

Abstract
Interferon-γ (IFN-γ), a proinflammatory cytokine, has been implicated in the pathogenesis of a number of forms of heart disease including myocarditis and congestive heart failure. In fact, overexpression of IFN-γ in mice causes dilated cardiomyopathy. However, the direct effects of IFN-γ on cardiac myocytes and the mechanism by which it causes cardiac dysfunction have not been described. Here, we present the molecular pathology of IFN-γ exposure and its effect on myofibrillar proteins in isolated neonatal rat ventricular myocytes. Treatment with IFN-γ caused cardiac myocyte atrophy attributable to a specific decrease in myosin heavy chain protein. This selective degradation of myosin heavy chain was not accompanied by a decrease in total protein synthesis or by an increase in total protein degradation. IFN-γ increased both proteasome and immunoproteasome activity in cardiac myocytes and their inhibition blocked myosin heavy chain loss and myocyte atrophy, whereas inhibition of the lysosome or autophagosome did not. Collectively, these results provide a mechanism by which IFN-γ causes cardiac pathology in the setting of chronic inflammatory diseases.
Both infectious and noninfectious agents including viruses, parasites, and some chemotherapeutic drugs can elicit a significant immune response in the heart. The resulting inflammation, which can persist long after resolution of the cardiac insult, causes myocarditis and is implicated in approximately 20% of sudden deaths in children and young adults.1 The most common cause of myocarditis worldwide is the parasitic protozoan Trypanosoma cruzi, which causes Chagas disease and is the leading cause of congestive heart failure in South America.2 Approximately 80% of patients with Chagas disease develop cardiac complications and 20% require heart transplantation.3 Coxsackievirus type B3, HIV, and adenovirus are the predominant causes of myocarditis in developed countries.4 Through direct infection of cardiac tissue and a subsequent persistent inflammatory response in the heart, a subset of these patients will develop chronic myocarditis, which is characterized by fibrosis and myocyte atrophy, and, ultimately, can lead to dilated cardiomyopathy (DCM).5,6 Approximately 50% of patients with DCM die within 2 years of diagnosis.7 In one study of asymptomatic HIV-positive patients, for example, 80% had DCM, with most (83%) showing concomitant myocarditis.4,8–10 Myocarditis therefore is associated with many different diseases, yet often is overlooked clinically, likely owing to its asymptomatic course in most patients and its association with cardiomyopathies.
In cardiac infection, natural killer cells and macrophages infiltrate the myocardium and promote recruitment of T lymphocytes, which induces a robust local increase in proinflammatory cytokine production. Interferon-γ (IFN-γ), the predominant inflammatory cytokine in myocarditis, is up-regulated significantly in the myocardium of patients with Chagas disease2 and is implicated in the pathogenesis of myocarditis.11 During infection, IFN-γ up-regulates expression of major histocompatibility complexes I and II on infected and uninfected cells, thus enhancing the diversity of peptides displayed on these cells.12 Inflammation that is associated with up-regulation of IFN-γ and other cytokines often persists in patients for months or years after the clearance of infection. Although initially protective, chronic inflammation is correlated with the development of DCM and heart failure in some patients.13 However, the mechanism by which inflammation causes DCM and heart failure is not well understood.
A causative cytotoxic role for IFN-γ is supported by studies in mice: constitutive overexpression of IFN-γ in the liver, which increases serum IFN-γ levels, causes cardiac myofiber atrophy and DCM, similar to that which develops in some patients with myocarditis.14,15 Interestingly, systemic overexpression of IFN-γ only affected the heart and did not affect skeletal or smooth muscle. However, the direct effects of IFN-γ on cardiac myocytes were not determined. Here, we examine the effects of IFN-γ on cardiac myocytes in isolation from the complicated systemic and local effects of infection and suggest a mechanism for the specificity of cytotoxicity in the heart.
Materials and Methods
Cell Culture
Animal use was in accordance with protocols approved by the Institutional Animal Care and Use Committee at the University of Colorado at Boulder and conformed with guidelines published by the US NIH. Neonatal rat ventricular myocytes (NRVMs) were isolated and cultured as previously described.16 Approximately 50 to 75 hearts were pooled per preparation. Each experiment was performed in triplicate and then repeated at least two times, each with distinct NRVM populations. After an 18-hour recovery period, cells were treated with PBS and 0.2 or 200 U/mL mouse recombinant IFN-γ (Calbiochem/EMD Chemicals, Gibbstown, NJ) for 24 or 48 hours. These doses were chosen to be physiologic and supraphysiologic, respectively. The supraphysiologic dose is similar to those used by others to study the in vitro effect of IFN-γ on cardiomyocytes.17–19 Proteasome activity was blocked pharmacologically by treating cells with 1 μmol/L MG-132 (Boston Biochem, Cambridge, MA) or 5 nmol/L bortezomib (Millennium Pharmaceuticals, Cambridge, MA) for 48 hours. To inhibit lysosome activity, cells were treated with 10 mmol/L NH4Cl (Sigma, St. Louis, MO) or 2.5 mmol/L 3-methyladenine (Sigma). NRVMs also were treated with 100 nmol/L or 10 μmol/L dexamethasone or 20 ng/mL tumor necrosis factor α (TNF-α) for 48 hours in parallel experiments.
Immunocytochemistry and Cell Area Measurement
NRVMs cultured on coverslips were fixed in 4% paraformaldehyde for 20 minutes, permeabilized with 0.1% Triton X-100 (Fisher Scientific, Pittsburgh, PA), blocked with 10% horse serum, and incubated overnight at 4°C with anti–α-actinin antibodies (1:200; Sigma). Cells then were incubated with appropriate fluorophore-conjugated secondary antibodies for 30 minutes at room temperature and visualized using fluorescence microscopy. Digitized images of cells (three fields in triplicate, 60 to 80 cells total per condition) were obtained and the cell area was measured using ImageJ software version 1.43t (NIH, Bethesda, MD).
