafish gastrulation. Nature 405, 76-81 [DOI] [PubMed] [Google Scholar]
- Henderson D. J., Conway S. J., Greene N. D., Gerrelli D., Murdoch J. N., Anderson R. H., Copp A. J. (2001). Cardiovascular defects associated with abnormalities in midline development in the Loop-tail mouse mutant. Circ. Res. 89, 6-12 [DOI] [PubMed] [Google Scholar]
- Ho H. Y., Susman M. W., Bikoff J. B., Ryu Y. K., Jonas A. M., Hu L., Kuruvilla R., Greenberg M. E. (2012). Wnt5a-Ror-Dishevelled signaling constitutes a core developmental pathway that controls tissue morphogenesis. Proc. Natl. Acad. Sci. USA 109, 4044-4051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joeng K. S., Schumacher C. A., Zylstra-Diegel C. R., Long F., Williams B. O. (2011). Lrp5 and Lrp6 redundantly control skeletal development in the mouse embryo. Dev. Biol. 359, 222-229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly O. G., Pinson K. I., Skarnes W. C. (2004). The Wnt co-receptors Lrp5 and Lrp6 are essential for gastrulation in mice. Development 131, 2803-2815 [DOI] [PubMed] [Google Scholar]
- Kibar Z., Vogan K. J., Groulx N., Justice M. J., Underhill D. A., Gros P. (2001). Ltap, a mammalian homolog of Drosophila Strabismus/Van Gogh, is altered in the mouse neural tube mutant Loop-tail. Nat. Genet. 28, 251-255 [DOI] [PubMed] [Google Scholar]
- Kibar Z., Torban E., McDearmid J. R., Reynolds A., Berghout J., Mathieu M., Kirillova I., De Marco P., Merello E., Hayes J. M., et al. (2007). Mutations in VANGL1 associated with neural-tube defects. N. Engl. J. Med. 356, 1432-1437 [DOI] [PubMed] [Google Scholar]
- Kibar Z., Bosoi C. M., Kooistra M., Salem S., Finnell R. H., De Marco P., Merello E., Bassuk A. G., Capra V., Gros P. (2009). Novel mutations in VANGL1 in neural tube defects. Hum. Mutat. 30, E706-E715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kibar Z., Salem S., Bosoi C. M., Pauwels E., De Marco P., Merello E., Bassuk A. G., Capra V., Gros P. (2011). Contribution of VANGL2 mutations to isolated neural tube defects. Clin. Genet. 80, 76-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim G. H., Her J. H., Han J. K. (2008). Ryk cooperates with Frizzled 7 to promote Wnt11-mediated endocytosis and is essential for Xenopus laevis convergent extension movements. J. Cell Biol. 182, 1073-1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lienkamp S., Ganner A., Walz G. (2012). Inversin, Wnt signaling and primary cilia. Differentiation 83, S49-S55 [DOI] [PubMed] [Google Scholar]
- Liu C., Bakeri H., Li T., Swaroop A. (2012). Regulation of retinal progenitor expansion by Frizzled receptors: implications for microphthalmia and retinal coloboma. Hum. Mol. Genet. 21, 1848-1860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyuksyutova A. I., Lu C. C., Milanesio N., King L. A., Guo N., Wang Y., Nathans J., Tessier-Lavigne M., Zou Y. (2003). Anterior-posterior guidance of commissural axons by Wnt-frizzled signaling. Science 302, 1984-1988 [DOI] [PubMed] [Google Scholar]
- Mathew D., Ataman B., Chen J., Zhang Y., Cumberledge S., Budnik V. (2005). Wingless signaling at synapses is through cleavage and nuclear import of receptor DFrizzled2. Science 310, 1344-1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menezes R., Letra A., Kim A. H., Küchler E. C., Day A., Tannure P. N., Gomes da Motta L., Paiva K. B., Granjeiro J. M., Vieira A. R. (2010). Studies with Wnt genes and nonsyndromic cleft lip and palate. Birth Defects Res. A Clin. Mol. Teratol. 88, 995-1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikels A. J., Nusse R. (2006). Purified Wnt5a protein activates or inhibits beta-catenin-TCF signaling depending on receptor context. PLoS Biol. 4, e115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molday R. S., MacKenzie D. (1983). Monoclonal antibodies to rhodopsin: characterization, cross-reactivity, and application as structural probes. Biochemistry 22, 653-660 [DOI] [PubMed] [Google Scholar]
- Mostowska A., Hozyasz K. K., Biedziak B., Wojcicki P., Lianeri M., Jagodzinski P. P. (2012). Genotype and haplotype analysis of WNT genes in non-syndromic cleft lip with or without cleft palate. Eur. J. Oral Sci. 120, 1-8 [DOI] [PubMed] [Google Scholar]
