

Abstract
Parasitic diseases continue to have a devastating impact on human populations worldwide. Lack of effective treatments, the high cost of existing ones, and frequent emergence of resistance to these agents provide a strong argument for the development of novel therapies. Here we report the results of a hybrid approach designed to obtain a dual acting molecule that would demonstrate activity against a variety of parasitic targets. The antimalarial drug amodiaquine has been covalently joined with a nitric oxide-releasing furoxan to achieve multiple mechanisms of action. Using in vitro and ex vivo assays, the hybrid molecule shows activity against three parasites – Plasmodium falciparum, Schistosoma mansoni, and Ancylostoma ceylanicum.
Introduction
Despite increasing focus and research, parasitic diseases caused by protozoa or helminths remain a tremendous worldwide threat to the health of human populations. For example, the WHO estimates that one in every four people is infected with parasitic worms,1 and over 400 million people are infected with either malaria,2 schistosomiasis,3 or hookworm.4 A variety of approaches have been pursued in order to prevent or treat these diseases, including vector control, vaccination, and therapeutic treatment. Therapeutic treatment often involves the use of small molecules – current agents include praziquantel (1, Fig. 1) for schistosomiasis5 and albendazole (ABZ, 2, Fig. 1) for hookworm.6 Malaria has seen numerous treatments involving orally administered small molecules. Most recently, the use of artemisinin (3, Fig. 1) combination therapy (ACT) has become the standard of care for treatment of malarial infections caused by the parasite Plasmodium falciparum, and considerable advancement has been made in the way of developing highly potent artemisinin derivatives.7,8 However, a number of other molecules have been used; among these are chloroquine (4, CQ, Fig. 1) and amodiaquine (5, AQ, Fig. 1). Widespread resistance to CQ has developed9,10 and although AQ is sometimes still used as a viable treatment, the potential for toxic side effects exists due to the 4-aminophenol structural feature.11
Fig. 1.
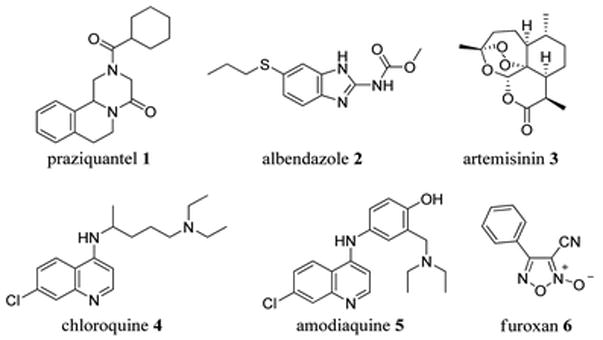
Small molecule anti-parasitic therapies.
These molecules share the same quinoline core, which interestingly is present in many of the most potent antimalarials reported to date.9 Furthermore, the core has shown promise in chemotherapy of a variety of other diseases, most notably schistosomiasis.12 Many modifications to the quinoline core have been pursued over the years, including hybridization with other active entities, in order to maintain or rescue the innate activity.13,14
The concept of drug hybridization is not new – the idea of combining two therapeutically relevant moieties has been frequently explored.15 The desired outcome is to either combine “complementary structural features” to enhance affinity for a single target15 or combine portions of molecules that have either distinct mechanisms of action or distinct targets.16 Following the latter rationale, hybrid partners receiving recent attention are those that release nitric oxide (NO).16 Nitric oxide operates in a variety of physiological functions,17 and activity against parasitic diseases has recently emerged as another role for this gaseous molecule.18–20 Specifically, NO signaling appears to be involved in regulation of the immune response against P. falciparum.21,22
Many functional groups that release NO (NO donors) are known, but the 3-cyano-4-phenyl-1,2,5-oxadiazole 2-oxide (6, Fig. 1, referred to as “phenyl furoxan” or just “furoxan” from this point forward) has shown activity against malaria, schistosomiasis, and hookworm.19,20,23,24 The versatility of the quinoline core (specifically, amodiaquine) and the recent attention given to the furoxan moiety spurred us to employ a hybrid approach (Fig. 2), with the goal of achieving a small molecule active against a variety of parasitic diseases.
Fig. 2.
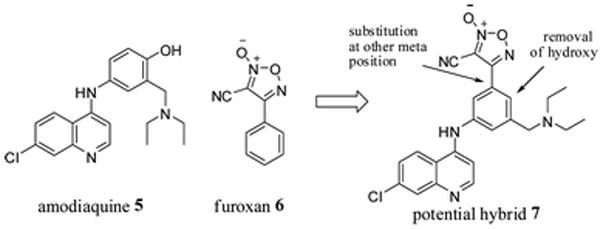
Proposed structure of a furoxan–amodiaquine hybrid.
Results and discussion
Synthesis
Extensive structure–activity relationships (SAR) between aminoquinolines (such as amodiaquine) and antimalarial activity have been established.13,14 From these studies it was clear that the furoxan should be attached to the pendant aromatic ring of these agents. Substitution at the other meta position of amodiaquine (relative to the nitrogen, Fig. 2) allows removal of the hydroxyl group, a desirable change for both ease of synthesis and to avoid potential metabolic liabilities.11 Furthermore, direct attachment to the aromatic ring would mimic the furoxans that have shown the most potent activity against S. mansoni.19,20
The synthesis began with commercially available 2,7-dichloroquinoline (8), which was reacted with (3-amino-5-bromo-phenyl)methanol (9) in the presence of hydrochloric acid (HCl) to form the 2-substituted aminoquinoline via an addition–elimination reaction. The product was heated under reflux with HCl in a sealed tube to convert the benzyl alcohol to a benzyl chloride,23 and subsequent reaction with tert-butyl amine in the presence of pyridine (py) installed the necessary basic nitrogen group, affording compound 10. tert-Butyl amine was chosen over diethylamine based on previous SAR.13,14 The Heck reaction was then performed, using ethyl acrylate, palladium(II) acetate, 1,4-bis(diphenylphosphino)propane (dppp), and potassium carbonate in DMF. The reaction mixture was heated in the microwave for several hours at 130 °C, affording the cinnamyl ester 11. Addition of two equivalents of DIBALH reduced the ester to the allyl alcohol, and subsequent cyclization by treatment with sodium nitrite in acetic acid gave desired furoxan primary alcohol 12.25 Not surprisingly, the secondary amine was also nitrosylated, which turned out to be a sufficient protecting group for the remainder of the synthesis as it was easily removed in the final step (vide infra). Ideally the primary alcohol would be oxidized to the aldehyde and then condensed with hydroxylamine to provide the oxime, however we were unable to find oxidation conditions that are compatible with the numerous nitrogen atoms that are present in the molecule. Alternatively, the alcohol was reacted with N-(tert-butyldimethyl-silyloxy) benzenesulfonamide (TBSONHTs) under Mitsunobu conditions, and subsequently treated with cesium fluoride to provide the requisite furoxan oxime 13.25,26 Rapid dehydration of the oxime 13 to provide the nitrile 14 was achieved with thionyl chloride, and treatment with HCl in dioxane removed the N-nitroso group to provide the desired furoxan–amodiaquine hybrid 15 (Scheme 1).