Cell Volume Measurement
After 48 hours of IFN-γ treatment, NRVMs were trypsinized and suspended in PBS containing 1% bovine calf serum and 10 mmol/L EDTA. The cell volume per sample was measured in a Coulter Counter Analyzer (Beckman Coulter, Brea, CA). Each sample was analyzed in triplicate and experiments were repeated twice. Average volumes were calculated for each treatment group and values were normalized to vehicle-treated myocytes.
Myofibrillar Gels and Western Blots
For myosin heavy chain (MyHC) analysis, NRVMs were lysed in high-salt myosin extraction buffer.20 Lysates were centrifuged at 13,500 × g for 15 minutes at 4°C. The supernatant was removed and protein concentration was determined using a detergent-compatible protein assay (Bio-Rad Laboratories, Hercules, CA). Protein (2.5 μg per sample) was loaded onto myofibrillar gels, which were composed of 15% acrylamide (124:1 acrylamide: bis), 9.79% glycerol, 0.73 mol/L Tris pH 9.3, and 0.1% SDS. Gels were run at 16 mA for 6 hours at 4°C, silver stained (Bio-Rad Silver Stain Plus; Bio-Rad Laboratories), and digitally scanned. Antibodies for immunoblots were as follows: anti–α-MyHC (BA-G5 hybridoma, 1:5; ATCC, Manassas, VA), anti–β-MyHC (1:400; Vector Laboratories, Burlingame, CA), and anti–light chain 3b (1:1000; Cell Signaling, Boston, MA). Immunoreactivity was visualized using an enhanced chemiluminescence detection system (Perkin Elmer, Waltham, MA) and band intensities were quantified using ImageJ software.21
RNA Analysis
Total RNA was isolated from NRVM cultures using a TRIzol-based reagent (Molecular Research Center, Inc., Cincinnati, OH). First-strand cDNA was synthesized using reverse transcriptase (Invitrogen, Carlsbad, CA) and random hexamer primers. Gene expression then was measured using quantitative real-time polymerase chain reaction (qPCR) using a 7500 Real-Time PCR system (Applied Biosystems, Carlsbad, CA). Primer sequences were as follows: atrogin-1 forward, 5′-GAACATCATGCAGAGGCTGA-3′ and reverse, 5′-CGAGTCACTTCTGGCCGATG-3′; and muscle ring finger protein 1 (MuRF-1) forward, 5′-GTGAGGTTGCCCCTTTGCAA-3′ and reverse, 5′-CGACTCATTGACGTAGAGGT-3′.
Caspase and Proteasome Activity Assays
Caspase-3 activity in cardiac lysates was determined by monitoring the rate of cleavage of a synthetic fluorogenic caspase-3–specific substrate (Ac-Asp-Glu-Val-Asp-AMC; Calbiochem/EMD Chemicals) as previously described.22 Proteasome activity was determined as described,23 except ATP was not added to the reaction mixture because optimization of conditions revealed that the addition of ATP significantly decreased activity. Activity was determined using 15 μg protein and 20 μmol/L fluorogenic substrate [proteasome: Suc-LLVY-AMC (Boston Biochem) or immunoproteasome: Ac-PAL-AMC (a gift from Millennium Pharmaceuticals, Inc.)]. To ensure specificity of the assay, 5 nmol/L bortezomib (a gift from Millennium Pharmaceuticals, Inc.), which more specifically and potently inhibits proteasomes, was used in parallel experiments.24 Fluorescent emission resulting from cleavage of the appropriate substrate was recorded every 2 minutes for 1 hour at 37°C using a Synergy 2 multimode microplate reader (BioTek, Winooski, VT). Proteasome activity was calculated as the slope of the linear portion of the line produced by emission of cleaved substrate over time for each sample normalized to protein concentration.
Pulse and Pulse-Chase Assays
To evaluate protein synthesis, NRVMs were treated with 200 U/mL IFN-γ for 46 hours. Cells were washed and incubated with [3H]-tyrosine and IFN-γ for 2 hours. Cells were collected in 10% trichloroacetic acid and the incorporated radioactivity was determined using a scintillation counter. Protein synthesis is represented as the number of counts normalized to protein concentration per well. Protein degradation rates in NRVMs were determined as previously described,25 with modification; after the initial 24-hour incubation period with [3H]-tyrosine, cells were treated with IFN-γ for 24 hours in medium containing 2 mmol/L unlabeled tyrosine to prevent reincorporation of radiolabeled tyrosine. In some experiments, cells also were treated with either proteasome (1 μmol/L MG-132) or lysosomal (10 mmol/L NH4Cl) inhibitors for 2 hours before collecting media samples. For autoradiography experiments, cells were incubated for 24 hours with [35S]-methionine and then treated with IFN-γ for 24 hours in the presence of 2 mmol/L unlabeled methionine. Cellular proteins were extracted in myosin extraction buffer, and run on myofibrillar gels as described earlier. Gels were dried and digitally scanned autoradiograms were analyzed densitometrically using ImageJ software. All experiments were performed three times and measurements were performed in triplicate.
Data and Statistical Analysis
Data are presented as means ± SEM calculated from averages obtained from at least three NRVM isolations. Differences between groups were evaluated for statistical significance using the Student's t-tests. P values less than 0.05 were considered significant.
Results
IFN-γ Causes Myocyte Atrophy and a Specific Decrease in Cardiac MyHC in Vitro
Transgenic mice with constitutively increased levels of circulating IFN-γ show chronic myocarditis with significant cardiac myofiber atrophy14 consistent with cardiac pathology observed in a subset of patients with myocarditis.6 To determine whether this phenotype occurs in cardiac myocytes in culture, NRVMs were treated with 200 U/mL IFN-γ for 48 hours. NRVMs were used to isolate the direct effects of IFN-γ on cardiomyocytes without the complicated immune responses to systemic infection. NRVMs are a well-described model system because they respond similarly to intact hearts to many hypertrophic agonists and cardiotoxic agents.26–28 They also can be efficiently infected with viruses or viral particles that are implicated in myocarditis and respond to the cytokines that are up-regulated during these infections.26 The predominant morphologic effect of IFN-γ on NRVMs was a qualitative reduction in myocyte size (Figure 1A). Quantification revealed that treatment with IFN-γ caused a significant decrease in myocyte area (reduced by 26% ± 2%) (Figure 1B) and volume (reduced by 9% ± 1.5%) (Figure 1C).