- Murdoch J. N., Doudney K., Paternotte C., Copp A. J., Stanier P. (2001). Severe neural tube defects in the loop-tail mouse result from mutation of Lpp1, a novel gene involved in floor plate specification. Hum. Mol. Genet. 10, 2593-2601 [DOI] [PubMed] [Google Scholar]
- Nakaya M. A., Biris K., Tsukiyama T., Jaime S., Rawls J. A., Yamaguchi T. P. (2005). Wnt3a links left-right determination with segmentation and anteroposterior axis elongation. Development 132, 5425-5436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oishi I., Suzuki H., Onishi N., Takada R., Kani S., Ohkawara B., Koshida I., Suzuki K., Yamada G., Schwabe G. C., et al. (2003). The receptor tyrosine kinase Ror2 is involved in non-canonical Wnt5a/JNK signalling pathway. Genes Cells 8, 645-654 [DOI] [PubMed] [Google Scholar]
- Pandur P., Läsche M., Eisenberg L. M., Kühl M. (2002). Wnt-11 activation of a non-canonical Wnt signalling pathway is required for cardiogenesis. Nature 418, 636-641 [DOI] [PubMed] [Google Scholar]
- Pinson K. I., Brennan J., Monkley S., Avery B. J., Skarnes W. C. (2000). An LDL-receptor-related protein mediates Wnt signalling in mice. Nature 407, 535-538 [DOI] [PubMed] [Google Scholar]
- Qian D., Jones C., Rzadzinska A., Mark S., Zhang X., Steel K. P., Dai X., Chen P. (2007). Wnt5a functions in planar cell polarity regulation in mice. Dev. Biol. 306, 121-133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson A., Escuin S., Doudney K., Vekemans M., Stevenson R. E., Greene N. D., Copp A. J., Stanier P. (2012). Mutations in the planar cell polarity genes CELSR1 and SCRIB are associated with the severe neural tube defect craniorachischisis. Hum. Mutat. 33, 440-447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato A., Yamamoto H., Sakane H., Koyama H., Kikuchi A. (2010). Wnt5a regulates distinct signalling pathways by binding to Frizzled2. EMBO J. 29, 41-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleiffarth J. R., Person A. D., Martinsen B. J., Sukovich D. J., Neumann A., Baker C. V., Lohr J. L., Cornfield D. N., Ekker S. C., Petryk A. (2007). Wnt5a is required for cardiac outflow tract septation in mice. Pediatr. Res. 61, 386-391 [DOI] [PubMed] [Google Scholar]
- Seo J. H., Zilber Y., Babayeva S., Liu J., Kyriakopoulos P., De Marco P., Merello E., Capra V., Gros P., Torban E. (2011). Mutations in the planar cell polarity gene, Fuzzy, are associated with neural tube defects in humans. Hum. Mol. Genet. 20, 4324-4333 [DOI] [PubMed] [Google Scholar]
- Song L., Li Y., Wang K., Wang Y. Z., Molotkov A., Gao L., Zhao T., Yamagami T., Wang Y., Gan Q., et al. (2009). Lrp6-mediated canonical Wnt signaling is required for lip formation and fusion. Development 136, 3161-3171 [DOI] [PubMed] [Google Scholar]
- Song H., Hu J., Chen W., Elliott G., Andre P., Gao B., Yang Y. (2010). Planar cell polarity breaks bilateral symmetry by controlling ciliary positioning. Nature 466, 378-382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L., Li Y., Wang K., Zhou C. J. (2010). Cardiac neural crest and outflow tract defects in Lrp6 mutant mice. Dev. Dyn. 239, 200-210 [DOI] [PubMed] [Google Scholar]
- Speese S. D., Ashley J., Jokhi V., Nunnari J., Barria R., Li Y., Ataman B., Koon A., Chang Y. T., Li Q., et al. (2012). Nuclear envelope budding enables large ribonucleoprotein particle export during synaptic Wnt signaling. Cell 149, 832-846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strong L. C., Hollander W. F. (1949). Hereditary loop-tail in the house mouse accompanied by imperforate vagina and with lethal craniorachischisis when homozygous. J. Hered. 40, 329-334 [Google Scholar]
- Sumanas S., Ekker S. C. (2001). Xenopus frizzled-7 morphant displays defects in dorsoventral patterning and convergent extension movements during gastrulation. Genesis 30, 119-122 [DOI] [PubMed] [Google Scholar]
- Sumanas S., Kim H. J., Hermanson S., Ekker S. C. (2001). Zebrafish frizzled-2 morphant displays defects in body axis elongation. Genesis 30, 114-118 [DOI] [PubMed] [Google Scholar]