Scheme 1.
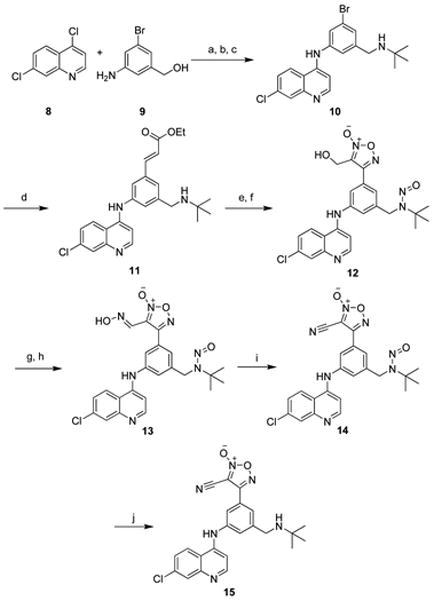
Reagents and conditions:
(a) HCl, MeOH, 89% (b) conc. HCl, reflux in sealed tube, 6 h, 77% (c) tBuNH2, py, DMF, 80 °C, 80% (d) ethyl acrylate, K2CO3, Pd(OAc)2, dppp, DMF, MW, 130 °C, 7 h, 81% (e) DIBALH, CH2Cl2, −78 °C, 74% (f) NaNO2, AcOH, 57% (crude) (g) TBSONHTs, DEAD, PPh3, THF, 62% (h) CsF, MeCN, 48% (i) SOCl2, 0 °C, DMF, 99% and (j) 4.0 M HCl in dioxane, 99%.
Biological evaluation
Antimalarial activity
The target furoxan–AQ hybrid 15, along with several synthetic intermediates, were screened in a SYBR green I dye DNA staining assay.27,28 Several previously synthesized furoxans that had shown activity against S. mansoni19 were also included. Human erythrocytes infected with P. falciparum were monitored for parasite proliferation, and only the chloroquine-sensitive 3d7 clonal line was initially screened to determine any promising results.28 The assay was also run in a delayed death format29 where parasites are incubated for 96 hours in order to detect activity against parasitic progeny, luciferase being the viability reporter in this case. Use of both assays allowed for conclusions to be drawn about the long term activity of the molecules.
Analysis of the activity data shown in Table 1 leads to several conclusions. First, artemisinin (3) is quite potent in both assay formats (as expected), while amodiaquine (5) and the phenyl furoxan (6) were essentially inactive in delayed death. Over 25 furoxan analogs were screened and produced similar results (see Table S1†). Several intermediates were very active in the standard assay format but lost some activity in the delayed death format. For example, the activity of the oxime intermediate 13 decreased nearly 50-fold between the two assays (0.024 μM and 1.07 μM, respectively). The N-nitroso precursor 14 showed reduced activity in the standard assay (0.113 μM IC50) and was essentially inactive in the delayed death format (>15 μM IC50). This is likely due to the highly accepted rationale that a basic nitrogen atom at this location is essential to promote accumulation of the molecule in the parasitic digestive vacuole.13,14 The amodiaquine–furoxan hybrid target 15 showed significant potency in the standard assay and in the delayed death format (40 nM and 630 nM IC50, respectively). Overall, the hybrid 15 showed acceptable potency and provides a basis for which further SAR could be explored.
Table 1.
Antimalarial IC50 data (μM) determined from SYBR green assay against the 3d7 parasite line and luciferase-reporting delayed death assay (3d7 del)
| Compound | 3d7 (μM) | 3d7 del (μM) |
|---|---|---|
| 3 | 0.011 | 0.012 |
| 5 | 0.015 | >10 |
| 6 | 0.059 | >15 |
| 10 | 0.007 | 0.065 |
| 13 | 0.024 | 1.07 |
| 14 | 0.113 | >15 |
| 15 | 0.040 | 0.630 |
Antischistosomiasis activity
The target hybrid 15 was further screened against S. mansoni, using both a thioredoxin glutathione reductase (TGR) biochemical assay as well as an ex vivo parasite killing assay.19,30 It has been previously demonstrated that TGR is a likely target of the furoxan.19 TGR is a flavoenzyme containing a selenocysteine and is required to maintain redox balance in the parasitic cellular environment,31 and it has been postulated that the furoxan achieves activity by releasing NO which inhibits TGR via an S-nitrosylation (or Senitrosylation) mechanism.19 The inhibitory activity of hybrid 15 against TGR was evaluated as compared with furoxan 6 (Fig. 3). The hybrid 15 had comparable activity to the previously evaluated phenyl furoxan 6.
Fig. 3.
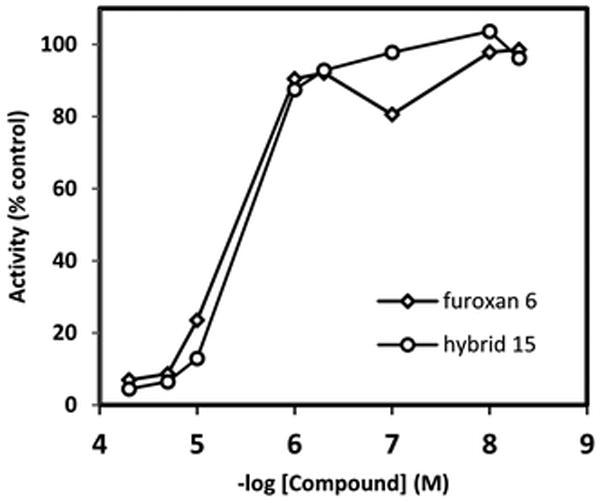
TGR activity inhibition for furoxan 6 and hybrid 15.