Figure 1.
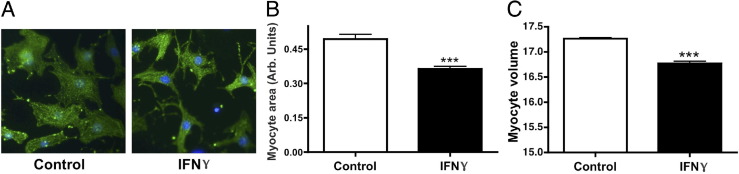
IFN-γ induces atrophy of NRVMs. A: Representative images of NRVMs treated with vehicle (control) or IFN-γ immunolabeled with antibodies raised against α-actinin. B: Quantification of myocyte area after treatment with vehicle or IFN-γ. C: Quantification of NRVM volume after treatment with vehicle or IFN-γ. ***P < 0.001 compared with control.
We hypothesized that the reduction in cell size in response to IFN-γ is attributable to a decrease in MyHC, the most abundant sarcomeric protein in cardiac muscle by mass.29 NRVMs were treated with IFN-γ or vehicle, and MyHC was solubilized and resolved on myofibrillar gels. Treatment with IFN-γ caused a decrease in total MyHC by 40% ± 13% at 24 hours and 58% ± 6% at 48 hours (Figure 2A) compared with untreated controls at the respective time points. This effect also was observed at a lower physiological concentration of IFN-γ (0.2 U/mL) (Figure 2B).
Figure 2.
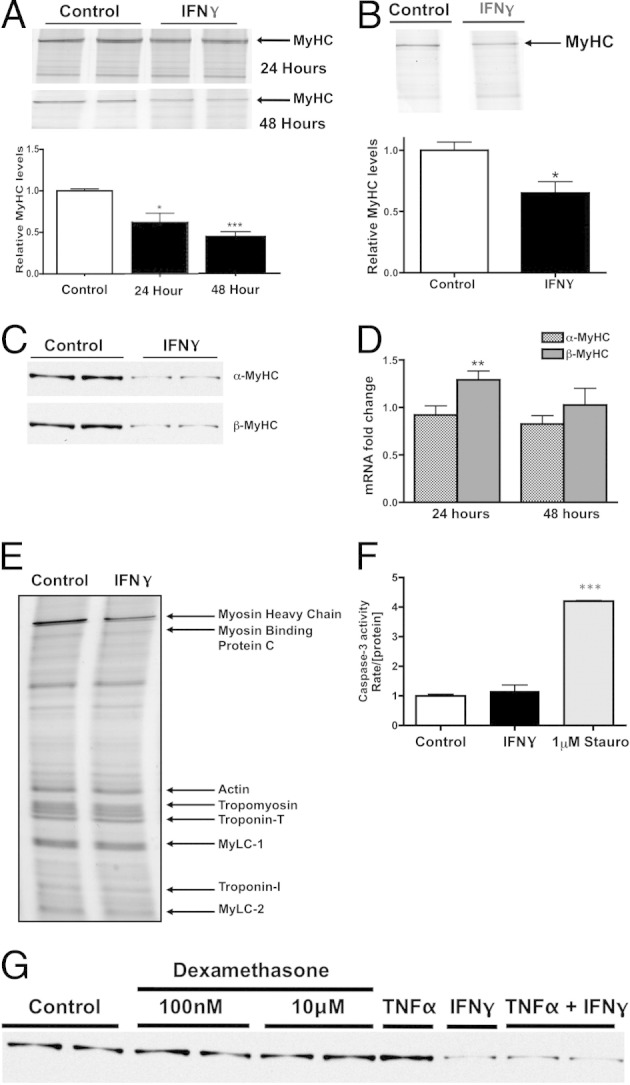
IFN-γ induces a specific decrease in MyHC protein. A: Top panel: Representative myofibrillar gel of protein lysates obtained from NRVMs treated with vehicle (control lanes) or 200 U/mL IFN-γ for 24 or 48 hours. Bottom panel: Densitometric quantification of MyHC band intensities, normalized to vehicle-treated cells. B: Top panel: Representative myofibrillar gel of protein lysates treated with vehicle (control lane) or 0.2 U/mL IFN-γ for 24 hours. Bottom panel: Densitometric quantification of MyHC band intensities, normalized to vehicle-treated cells. C: Representative Western blots using antibodies raised against α-MyHC and β-MyHC in NRVMs treated with vehicle or IFN-γ. D: Quantification of α-MyHC and β-MyHC gene expression in NRVMs treated with IFN-γ for 24 or 48 hours. E: Representative myofibrillar gel of protein lysates obtained from NRVMs treated with vehicle or IFN-γ. Bands representing sarcomeric proteins are as indicated. MyLC, myosin light chain. F: Caspase activity in NRVMs treated with vehicle, IFN-γ, or 1 μmol/L Staurosporine (Stauro) for 48 hours. The rate of caspase activity was normalized to the concentration of protein in each sample (n = 2 preparations). G: Representative Western blot using antibodies raised against cardiac MyHC in NRVMs treated with IFN-γ, TNF-α, dexamethasone, or vehicle. Samples are on the same gel but the last four lanes are noncontiguous. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with control.
Because MyHC is the most abundant protein by mass in NRVMs, we determined the effect of IFN-γ on the two MyHC isoforms in cardiac muscle: α-MyHC and β-MyHC. Unlike adult rodent hearts, in which α-MyHC predominantly is expressed, equal amounts of α-MyHC and β-MyHC are expressed in NRVMs.30 Western blots for the two cardiac MyHC isoforms revealed an equivalent decrease in both α-MyHC and β-MyHC protein at 48 hours (Figure 2C). We concluded that this decrease in MyHC is mediated posttranscriptionally because α-MyHC and β-MyHC mRNA levels did not decrease significantly with IFN-γ treatment compared with vehicle-treated NRVMs (Figure 2D). Interestingly, mRNA levels of β-MyHC actually increased after 24 hours of treatment. These data suggest that the mechanism by which IFN-γ induces atrophy is distinct from that of other cytokines that are up-regulated with cancer and cause repression of sarcomeric mRNA levels.31
To extend our analyses to other myofibrillar proteins that comprise the sarcomere, we analyzed the myofibrillar fractions of vehicle- and IFN-γ-treated NRVMs. SDS-PAGE gels revealed that despite the decrease in MyHC, all nonmyosin myofibrillar proteins (myosin binding protein, actin, troponin, tropomyosin, and myosin light chains 1 and 2) were unaffected (Figure 2E). Myosin-to-actin ratios therefore were decreased consistently (51.5% ± 3.6%) whereas all other myofibrillar protein ratios did not significantly change, indicating a possible protein-specific catabolic mechanism by which atrophy occurs.