- Takada S., Stark K. L., Shea M. J., Vassileva G., McMahon J. A., McMahon A. P. (1994). Wnt-3a regulates somite and tailbud formation in the mouse embryo. Genes Dev. 8, 174-189 [DOI] [PubMed] [Google Scholar]
- Torban E., Patenaude A. M., Leclerc S., Rakowiecki S., Gauthier S., Andelfinger G., Epstein D. J., Gros P. (2008). Genetic interaction between members of the Vangl family causes neural tube defects in mice. Proc. Natl. Acad. Sci. USA 105, 3449-3454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Amerongen R., Nusse R. (2009). Towards an integrated view of Wnt signaling in development. Development 136, 3205-3214 [DOI] [PubMed] [Google Scholar]
- Wang Y., Thekdi N., Smallwood P. M., Macke J. P., Nathans J. (2002). Frizzled-3 is required for the development of major fiber tracts in the rostral CNS. J. Neurosci. 22, 8563-8573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Badea T., Nathans J. (2006a). Order from disorder: Self-organization in mammalian hair patterning. Proc. Natl. Acad. Sci. USA 103, 19800-19805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Guo N., Nathans J. (2006b). The role of Frizzled3 and Frizzled6 in neural tube closure and in the planar polarity of inner-ear sensory hair cells. J. Neurosci. 26, 2147-2156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Hamblet N. S., Mark S., Dickinson M. E., Brinkman B. C., Segil N., Fraser S. E., Chen P., Wallingford J. B., Wynshaw-Boris A. (2006c). Dishevelled genes mediate a conserved mammalian PCP pathway to regulate convergent extension during neurulation. Development 133, 1767-1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessels M. W., Willems P. J. (2010). Genetic factors in non-syndromic congenital heart malformations. Clin. Genet. 78, 103-123 [DOI] [PubMed] [Google Scholar]
- Winklbauer R., Medina A., Swain R. K., Steinbeisser H. (2001). Frizzled-7 signalling controls tissue separation during Xenopus gastrulation. Nature 413, 856-860 [DOI] [PubMed] [Google Scholar]
- Xu Q., Wang Y., Dabdoub A., Smallwood P. M., Williams J., Woods C., Kelley M. W., Jiang L., Tasman W., Zhang K., et al. (2004). Vascular development in the retina and inner ear: control by Norrin and Frizzled-4, a high-affinity ligand-receptor pair. Cell 116, 883-895 [DOI] [PubMed] [Google Scholar]
- Yamaguchi T. P., Bradley A., McMahon A. P., Jones S. (1999). A Wnt5a pathway underlies outgrowth of multiple structures in the vertebrate embryo. Development 126, 1211-1223 [DOI] [PubMed] [Google Scholar]
- Yao T., Yang L., Li P. Q., Wu H., Xie H. B., Shen X., Xie X. D. (2011). Association of Wnt3A gene variants with non-syndromic cleft lip with or without cleft palate in Chinese population. Arch. Oral Biol. 56, 73-78 [DOI] [PubMed] [Google Scholar]
- Ye X., Wang Y., Cahill H., Yu M., Badea T. C., Smallwood P. M., Peachey N. S., Nathans J. (2009). Norrin, frizzled-4, and Lrp5 signaling in endothelial cells controls a genetic program for retinal vascularization. Cell 139, 285-298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X., Wang Y., Rattner A., Nathans J. (2011). Genetic mosaic analysis reveals a major role for frizzled 4 and frizzled 8 in controlling ureteric growth in the developing kidney. Development 138, 1161-1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H., Copley C. O., Goodrich L. V., Deans M. R. (2012). Comparison of phenotypes between different vangl2 mutants demonstrates dominant effects of the Looptail mutation during hair cell development. PLoS ONE 7, e31988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H., Smallwood P. M., Wang Y., Vidaltamayo R., Reed R., Nathans J. (2010). Frizzled 1 and frizzled 2 genes function in palate, ventricular septum and neural tube closure: general implications for tissue fusion processes. Development 137, 3707-3717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C. J., Wang Y. Z., Yamagami T., Zhao T., Song L., Wang K. (2010). Generation of Lrp6 conditional gene-targeting mouse line for modeling and dissecting multiple birth defects/congenital anomalies. Dev. Dyn. 239, 318-326 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.