Additionally, both compounds were screened in an ex vivo worm killing assay. Adult S. mansoni worms were incubated in culture medium following isolation from mice hosts. Compounds 6 and 15 were dissolved in DMSO and administered to the cultured parasites at varying concentrations. Worm mobility and survival was observed under a stereomicroscope. At the lower dose of 10 μM, the hybrid 15 was ineffective, while the furoxan 6 showed minimal activity. However, the hybrid 15 achieved potent inhibition at 50 μM, as shown in Fig. 4, achieving killing (complete loss of motility) of the parasites in 24 hours. The furoxan 6 has been previously screened at 50 μM and showed similar activity (not shown), while amodiaquine has been previously shown to be inactive against S. mansoni.32 This potency likely could be improved with additional structure–activity relationship (SAR) studies.
Fig. 4.
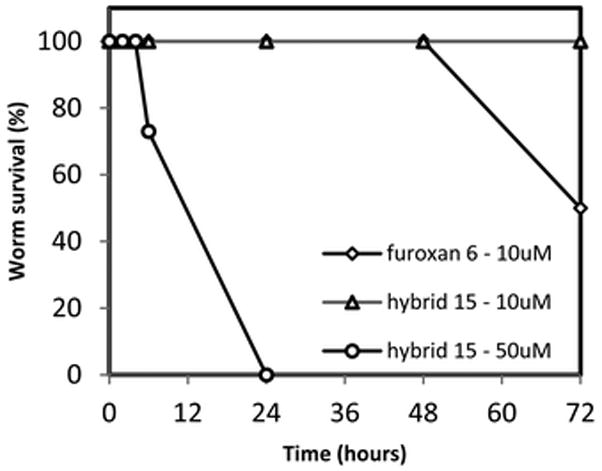
Ex vivo S. mansoni parasite killing assay.
Hookworm activity
Finally, the target hybrid 15 was evaluated for nematicidal activity against A. ceylanicum. This was done using an ex vivo adult worm survival assay.24,33 The furoxans as a class have recently shown activity against hookworm.24 Compounds of interest were dissolved in DMSO and administered to cultures of adult worms. Parasite motility and morphology were observed using a light microscope. Parasites were considered dead upon complete loss of motility. For these experiments, the drug albendazole (ABZ, 2, Fig. 1) was included as a positive control and DMSO was used as a carrier control. As shown in Fig. 5, treatment of adult hookworms with hybrid 15 at 100 μM resulted in a greater percentage of parasite death over a 120 hour time course when compared with the current standard of care, albendazole. The effect of worm killing was significantly different at 72 h (P < 0.05). Further structural modifications to the hybrid 15 chemotype could improve the nematicidal activity.
Fig. 5.
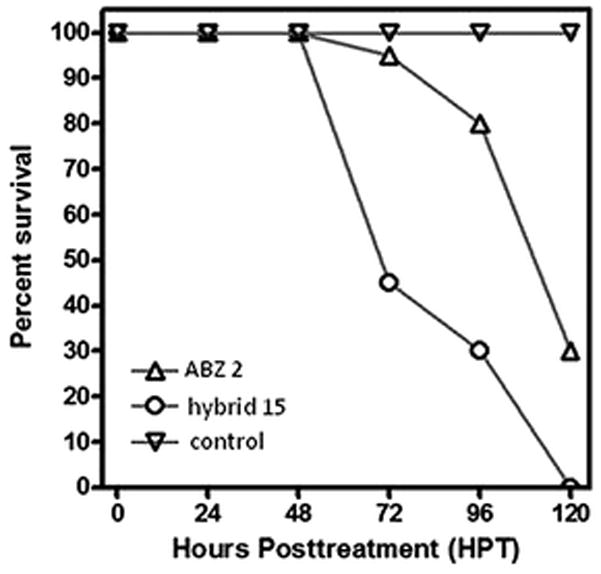
Ex vivo hookworm parasite killing assay of ABZ 2 at 100 μM and hybrid 15 at 100 μM.
Conclusions
As parasitic diseases continue to threaten human populations, advanced therapeutics should be constantly sought to improve the standard of care for those infected. Multiple parasites are often endemic to the same region, and therefore it is desirable to develop a therapeutic that is active against multiple parasites. Here we show a novel hybrid molecule that covalently joins the NO releasing furoxan moiety with the privileged quinoline molecule amodiaquine. To our gratification, the hybrid molecule 15 was active against three major parasites; P. falciparum, S. mansoni, and A. ceylanicum. As the forerunner in this novel molecular series, further exploration of the structure–activity relationships may likely lead to an increase in the activity against one or all of the target diseases.
Materials and methods
Chemistry
All reactions were performed under argon in oven-dried or flame-dried glassware. Microwave reactions were performed in a Biotage Initiator microwave. Dichloromethane was dispensed from an LC Technology Solutions SPBT-1 bench top solvent purification system. All commercially available reagents were purchased from Sigma Aldrich and used as received. All experiments were monitored by analytical thin layer chromatography (TLC) performed on Silicycle silica gel 60 Å glass supported plates with 0.25 mm thickness. Flash chromatography was performed with EMD silica gel (40–63 μM). Yields are not optimized. Infrared (IR) spectra were recorded on a Perkin Elmer 1600 FT-IR spectrometer. Nuclear magnetic resonance (NMR) spectra were recorded on a BrukerAvance 400 MHz FT-NMR spectrometer (400 MHz for 1H, 100 MHz for 13C). The following abbreviations are used in the Experimental section for the description of 1H NMR spectra: singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m), broad singlet (bs), doublet of doublets (dd), doublet of triplets (dt), and doublet of quartets (dq). Low resolution mass spectra (electrospray ionization) were acquired on an Agilent Technologies 6130 quadrupole spectrometer coupled to an Agilent Technologies 1200 series HPLC. High resolution mass spectrum-electron ionization spray (HRMS-ESI) were obtained on an Agilent Technologies 1200 series Dual Absorbance Detector HPLC system equipped with a Phenomenex Luna 75 × 3 mm, C18, 3 μm column at 45 °C (UV detection at 220 nm, BW 8 nm, and 254 nm, BW 8 nm, flow rate: 0.8 mL min−1 (increasing), injection volume: 1.0 μL, sample solvent: 100% methanol, sample conc.: ~0.01 mg mL−1, mobile phase A: water with 0.1% acetic acid, mobile phase B: acetonitrile with 0.1% acetic acid) coupled to a Agilent 6210 time-of-flight mass spectrometer (ion source: Dual ESI, min range: 115 m/z, max range: 1400 m/z, scan rate: 0.9 seconds, gas temp: 340 °C, gas flow: 10 L min−1, nebulizer: 50 PSI, ion polarity: positive, VCap: 3500 V, fragmentor: 175 V, skimmer1: 65 V, OctopoleRFPeak: 250 V, ref mass: enabled (Agilent P/N G1969-85001). Data were analyzed using Agilent Masshunter Workstation Data Acquisition (v B.02.00, Patch 1,2,3) and Agilent Masshunter Qualitative Analysis (v B.02.00, Build 2.0.197.7, Patch 3). List of abbreviations are as follows: dichloromethane – DCM; dimethylformamide – DMF; sat. aq. NaCl – brine; ethyl acetate – EtOAc.