Although IFN-γ treatment has not been associated previously with reduced myocyte viability and the hearts of patients with chronic myocarditis do not show markers for apoptosis,32 we measured the activity of caspase-3 to test whether reduced MyHC is the result of cell death in our system. As expected, IFN-γ did not activate caspase-3 activity in NRVMs (Figure 2F). Thus, IFN-γ treatment caused both NRVM atrophy and a specific decrease in MyHC protein through a mechanism independent of apoptosis.
Increased levels of circulating cytokines and glucocorticoids observed in some chronic disease states are associated with significant skeletal or cardiac muscle atrophy and degradation of myofibrillar proteins through mechanisms that are not well understood.33,34 To determine whether the selective decrease in MyHC induced by IFN-γ in NRVMs is restricted to this cytokine, we treated cells with TNF-α35 or dexamethasone,36 which have been shown to induce muscle atrophy.35,36 Neither treatment caused a decrease in MyHC in NRVMs (Figure 2G). In addition, treatment with both TNF-α and IFN-γ did not increase MyHC loss greater than that observed with IFN-γ alone.
Cardiac Myocyte Atrophy Is Attributable to the Selective Degradation of MyHC
Myocyte atrophy may be attributable to decreased protein synthesis, an increase in protein degradation, or both. We first performed a pulse assay with [3H]-tyrosine to measure changes in total protein synthesis in the presence of IFN-γ. NRVMs were harvested after 48 hours of exposure to IFN-γ and 2 hours after incorporation of [3H]-tyrosine, which was quantified and represented the extent of protein synthesis. Total protein synthesis was unaffected by 48-hour IFN-γ treatment (Figure 3A). To examine whether increased protein degradation is responsible for the decrease in myocyte size, cardiac myocyte proteins were radiolabeled with [3H]-tyrosine and then treated with IFN-γ in media containing unlabeled tyrosine to prevent reincorporation. The extent of protein degradation was quantified by measuring [3H]-tyrosine in the medium 24 hours after IFN-γ treatment. There was no change in total protein degradation rates compared with vehicle-treated cells (Figure 3B).
Figure 3.
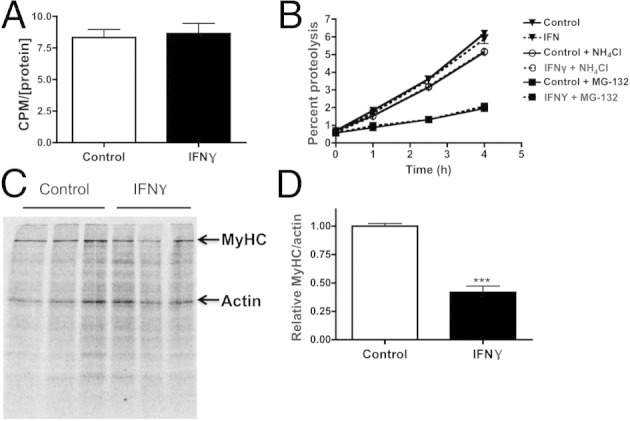
IFN-γ induces a specific decrease in MyHC without altering total protein synthesis or degradation. A: Quantification of total protein synthesis measured using [3H]-tyrosine pulse assay in NRVMs treated with vehicle (control) or IFN-γ. CPM, count per minute. B: Quantification of protein degradation by measuring [3H]-tyrosine released in the media 24 hours after vehicle, IFN-γ, NH4Cl, or MG-132 treatment. C: Myofibrillar autoradiogram of NRVM proteins pulse labeled with [35S]-methionine and treated with vehicle or IFN-γ. MyHC and actin bands are as indicated. D: Quantification of MyHC and actin band intensities (C) expressed as a ratio, normalized to vehicle-treated cells. ***P < 0.001 compared with control.
To increase the sensitivity of our study, NRVM proteins were radiolabeled with [35S]-methionine for 24 hours and then treated for 24 hours with IFN-γ in media containing excess unlabeled methionine. Similar to [3H]-tyrosine, [35S]-methionine becomes incorporated into newly synthesized proteins but has the advantage of allowing efficient visualization of these proteins on a gel using autoradiography. Labeled cell extracts run on myofibrillar gels revealed, once again, that MyHC is decreased selectively in the IFN-γ–treated myocytes (Figure 3C). IFN-γ induced a 48% ± 8% reduction in the MyHC-to-actin ratio (Figure 3D). These data do not support a mechanism involving alterations in total protein turnover, unlike other models of cardiac atrophy.37 We therefore performed a series of experiments to examine the proteolytic mechanisms that mediate the specific degradation of MyHC.
The Lysosome Does Not Contribute to IFN-γ–Induced MyHC Degradation
Several proteolytic mechanisms are responsible for regulating the degradation of the sarcomere in cardiac myocytes, namely the lysosomal and proteasomal pathways. We previously showed that the autophagosomal-lysosomal pathway is up-regulated in the atrophic hearts of tumor-bearing mice that have high circulating levels of proinflammatory cytokines.38 We first sought to determine the relative contributions of the lysosomal and ubiquitin proteasome pathways on basal levels of protein degradation in control and IFN-γ–treated NRVMs. We performed a pulse-chase assay as described earlier and treated cells with a lysosome inhibitor (NH4Cl) or proteasome inhibitor (MG-132). Inhibiting the lysosome in control cells reduced total protein degradation by 31% ± 2.1% (Figures 3B and 4A), whereas inhibiting the proteasome reduced total protein degradation by 72% ± 1.4% (Figures 3B and 5B), confirming their relative contribution to basal levels of protein degradation. As mentioned previously, treatment with IFN-γ did not affect total protein degradation rates or the relative contributions of each degradative pathway (Figure 3B).
Figure 4.

IFN-γ–induced specific degradation of MyHC is not attributable to the lysosome-autophagosome pathway. A: Quantification of proteolytic activity after inhibition of lysosome in NRVMs treated with vehicle (control) or lysosome inhibitor (NH4Cl), normalized to vehicle-treated NRVMs. B: Left panel: Representative myofibrillar gel of protein lysates from NRVMs treated with lysosome inhibitor (NH4Cl) and vehicle or IFN-γ. MyHC band is as indicated. Right panel: Densitometric quantification of MyHC band intensities, normalized to vehicle-treated cells. C: Representative Western blot using antibodies raised against light chain 3 (LC3)-II, a marker of autophagosome activity, or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) on lysates from NRVMs treated with vehicle, IFN-γ, IFN-γ plus an autophagosome inhibitor [3-methyladenine (3-MA)], or rapamycin (Rap). D: Left panel: Representative myofibrillar gel of protein lysates from NRVMs treated with 3-MA and vehicle or IFN-γ. MyHC band is as indicated. Right panel: Densitometric quantification of MyHC band intensities normalized to vehicle-treated cells. ***P < 0.001 compared with control.
Figure 5.
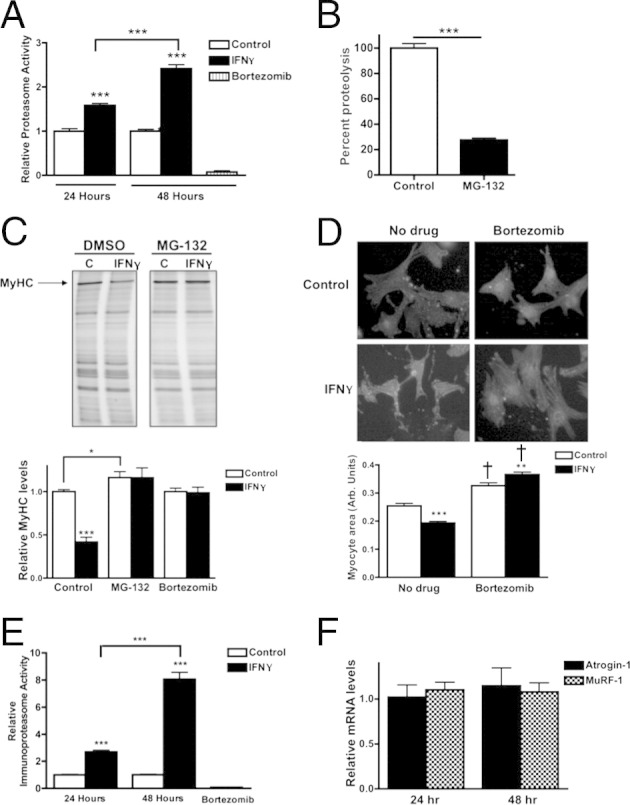
The proteasome is responsible for IFN-γ–induced degradation of MyHC in NRVMs. A: Proteasome activity in NRVMs treated with vehicle (control), IFN-γ, or bortezomib for 24 or 48 hours. Bars are normalized to control at each time point. B: Quantification of proteolytic activity after inhibition of the proteasome in NRVMs treated with MG-132 or vehicle, normalized to vehicle-treated NRVMs. C: Top panel: Representative myofibrillar gels of protein lysates from NRVMs treated with vehicle (C) or IFN-γ and vehicle (dimethyl sulfoxide, DMSO) or MG-132. The MyHC band is as indicated. Bottom panel: Densitometric quantification of MyHC. Values are normalized to vehicle-treated cells. D: Top panel: Representative images of NRVMs treated with vehicle or IFN-γ in the presence or absence of bortezomib. Cells were immunolabeled with antibodies raised against α-actinin. Bottom panel: Quantification of the NRVM area, indicated as arbitrary units. E: Immunoproteasome activity in NRVMs treated with vehicle (control), IFN-γ, or bortezomib for 24 or 48 hours. Bars are normalized to control at each time point. F: Quantification of expression of atrogin-1 or MuRF-1 normalized to 18S in NRVMs treated with IFN-γ for 24 or 48 hours. All values are normalized to expression in vehicle-treated myocytes. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with control or as indicated. †P < 0.001 compared with the respective no drug samples.
To further examine the role of the lysosomal pathway, we co-treated NRVMs with NH4Cl and IFN-γ and analyzed the levels of MyHC and other sarcomeric proteins. Although inhibition of the lysosome in cardiac myocytes reduced proteolysis (Figure 4A), it did not prevent MyHC loss induced by IFN-γ (Figure 4B), indicating that the lysosome is not involved in MyHC degradation in this model, but rather is involved in basal levels of protein turnover required to maintain cardiac myocyte function.39 To determine whether components of the autophagosomal pathway are up-regulated during cardiac myocyte atrophy, we quantified the levels of microtubule-associated light chain 3 II, a protein specifically localized to autophagic vacuoles and a marker of increased autophagic activity.40 These experiments revealed that IFN-γ did not increase light chain 3 II levels (Figure 4C), whereas rapamycin, a known inducer of autophagy, did. To examine autophagosomal activity in our model more directly, autophagy was inhibited with 3-methyladenine, which blocks autophagosome formation.41 Co-treatment with IFN-γ and 3-methyladenine did not affect light chain 3 II levels or reduce MyHC degradation induced by IFN-γ alone (Figure 4, C and D). Taken together, these results show that the autophagosomal-lysosomal pathway is not involved in IFN-γ–induced cardiac MyHC degradation in NRVMs.