Synthesis of N-(3-bromo-5-((tert-butylamino)methyl)phenyl)-7-chloroquinolin-4-amine (10)
To a round bottom flask were added 4,7-dichloroquinoline (8, 2.29 g, 11.56 mmol), conc. hydrochloric acid (0.348 mL, 11.45 mmol, 1.0 eq.), and MeOH (30 mL). The reaction mixture was stirred at rt for 10 min until the starting material was completely in solution. At that time, (3-amino-5-bromophenyl)methanol (9, 2.31 g, 11.45 mmol, 1 eq.) was added in one portion and the resulting reaction mixture was stirred at rt for 16 h. After ~15 min a precipitate began to form. After 16 h, (3-bromo-5-(7-chloroquinolin-4-ylamino)-phenyl)-methanol hydrochloride was filtered as a yellow solid: yield (4.09 g, 10.22 mmol, 89%); 1H NMR (400 MHz, DMSO-d6) δ 15.01 (brs, 1H), 11.28 (brs, 1H), 8.90 (d, J = 9.14 Hz, 1H), 8.57 (d, J = 7.02 Hz, 1H), 8.21 (d, J = 2.07 Hz, 1H), 7.88 (dd, J = 9.11, 2.10 Hz, 1H), 7.60 (t, J = 1.72 Hz, 1H), 7.55 (s, 1H), 7.45 (s, 1H), 6.89 (d, J = 6.97 Hz, 1H), 4.57 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 155.1, 147.6, 144.1, 139.6, 139.0, 138.9, 128.3, 128.3, 127.9, 126.7, 126.5, 122.5, 122.4, 119.7, 116.6, 62.2; ESI-HRMS m/z (M + H)+ for C16H12BrClN2O calc. = 364.9873, found = 364.9876, HPLC tr = 2.78 min (98%).
(3-Bromo-5-(7-chloroquinolin-4-yl-amino)phenyl)methanol hydrochloride (4.60 g, 11.50 mmol) was taken directly from the previous reaction and transferred to a high pressure reaction vessel. Conc. hydrochloric acid (35 mL) was added, the vessel was sealed, and the reaction mixture was heated to 100 °C for 16 h. The solvent was removed under reduced pressure, the residue was triturated with ether, and the resulting fine yellow solid was filtered via Buchner funnel. The solid was purified directly on silica. Gradient elution (1–20% MeOH (containing 10% NH4OH) in DCM) afforded N-(3-bromo-5-(chloro-methyl) phenyl)-7-chloro-quinolin-4-amine as a yellow amorphous solid: yield (3.37 g, 8.82 mmol, 77%); 1H NMR (400 MHz, CDCl3) δ 9.26 (s, 1H), 8.56 (d, J = 5.13 Hz, 1H), 8.38 (d, J = 9.09 Hz, 1H), 7.94 (d, J = 1.98 Hz, 1H), 7.62 (dd, J = 9.02, 2.19 Hz, 1H), 7.50 (s, 1H), 7.46 (s, 1H), 7.39 (s, 1H), 7.07 (d, J = 4.99 Hz, 1H), 4.78 (s, 2H) 13C NMR (100 MHz, CDCl3) δ 141.6, 134.6, 133.5, 126.5, 125.9, 124.9, 124.0, 122.5, 120.9, 49.1, 45.4; ESI-HRMS m/z (M + H)+ for C16H11BrCl2N2 calc. = 382.9532, found = 382.9537, HPLC tr = 3.00 min (99%).
To a round bottom flask were added N-(3-bromo-5-(chlor-omethyl)phenyl)-7-chloroquinolin-4-amine (1.758 g, 4.60 mmol), DMF (15 mL), and py (1.12 mL, 13.8 mmol, 3 eq.). tert-Butyl-amine (1.46 mL, 13.8 mmol, 3 eq.) was added and the reaction mixture was stirred at 80 °C for 16 h. The reaction was complete by LCMS and TLC. The reaction mixture was diluted with EtOAc and washed with 3 N LiCl and brine. The organic layer was dried on MgSO4, filtered, the solvent was removed under reduced pressure. The residue was purified directly on silica. Gradient elution (2–18% MeOH (containing 10% NH4OH) in DCM) afforded the desired compound 10 as a yellow flaky solid: yield (1.54 g, 3.67 mmol, 80%); 1H NMR (400 MHz, CDCl3) 9.16 (s, 1H), 8.53 (d, J = 5.28 Hz, 1H), 8.39 (d, J = 9.05 Hz, 1H), 7.92 (s, 1H), 7.60 (d, J = 8.95 Hz, 1H), 7.37 (brs, 2H), 7.31 (s, 1H), 7.02 (d, J = 5.23 Hz, 1H), 3.68 (brs, 2H), 3.17 (d, J = 5.23 Hz, 1H), 1.08 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 152.5, 152.49, 150.04, 147.6, 142.3, 134.5, 128.2, 128.2, 124.9, 122.0, 120.57, 119.0, 118.9, 103.2, 103.1, 29.3; ESI-HRMS m/z (M + H)+ for C20H21BrClN3 calc. 420.0659, found = 420.0657, HPLC tr = 2.59 min (97%).