IFN-γ Causes Cardiac MyHC Degradation by the Ubiquitin Proteasome System and an Increase in Immunoproteasome Activity in Vitro
The ubiquitin proteasome system (UPS) is primarily responsible for protein turnover in the heart and is necessary for the maintenance of sarcomeric stoichiometry.42 The UPS also is responsible for mediating detrimental cardiac effects in other chronic diseases with high levels of circulating inflammatory cytokines.43 Because we found that the UPS is the primary proteolytic pathway active in NRVMs, we performed a fluorometric proteasome activity assay to examine the role of the UPS in IFN-γ–induced myocyte atrophy. A time-dependent increase in UPS activity after 24 and 48 hours of IFN-γ treatment was observed (1.6-fold higher and 2.4-fold higher activity, respectively) (Figure 5A). To measure basal protein degradation and confirm the role of the UPS in untreated myocytes, NRVMs were treated with MG-132. As mentioned earlier, proteolysis was reduced significantly by inhibition of the proteasome (72% reduction in total protein degradation compared with untreated cells) (Figures 3B and 5B). To determine whether proteasome inhibition prevents MyHC degradation, NRVMs were co-treated with IFN-γ and MG-132. MG-132 treatment completely prevented MyHC loss in IFN-γ–treated NRVMs and increased MyHC levels slightly above that of control myocytes (16% ± 6%)(Figure 5C). Treatment with bortezomib, a more potent and specific inhibitor of chymotrypsin activity of the proteasome,24 also fully prevented the degradation of MyHC in IFN-γ–treated cells (Figure 5C), supporting the conclusion that the proteasome is involved in IFN-γ–mediated MyHC degradation. To determine whether proteasome inhibition prevents IFN-γ–induced myocyte atrophy, cross-sectional area was measured after 48 hours of IFN-γ treatment with or without bortezomib. Bortezomib treatment prevented myocyte atrophy and caused a significant increase in myocyte size over control values (Figure 5D). Inhibition of the UPS with bortezomib causes a significant increase in cellular protein, which recently was shown to manifest as cellular hypertrophy.44,45
Viral infection up-regulates a second type of proteasome, the immunoproteasome.46 The inner proteolytic core of the proteasome is composed of two rings of β-subunits that contain chymotryptic, tryptic, and caspase-like activity.47 IFN-γ causes the replacement of three of these β-subunits with corresponding immunoforms: LMP2, LMP7, and MECL-1.48 The incorporation of these inducible subunits collectively forms the immunoproteasome, which favors formation of peptide fragments for display on major histocompatibility complexes during an immune response. Indeed, coxsackievirus type B3–infected mice that have high levels of IFN-γ in the heart show significant up-regulation of immunoproteasome in cardiac tissue.49 Our previous studies on proteasome activity did not distinguish between proteasome and the inducible immunoproteasome. We therefore measured immunoproteasome activity to delineate these processes. By using a substrate that has 10-fold higher specificity for the immunoproteasome over the proteasome, we found a 2.7-fold increase after 24 hours of IFN-γ treatment and a striking eightfold increase in immunoproteasome activity after 48 hours (Figure 5E), suggesting that the immunoproteasome plays a primary role in IFN-γ–induced MyHC degradation and atrophy in NRVMs.
To further probe the proteasome pathway, we quantified the expression of atrogin-1 and MuRF-1, which are muscle-specific E3-ubiquitin ligases that mark specific substrates for degradation by the proteasome and are transcriptionally up-regulated during skeletal muscle atrophy.50 Atrogin-1 and MuRF-1 mRNA levels in cardiac myocytes were measured after 24 and 48 hours of IFN-γ treatment, but neither gene was induced at any time point (Figure 5F). Our data suggest that these E3 ligases are not involved in IFN-γ–induced MyHC degradation, which is consistent with our findings in atrophic mouse hearts.38 However, the levels of atrogin-1 and MuRF-1 transcripts are approximately three times higher in heart than in skeletal muscle,38 suggesting that basal levels of these ligases may be sufficient for increased MyHC degradation induced by IFN-γ.
Discussion
Proinflammatory cytokines are up-regulated in response to cardiotropic infections including those caused by viruses and parasitic protozoans. Despite clearance of the active infection by the immune system, proinflammatory cytokines can persist in the heart for months or years, leading to chronic myocarditis and, in some cases, severe cardiomyopathy.6 Defining the mechanism of the cytotoxic cardiac effects of IFN-γ in animal models is complicated by the acute pathology caused by active cardiotropic infection. Viral proteins, for example, can induce myocyte damage directly by production of enzymes that are capable of degrading cardiac myocyte proteins such as dystrophin.51,52 Mice with constitutively increased serum levels of IFN-γ, one of the predominant cytokines in chronic myocaditis, have cardiac dysfunction including left venticular dilation and premature death, likely caused by heart failure.14 However, a coincident increase in serum TNF-α complicates the interpretation of these studies. We therefore sought to isolate the effects of IFN-γ from the complicated immune response that is mounted both during and after active cardiotropic infection and from those of viral or protozoan proteins.
The Specific Degradation of MyHC Mediates IFN-γ–Induced Cardiac Myocyte Atrophy
Cardiac atrophy occurs in a transgenic murine model of HIV infection, in rats infected with T. cruzi, and in mice overexpressing IFN-γ.53,54 Our studies show that cardiac myocyte atrophy induced by IFN-γ is caused by the specific degradation of MyHC rather than a general degradative process. Interestingly, myocyte atrophy occurred without a loss of total protein or an increase in total protein degradation, thus it is possible that the reduction in cell size and volume could reflect a change in conformation resulting from the loss of MyHC.
Interestingly, all other myofibrillar proteins studied were unaffected. Selective degradation of myocyte proteins has received relatively little attention as a mechanism by which cardiac atrophy occurs. Importantly, this process occurs in the absence of immune cell involvement and therefore does not require release of TNF-α from dendritic cells or macrophages, which appears to be responsible at least in part for the cardiac dysfunction observed when IFN-γ is overexpressed in mice.55 In addition, although TNF-α and IFN-γ are both required for MyHC loss in skeletal muscle in vitro,31 we found that TNF-α did not have any effect on MyHC loss either alone or in combination with IFN-γ, which implies that IFN-γ does not mediate its effects through TNF-α in this model.