Synthesis of (E)-ethyl-3-(3-((tert-butylamino)methyl)-5-(7-chloroquinolin-4-ylamino)phenyl)-acrylate (11)
To a 20 mL microwave vial was added DMF (14 mL), and the solvent was degassed with an N2 balloon. After 20 min, N-(3-bromo-5-((tert-butylamino)methyl)phenyl)-7-chloroquinolin-4-amine (10, 1.54 g, 3.67 mmol), K2CO3 (1.52 g, 11.0 mmol, 3 eq.), diacetoxypalladium (Pd(OAc)2, 0.041 g, 0.183 mmol 0.05 eq.), and 1,3-bis(diphenyl-phosphino)propane (0.151 g, 0.367 mmol, 0.1 eq.) were added, and the reaction mixture was preheated in an oil bath at 50 ~C for 20 min. At that time, ethyl acrylate (1.9 mL, 18.3 mmol, 5 eq.) was added and the reaction mixture was heated in the microwave at 130 °C for 7 h. The reaction was complete and was diluted with EtOAc and washed with 3 N LiCl and brine. The organic layer was dried on MgSO4, filtered, the solvent was removed under reduced pressure. The residue was purified directly on silica. Gradient elution (2–6% MeOH (containing 10% NH4OH) in DCM) afforded the desired compound 11 as a yellow amorphous solid: yield (1.295 g, 2.96 mmol, 81%); 1H NMR (400 MHz, CDCl3) δ 8.44 (d, J = 5.30 Hz, 1H), 8.04 (d, J = 8.91 Hz, 1H), 7.89 (s, 1H), 7.50 (d, J = 15.98 Hz, 1H), 7.21–7.35 (m, 4H), 6.87 (d, J = 5.24 Hz, 1H), 6.31 (d, J = 15.98 Hz, 1H), 5.25 (s, 1H), 4.18 (q, J = 7.07 Hz, 2H), 3.71 (s, 2H), 1.25 (t, J = 7.11 Hz, 3H), 1.17 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 166.9, 151.6, 149.5, 147.8, 143.9, 142.5, 140.6, 135.9, 135.4, 128.38, 126.0, 124.1, 122.4, 120.5, 118.9, 118.4, 102.7, 102.4, 60.7, 51.9, 46.6, 28.7, 14.3; ESI-HRMS m/z (M + H)+ for C25H29ClN3O2 calc. = 438.1943, found = 438.1938, HPLC tr = 2.68 min (99%).
Synthesis of 4-(3-((tert-butyl(nitroso)amino)methyl)-5-(7-chlor-oquinolin-4-ylamino)phenyl)-3-(hydroxymethyl)-1,2,5-oxadiazole 2-oxide (12)
To a round bottom flask were added (E)-ethyl 3-(3-((tert-butylamino)-methyl)-5-(7-chloroquinolin-4-ylamino)phenyl) acrylate (11, 1.19 g, 2.72 mmol) and DCM (19 mL), followed by diisobutylaluminum hydride (DIBALH, 1.0 M in DCM, 8.15 mL, 8.15 mmol, 3 eq.) at −78 °C. The reaction mixture was stirred for 10 h and checked by LCMS. The reaction was not complete, and additional DIBALH (5.43 mL, 5.43 mmol, 2 eq.) was added. The reaction mixture was stirred for an addition 3 h. Upon completion, py (2.198 mL, 27.2 mmol) was added, followed by water. The mixture was diluted with EtOAc, and the organic layer was dried on MgSO4, filtered, and the solvent was removed under reduced pressure. The residue was purified directly on silica. Gradient elution (5–20% MeOH(containing 10% NH4OH) in DCM) afforded (E)-3-(3-((tert-butylamino) methyl)-5-(7-chloro-quinolin-4-ylamino)-phenyl)-prop-2-en-1-ol as a yellow amorphous solid: yield (0.800 g, 2.02 mmol, 74%); 1H NMR (400 MHz, CDCl3) δ 7.94 (d, J = 9.00 Hz, 2H), 7.86 (d, J = 2.10 Hz, 2H), 7.82 (brs, 1H), 7.25 (dd, J = 9.00, 2.10 Hz, 2H), 7.07 (s, 1H), 7.02 (d, J = 4.26 Hz, 2H), 6.79 (d, J = 5.43 Hz, 1H), 6.43 (d, J = 15.89 Hz, 1H), 6.23 (dt, J = 15.85, 5.38 Hz, 1H), 4.20 (d, J = 4.94 Hz, 2H), 3.61 (s, 2H), 1.13 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 151.4, 149.1, 148.5, 142.5, 140.0, 138.5, 135.3, 130.3, 129.4, 127.8, 125.7, 122.8, 122.3, 121.9, 119.2, 118.2, 102.1, 62.5, 51.1, 49.9, 46.9, 28.8; ESI-HRMS m/z (M + H)+ for C23H27ClN3O calc. = 396.1837, found = 396.1835, HPLC tr = 2.53 min (99%).
To a round bottom flask were added (E)-3-(3-((tert-butylamino)methyl)-5-(7-chloroquinolin-4-ylamino)phenyl)prop-2-en-1-ol (19, 0.500 g, 1.26 mmol) and AcOH (10 mL), followed by sodium nitrite (NaNO2, 0.49 g, 5.05 mmol, 4 eq.). The reaction mixture was sealed and stirred at rt for 10 h. Additional portions of NaNO2 and AcOH were added periodically. Upon completion, the solvent removed under reduced pressure and the residue was purified on silica. Gradient elution (1–6% MeOH (containing 10% NH4OH) in DCM) afforded an inseparable mixture of N-oxide regioisomers as a yellow amorphous solid: yield (0.260 g, 0.54 mmol, 57%); the mixture was taken forward without further purification or characterization.
Synthesis of 4-(3-((tert-butyl(nitroso)amino)methyl)-5-(7-chloro-quinolin-4-ylamino)phenyl)-3-((hydroxyimino)methyl)-1,2,5-oxadiazole 2-oxide (13)
To a 1 dram vial were added 4-(3-((tert-butyl(nitroso)-amino)methyl)-5-(7-chloroquinolin-4-yl-amino)phenyl)-3-(hydroxymethyl)-1,2,5-oxadiazole 2-oxide (12, 0.118 g, 0.244 mmol), N-(tert-butyldimethylsilyloxy)-4-methylbenzene-sulfonamide (TBSONHTs, 0.147 g, 0.489 mmol), and triphenylphosphine (0.077 g, 0.293 mmol) in THF (5 mL). Diisopropylazodiacarboxylate (DIAD, 0.057 mL, 0.293 mmol) was added dropwise, and the reaction mixture was heated at 55 °C for 3 days. The reaction was not progressing further, so the solvent was removed under reduced pressure, and the residue was purified directly on silica. The solvent was removed under reduced pressure, and the residue was purified directly on silica. Gradient elution (0.5–5% MeOH (containing 10% NH4OH) in DCM) afforded 4-(3-((tert-butyl(nitroso)amino)methyl)-5-(7-chloro-quinolin-4-ylamino)-phenyl)-3-((N-(tert-butyldimethylsilyloxy)-4-methylphenyl-sulfonamido)methyl)-1,2,5-oxadiazole 2-oxide as a colorless, amorphous solid: yield (116 mg, 0.151 mmol, 62%); 1H NMR (400 MHz, CDCl3) δ 8.61 (d, J = 5.09 Hz, 1H), 8.00 (d, J = 2.01 Hz, 1H), 7.95 (d, J = 9.00 Hz, 1H), 7.78 (s, 2H), 7.76 (s, 1H), 7.48 (s, 1H), 7.46–7.39 (m, 4H), 7.15 (s, 1H), 7.10 (d, J = 5.23 Hz, 1H), 4.93 (s, 2H), 4.14 (brs, 2H), 2.50 (s, 3H), 1.61 (s, 9H), 0.87 (s, 9H), −0.03 (brs, 6H); 13C NMR (100 MHz, CDCl3) δ 155.9, 151.8, 149.7, 146.3, 146.1, 141.6, 138.9, 135.5, 130.0, 129.8, 128.7, 128.1, 127.3, 126.5, 121.7, 120.9, 120.7, 119.4, 118.5, 110.2, 62.2, 49.1, 44.5, 29.2, 25.8, 21.8, 17.8, −4.8; ESI-HRMS m/z (M + H)+ for C36H45ClN7O6SSi calc. = 766.2604, found = 766.2618, HPLC tr = 3.57 min (96%).
To a 1 dram vial were added 4-(3-((tert-butyl(nitroso)amino) methyl)-5-(7-chloroquinolin-4-ylamino)-phenyl)-3-((N-(tert-butyl-dimethylsilyloxy)-4-methyl-phenyl-sulfonamido)methyl)-1,2,5-oxadiazole 2-oxide (0.052 g, 0.068 mmol) and acetonitrile (0.679 mL), followed by cesium fluoride (CsF, 0.021 g, 0.136 mmol). The reaction mixture was stirred at rt for 30 min. Upon completion, the reaction mixture was acidified with 10% HCl and extracted with EtOAc. The organic layer was dried over MgSO4, filtered, and the solvent was removed under reduced pressure. The residue was purified directly on silica. Gradient elution (0.5–5% MeOH (containing 10% NH4OH) in DCM) afforded the desired compound 13 as a yellow, amorphous solid mixture of E–Z isomers: yield (16 mg, 0.032 mmol, 48%); ESI-HRMS m/z (M + H)+ for C23H22ClN7O4 calc. = 496.1495, found = 496. 1496, HPLC tr = 3.04 min (99%).
Synthesis of 4-(3-((tert-butyl(nitroso)amino)methyl)-5-(7-chlor-oquinolin-4-ylamino)phenyl)-3-cyano-1,2,5-oxadiazole 2-oxide (14)
To a conical microwave vial were added 4-(3-((tert-butyl-(nitroso)amino)methyl)-5-(7-chloroquinolin-4-ylamino)-phenyl)-3-((hydroxyimino)methyl)-1,2,5-oxadiazole 2-oxide (13, 0.02 g, 0.040 mmol) and DMF (0.403 mL). The stirring solution was cooled to 0 °C, and thionyl chloride (SOCl2, 0.012 mL, 0.161 mmol) was added. The ice bath was removed, and the reaction was checked after 10 min. The reaction was complete by TLC and LCMS. The reaction mixture was diluted with EtOAc and washed with sat. aq. NaHCO3, 3 N LiCl, and brine. The organic layer was extracted, dried over MgSO4, filtered, and the solvent was removed under reduced pressure. The residue was purified on reverse phase column, affording the desired compound 14 as a colorless, amorphous solid: yield (18.5 mg, 0.039 mmol, 96%); 1H NMR (400 MHz, methanol-d4) δ 8.42 (d, J = 5.38 Hz, 1H), 8.23 (d, J = 9.05 Hz, 1H), 7.84 (s, 1H), 7.75 (brs, 1H), 7.48 (d, J = 8.95 Hz, 1H), 7.32 (s, 1H), 7.30 (s, 1H), 7.09 (d, J = 5.43 Hz, 2H), 4.97 (s, 2H), 1.62 (s, 9H); 13C NMR (100 MHz, methanol-d4) δ 164.9, 155.86, 152.5, 150.1, 143.3, 141.0, 137.26, 127.9, 127.4, 127.1, 125.3, 124.9, 121.7, 120.2, 119.8, 119.7, 108.0, 104.2, 63.8, 45.9, 29.4; ESI-HRMS m/z (M + H)+ for C23H20ClN7O3 calc. = 478.1389, found = 478.1387, HPLC tr = 3.13 min (98%).
Synthesis of 4-(3-((tert-butylamino)methyl)-5-(7-chlor-oquinolin-4-ylamino)phenyl)-3-cyano-1,2,5-oxadiazole 2-oxide (hybrid 15)
To a 1 dram vial were added 4-(3-((tert-butyl-(nitroso) amino)methyl)-5-(7-chloroquinolin-4-ylamino)phenyl)-3-cyano-1,2,5-oxadiazole 2-oxide (14, 9.25 mg, 0.019 mmol) and MeOH (0.5 mL), followed by 4.0 M HCl in dioxane (0.097 mL, 0.387 mmol). The reaction mixture was stirred at rt for 30 min, and complete conversion was achieved as seen by LCMS and TLC. The reaction mixture was submitted directly for purification on reverse phase column, affording the desired hybrid compound 15 as a colorless, amorphous solid: yield (8.5 mg, 0.019 mmol, 98%); 1H NMR (400 MHz, CDCl3) δ 8.40 (d, J = 4.99 Hz, 1H), 8.13 (d, J = 8.97 Hz, 1H), 7.86 (brs, 1H), 7.74 (brs, 1H), 7.62 (brs, 1H), 7.49 (s, 1H), 7.39 (d, J = 8.53 Hz, 1H), 7.03 (d, J = 5.24 Hz, 1H), 3.87 (s, 2H), 1.28 (s, 9H); 13C NMR (100 MHz, DMSO-d6) δ 163.9, 154.9, 149.7, 149.6, 141.9, 134.1, 125.5, 124.9, 124.9, 118.8, 107.3, 98.39, 48.59; ESI-HRMS m/z (M + H)+ for C23H21ClN6O2 calc. = 449.1487, found = 449.1489, HPLC tr = 2.67 min (99%).