The E3 ligases atrogin-1 and MuRF-1 were not up-regulated in cardiomyocytes treated with IFN-γ despite the role of the proteasome in the degradation of MyHC. In addition to atrogin-1 and MuRF-1, other E3 ligases as well as E2 ligases and protein modifications can direct the specificity of the UPS.56 In diabetic cardiomyopathy, for example, MuRF-1 and atrogin-1 are unchanged but the C-terminus of HSC70 interacting protein ubiquitin ligase regulates myocyte injury during hyperglycemia.57 In addition, because expression of MuRF-1 and atrogin-1 are approximately threefold higher in cardiac muscle than in skeletal muscle,38 it is possible that the baseline levels of these ligases are sufficient to forgo the need for transcriptional up-regulation.
Sarcomeric proteins have been thought to be insensitive to degradation by the UPS because of limited accessibility of the proteins by E3 ligases.58 However, our data suggest that MyHC is degraded by the proteasome without complete sarcomeric disintegration because other myofibrillar proteins that are required for sarcomere stability are not degraded. In addition, the myofibrillar protein purification method used in these studies isolates proteins incorporated into the sarcomere rather than completely unincorporated or newly synthesized MyHC that might be susceptible to targeting by E3 ligases.20 Interestingly, it recently was reported that thick filament proteins such as myosin binding protein-C and myosin light chain may be ubiquitinated within the sarcomere.58 Other E3 ligases such as Ozz also selectively recognize sarcomeric proteins, such as MyHC, rather than unincorporated sarcomeric proteins.59 These studies shed light on possible novel mechanisms by which MyHC degradation occurs, without up-regulation of conventional E3 ligases or complete dissolution of the sarcomere.
Molecular Mimicry and Autoimmunity as Secondary Effects of Chronically High Levels of Circulating IFN-γ
As in other cell types, IFN-γ treatment of NRVMs induces a dramatic increase in immunoproteasome activity, which has been shown to promote expression of antigenic peptides on major histocompatibility complexes. This up-regulation augments peptide presentation on both infected and uninfected cells.47,60 It has been suggested that the structural similarity between viral proteins expressed in heart tissue during cardiotropic infection and degraded MyHC peptides could cause inappropriate recognition of MyHC by the immune system or molecular mimicry.61 Indeed, the phenotype of mice that overexpress IFN-γ is remarkably similar to that of mice that are immunized with cardiac α-myosin.62 In addition, in patients, the presence of autoimmune antibodies against sarcomeric proteins is predictive of the development of DCM.63 IFN-γ–induced increases in immunoproteasome activity and MyHC degradation in cardiac myocytes identified in our studies could promote such an effect.
Conclusions
We show that exposure to the proinflammatory cytokine IFN-γ caused atrophy of cardiomyocytes and stimulated the selective degradation of cardiac MyHC protein in a proteasome-dependent manner. Although specific components of the constitutive UPS pathway were not up-regulated, IFN-γ caused a striking increase in immunoproteasome activity in cardiac myocytes. Immunoproteasomes have altered cleavage site preferences and protein cleavage rates to increase antigenic peptide presentation and therefore may be responsible for the specific degradation of MyHC in this model. In fact, mice immunized with cardiac α-MyHC peptide fragments develop severe myocarditis,63 implying a potential positive feedback loop whereby inflammation and high IFN-γ levels increase MyHC peptide fragment generation, resulting in a persistent immune response and further inflammation and IFN-γ secretion. Although caspases have been implicated in proteolysis during atrophy by shuttling proteins to the UPS,64 our data support a caspase-independent mechanism of MyHC degradation. Our data may be extensible to patients who receive IFN-γ as therapeutics for osteoporosis or chronic granulomatous disease.28 In addition, this in vitro system may be useful as a model to study the mechanisms responsible for the specific degradation of MyHC. The data presented here provide new insights into the molecular mechanisms responsible for the cardiac phenotypes observed in many inflammatory diseases.
Acknowledgments
We thank Ann Robinson for isolating the NRVMs used in these studies and Dr. Larry Dick (Millennium Pharmaceuticals) for his helpful discussion.
Footnotes
Supported by NIH grant 2R01HL050560 (L.A.L.), American Heart Association predoctoral fellowship08100327Z (P.F.C.), and American Heart Association postdoctoral fellowship11POST7780011 (P.A.H.). Bortezomib and Ac-PAL-AMC were provided by Dr. Larry Dick (Millennium Pharmaceuticals, Inc).