Biology
Malaria
Parasite Viability Assay for qHTS and Follow-Up.28 Briefly, 3 μL of culture medium was dispensed into 1536-well black clear-bottom plates (Aurora Biotechnologies) using a MultidropCombi (Thermo Fisher Scientific Inc.). Then, 23 nL of compounds in DMSO were added by a pin tool (Kalypsys), and 5 μL of P. falciparum-infected human red blood cells (0.3% parasitemia, 2.5% hematocrit final concentration) were added. The plates were incubated at 37 °C in a humidified incubator in 5% CO2 for 72 h, and 2 μL of lysis buffer (20 mM Tris–HCl, 10 mM EDTA, 0.16% saponin) weight to volume (w/v), 1.6% Triton-X (v/v), 10× SYBR Green I (supplied as 10 000× concentration by Invitrogen) were added to each well. The plates were mixed for 25 s with gentle shaking and incubated overnight at 22–24 °C in the dark. The following morning, fluorescence intensity at 485 nm excitation and 535 nm emission wavelengths were measured on an EnVision (Perkin Elmer) plate reader. The compound library was screened against each parasite line at seven fivefold dilutions beginning at 5.7 or 29 μM. The antimalarial drugs artemisinin, amodiaquine, and DMSO were included as positive and negative controls for each plate, respectively.
Schistosomiasis
Parasite preparation, culture and compound testing
Female Swiss-Webster mice infected with S. mansoni cercariae by percutaneous tail exposure were obtained from the Biomedical Research Institute. Parasites were obtained from mice 7 weeks after infection by perfusion with RPMI 1640 medium (Cellgro, Fisher).34 Worms were washed thoroughly several times in RPMI 1640 in a laminar flow hood. Parasites were then washed several times in complete RPMI 1640 (cRPMI) containing 25 mM HEPES (Cellgro, Fisher), 150 units per mL penicillin, 125 μg mL−1 streptomycin, and 10% fetal bovine serum (Atlanta Biologicals). Worms were then distributed into 6-well tissue culture plates at 15–20 worms per well and cultured overnight in 5 mL cRPMI in 5% CO2 at 37 °C. Following overnight culture in cRPMI, media was removed from each well and replaced with 5 mL fresh cRPMI. Hybrid 15 was dissolved in DMSO at 10 mM and tested at 50 μM, 10 μM, 5 μM, and 1 μM. The same volume of DMSO was added to each well. Negative control worms were treated with an equal volume of DMSO alone. Positive controls were treated with 10 μM furoxan 6 in DMSO. After compounds were added, worm mobility and survival was observed under a stereomicroscope (Zeiss Stemi 2000-C) for 10 seconds per worm at the indicated time points. Media were removed and replaced with fresh media and compounds every 48 hours. This study was approved by the Institutional Animal Care and Use Committee at Rush University Medical Center (IACUC number 08-058; DHHS animal welfare assurance number A3120-01).
TGR assays
TGR preparation and IC50 determination were performed as described.30 Briefly, assays were done in 96-well plates in 200 μL with 20 nM enzyme and 100 μM NADPH in 0.1 M potassium phosphate (pH 7.4), 10 mM EDTA. Compounds were dissolved in DMSO. The enzyme was incubated for 10 minutes at room temperature with NADPH and compounds at 50 μM, 20 μM, 10 μM, 1 μM, 500 nM, 100 nM, 10 nM, and 5 nM before the addition of 3 mM 5,5′-dithio-bis(2-nitrobenzoic acid). Enzyme activity was monitored by following the change in A412 due to the formation of 5-thio-2-nitrobenzoic acid during the first 3 minutes. Residual TGR activity was compared to controls incubated with equal volumes of DMSO. All assays were done in triplicate.
Hookworm
Adult worm killing assay
A. ceylanicum worms were removed from the small intestine of infected Syrian hamsters and cultured in hookworm culture medium (HCM) consisting of RPMI 1640, 50% FCS, 20 U/20 μg mL−1 penicillin/streptomycin, 10 μg mL−1 Fungizone as described.33 The susceptibility of A. ceylanicum to compounds was evaluated by culturing wells of adult worms (10 worms per well, 2 wells per treatment) in the presence of 100 μM concentration of each compound. Control wells contained equivalent volumes of DMSO and/or albendazole (Sigma, 100 μM), the current standard for treating hookworm infections. At 24 hour intervals the wells were evaluated by light microscopy to determine the percent live/dead worms.
Supplementary Material
Acknowledgments
BTM would like to thank the NIH Graduate Partnership Program for providing the opportunity to perform research across institutions. This work was funded by National Institute of Allergy and Infectious Diseases Grant AI065622 (DLW), the NIH Roadmap for Medical Research (CJT and JI), and a Career Development Award to JJV (K22 A08476). Schistosome materials for this work were supplied in part through NIH/NIAID contract HHSN272201000005I to Dr Fred Lewis at the Biomedical Research Institute, Rockville, MD. GHP acknowledges financial support from the NIH in the form of grant R37-AI-34885.
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/c2md20238g
Notes and references
- 1.World Health Organization. Parasitic Diseases. http://www.who.int/vaccine_research/diseases/soa_parasitic/en/index.html.
- 2.World Health Organization. Malaria. http://www.who.int/mediacentre/factsheets/fs094/en/
- 3.World Health Organization. Schistosomiasis. http://www.who.int/mediacentre/factsheets/fs115/en/
- 4.World Health Organization. Hookworm Disease. 2012 http://www.who.int/vaccine_research/diseases/soa_parasitic/en/index2.html.
- 5.Andrews P, Dycka J, Frank G. Ann Trop Med Parasitol. 1980;74:167–177. doi: 10.1080/00034983.1980.11687327. [DOI] [PubMed] [Google Scholar]
- 6.Bethony J, Brooker S, Albonico M, Geiger SM, Loukas A, Diemert D, Hotez PJ. Lancet. 2006;367:1521–1532. doi: 10.1016/S0140-6736(06)68653-4. [DOI] [PubMed] [Google Scholar]
- 7.Slack RD, Jacobine AM, Posner GH. Med Chem Commun. 2012;3:281–297. [Google Scholar]
- 8.Slack RD, Mott BT, Woodard LE, Tripathi A, Sullivan D, Nenortas E, Girdwood SCT, Shapiro TA, Posner GH. J Med Chem. 2012;55:291–296. doi: 10.1021/jm201214d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roepe PD. Future Microbiol. 2009;4:441–455. doi: 10.2217/fmb.09.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chinappi M, Via A, Marcatili P, Tramontano A. PLoS One. 2010;5:e14064. doi: 10.1371/journal.pone.0014064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bisby RH. Biochem Pharmacol. 1990;39:2051–2055. doi: 10.1016/0006-2952(90)90628-x. [DOI] [PubMed] [Google Scholar]
- 12.Soares JBRC, Menezes D, Vannier-Santos MA, Ferreira-Pereira A, Almeida GT, Venancio TM, Verjovski-Almeida S, Zishiri VK, Kuter D, Hunter R, Egan TJ, Oliveira MF. PLoS Neglected Trop Dis. 2009;3:e477. doi: 10.1371/journal.pntd.0000477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Egan TJ. Expert Opin Ther Pat. 2001;11:185–209. [Google Scholar]
- 14.Egan TJ. Mini-Rev Med Chem. 2001;1:113–123. doi: 10.2174/1389557013407188. [DOI] [PubMed] [Google Scholar]
- 15.Fraga CAM. Expert Opin Drug Discovery. 2009;4:605–609. doi: 10.1517/17460440902956636. [DOI] [PubMed] [Google Scholar]
- 16.Gasco A, Boschi D, Chegaev K, Cena C, Di Stilo A, Fruttero R, Lazzarato L, Rolando B, Tosco P. Pure Appl Chem. 2008;80:1693–1701. [Google Scholar]
- 17.Mathur V, Satrawala Y, Rajput MS, Vishvkarma A, Shah TD. Pharm Lett. 2010;2:244–257. [Google Scholar]
- 18.Bertinaria M, Guglielmo S, Rolando B, Giorgis M, Aragno C, Fruttero R, Gasco A, Parapini S, Taramelli D, Martins YC, Carvalho LJM. Eur J Med Chem. 2011;46:1757–1767. doi: 10.1016/j.ejmech.2011.02.029. [DOI] [PubMed] [Google Scholar]
- 19.Rai G, Sayed AA, Lea WA, Luecke HF, Chakrapani H, Prast-Nielsen S, Jadhav A, Leister W, Shen M, Inglese J, Austin CP, Keefer L, Arner ESJ, Simeonov A, Maloney DJ, Williams DL, Thomas CJ. J Med Chem. 2009;52:6474–6483. doi: 10.1021/jm901021k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sayed AA, Simeonov A, Thomas CJ, Inglese J, Austin CP, Williams DL. Nat Med. 2008;14:407–412. doi: 10.1038/nm1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Venturinin G, Colasanti M, Salvati L, Gradoni L, Ascenzi P. Biochem Biophys Res Commun. 2000;267:190–193. doi: 10.1006/bbrc.1999.1922. [DOI] [PubMed] [Google Scholar]
- 22.Nahrevanian H. Braz J Infect Dis. 2009;13:440–448. doi: 10.1590/s1413-86702009000600010. [DOI] [PubMed] [Google Scholar]
- 23.Guglielmo S, Bertinaria M, Rolando B, Crosetti M, Fruttero R, Yardley V, Croft SL, Gasco A. Eur J Med Chem. 2009;44:5071–5079. doi: 10.1016/j.ejmech.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 24.Treger RS, Cook AG, Rai G, Maloney DJ, Simeonov A, Jadhav A, Thomas CJ, Williams DL, Cappello M, Vermeire JJ. International Journal for Parasitology: Drugs and Drug Resistance. 2012;2:171–177. doi: 10.1016/j.ijpddr.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rai G, Thomas CJ, Leister W, Maloney DJ. Tetrahedron Lett. 2009;50:1710–1713. doi: 10.1016/j.tetlet.2009.01.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kitahara K, Toma T, Shimokawa J, Fukuyama T. Org Lett. 2008;10:2259–2261. doi: 10.1021/ol800677p. [DOI] [PubMed] [Google Scholar]
- 27.Bennett TN. Antimicrob Agents Chemother. 2004;52:6752. doi: 10.1128/AAC.48.5.1807-1810.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brown LE, Cheng KC-C, Wei W-G, Yuan P, Dai P, Trilles R, Ni F, Yuan J, MacArthur R, Guha R, Johnson RL, Su X-z, Dominguez MM, Snyder JK, Beeler AB, Schaus SE, Inglese J, Porco JAJ. Proc Natl Acad Sci U S A. 2011;108:6775–6780. doi: 10.1073/pnas.1017666108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fidock D, Ekland E. NIH Molecular Libraries Probe Production Network. Columbia University; 2010. R21 NS059500. [Google Scholar]
- 30.Kuntz AN, Davioud-Charvet E, Sayed AA, Califf LL, Dessolin J, Arner ESJ, Williams DL. PLoS Med. 2007;4:e206. doi: 10.1371/journal.pmed.0040206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alger HM, Williams DL. Mol Biochem Parasitol. 2002;121:129–139. doi: 10.1016/s0166-6851(02)00031-2. [DOI] [PubMed] [Google Scholar]
- 32.Keiser J, Chollet J, Xiao SH, Mei JY, Jiao PY, Utzinger J, Tanner M. PLoS Neglected Trop Dis. 2009;3:e350. doi: 10.1371/journal.pntd.0000350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cappello M, Bungiro RD, Harrison LM, Bischoff LJ, Griffitts JS, Barrows BD, Aroian RV. Proc Natl Acad Sci U S A. 2006;103:15154–15159. doi: 10.1073/pnas.0607002103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lewis F. In: Current Protocols in Immunology. Coligan JE, Kruisbeek AM, Margulies DH, Shevach EM, Strober W, editors. John Wiley & Sons; New York: 1998. pp. 19.11.11–19.11.28. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.