References
- 1.Zee-Cheng C.S., Tsai C.C., Palmer D.C., Codd J.E., Pennington D.G., Williams G.A. High incidence of myocarditis by endomyocardial biopsy in patients with idiopathic congestive cardiomyopathy. J Am Coll Cardiol. 1984;3:63–70. doi: 10.1016/s0735-1097(84)80431-3. [DOI] [PubMed] [Google Scholar]
- 2.Cunha-Neto E., Dzau V.J., Allen P.D., Stamatiou D., Benvenutti L., Higuchi M.L., Koyama N.S., Silva J.S., Kalil J., Liew C.C. Cardiac gene expression profiling provides evidence for cytokinopathy as a molecular mechanism in Chagas' disease cardiomyopathy. Am J Pathol. 2005;167:305–313. doi: 10.1016/S0002-9440(10)62976-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taqueti V.R., Mitchell R.N., Lichtman A.H. Protecting the pump: controlling myocardial inflammatory responses. Annu Rev Physiol. 2006;68:67–95. doi: 10.1146/annurev.physiol.68.040104.124611. [DOI] [PubMed] [Google Scholar]
- 4.Feldman A.M., McNamara D. Myocarditis. N Engl J Med. 2000;343:1388–1398. doi: 10.1056/NEJM200011093431908. [DOI] [PubMed] [Google Scholar]
- 5.Kearney M.T., Cotton J.M., Richardson P.J., Shah A.M. Viral myocarditis and dilated cardiomyopathy: mechanisms, manifestations, and management. Postgrad Med J. 2001;77:4–10. doi: 10.1136/pmj.77.903.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gupta S., Markham D.W., Drazner M.H., Mammen P.P. Fulminant myocarditis. Nat Clin Pract Cardiovasc Med. 2008;5:693–706. doi: 10.1038/ncpcardio1331. [DOI] [PubMed] [Google Scholar]
- 7.Goodwin J.F. Cardiomyopathies and specific heart muscle diseases: Definitions, terminology, classifications and new and old approaches. Postgrad Med J. 1992;68(Suppl 1):S3–S6. [PubMed] [Google Scholar]
- 8.Bowles N.E., Ni J., Kearney D.L., Pauschinger M., Schultheiss H.P., McCarthy R., Hare J., Bricker J.T., Bowles K.R., Towbin J.A. Detection of viruses in myocardial tissues by polymerase chain reaction: Evidence of adenovirus as a common cause of myocarditis in children and adults. J Am Coll Cardiol. 2003;42:466–472. doi: 10.1016/s0735-1097(03)00648-x. [DOI] [PubMed] [Google Scholar]
- 9.Bowles N.E., Kearney D.L., Ni J., Perez-Atayde A.R., Kline M.W., Bricker J.T., Ayres N.A., Lipshultz S.E., Shearer W.T., Towbin J.A. The detection of viral genomes by polymerase chain reaction in the myocardium of pediatric patients with advanced HIV disease. J Am Coll Cardiol. 1999;34:857–865. doi: 10.1016/s0735-1097(99)00264-8. [DOI] [PubMed] [Google Scholar]
- 10.Barbaro G., Di Lorenzo G., Grisorio B., Barbarini G. Incidence of dilated cardiomyopathy and detection of HIV in myocardial cells of HIV-positive patients: Gruppo Italiano per lo Studio Cardiologico dei Pazienti Affetti da AIDS. N Engl J Med. 1998;339:1093–1099. doi: 10.1056/NEJM199810153391601. [DOI] [PubMed] [Google Scholar]
- 11.Gluck B., Schmidtke M., Merkle I., Stelzner A., Gemsa D. Persistent expression of cytokines in the chronic stage of CVB3-induced myocarditis in NMRI mice. J Mol Cell Cardiol. 2001;33:1615–1626. doi: 10.1006/jmcc.2001.1416. [DOI] [PubMed] [Google Scholar]
- 12.Schroder K., Hertzog P.J., Ravasi T., Hume D.A. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 13.Kuhl U., Pauschinger M., Noutsias M., Seeberg B., Bock T., Lassner D., Poller W., Kandolf R., Schultheiss H.P. High prevalence of viral genomes and multiple viral infections in the myocardium of adults with “idiopathic” left ventricular dysfunction. Circulation. 2005;111:887–893. doi: 10.1161/01.CIR.0000155616.07901.35. [DOI] [PubMed] [Google Scholar]
- 14.Reifenberg K., Lehr H.A., Torzewski M., Steige G., Wiese E., Kupper I., Becker C., Ott S., Nusser P., Yamamura K., Rechtsteiner G., Warger T., Pautz A., Kleinert H., Schmidt A., Pieske B., Wenzel P., Munzel T., Lohler J. Interferon-gamma induces chronic active myocarditis and cardiomyopathy in transgenic mice. Am J Pathol. 2007;171:463–472. doi: 10.2353/ajpath.2007.060906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shioi T., Matsumori A., Sasayama S. Persistent expression of cytokine in the chronic stage of viral myocarditis in mice. Circulation. 1996;94:2930–2937. doi: 10.1161/01.cir.94.11.2930. [DOI] [PubMed] [Google Scholar]
- 16.Maass A.H., Buvoli M. Cardiomyocyte preparation, culture, and gene transfer. Methods Mol Biol. 2007;366:321–330. doi: 10.1007/978-1-59745-030-0_18. [DOI] [PubMed] [Google Scholar]
- 17.Lau S.L., Yuen M.L., Kou C.Y., Au K.W., Zhou J., Tsui S.K. Interferons induce the expression of IFITM1 and IFITM3 and suppress the proliferation of rat neonatal cardiomyocytes. J Cell Biochem. 2012;113:841–847. doi: 10.1002/jcb.23412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Patten M., Kramer E., Bunemann J., Wenck C., Thoenes M., Wieland T., Long C. Endotoxin and cytokines alter contractile protein expression in cardiac myocytes in vivo. Pflugers Arch. 2001;442:920–927. doi: 10.1007/s004240100612. [DOI] [PubMed] [Google Scholar]
- 19.Stephanou A., Brar B.K., Scarabelli T.M., Jonassen A.K., Yellon D.M., Marber M.S., Knight R.A., Latchman D.S. Ischemia-induced STAT-1 expression and activation play a critical role in cardiomyocyte apoptosis. J Biol Chem. 2000;275:10002–10008. doi: 10.1074/jbc.275.14.10002. [DOI] [PubMed] [Google Scholar]
- 20.Cosper P.F., Leinwand L.A. Myosin heavy chain is not selectively decreased in murine cancer cachexia. Int J Cancer. 2012;130:2722–2727. doi: 10.1002/ijc.26298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rasband W. US National Institutes of Health; Bethesda, Maryland: 1997–2011. ImageJ. [Google Scholar]
- 22.Stauffer B.L., Konhilas J.P., Luczak E.D., Leinwand L.A. Soy diet worsens heart disease in mice. J Clin Invest. 2006;116:209–216. doi: 10.1172/JCI24676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Powell S.R., Davies K.J., Divald A. Optimal determination of heart tissue 26S-proteasome activity requires maximal stimulating ATP concentrations. J Mol Cell Cardiol. 2007;42:265–269. doi: 10.1016/j.yjmcc.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adams J., Behnke M., Chen S., Cruickshank A.A., Dick L.R., Grenier L., Klunder J.M., Ma Y.T., Plamondon L., Stein R.L. Potent and selective inhibitors of the proteasome: dipeptidyl boronic acids. Bioorg Med Chem Lett. 1998;8:333–338. doi: 10.1016/s0960-894x(98)00029-8. [DOI] [PubMed] [Google Scholar]
- 25.Zhao J., Brault J.J., Schild A., Cao P., Sandri M., Schiaffino S., Lecker S.H., Goldberg A.L. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007;6:472–483. doi: 10.1016/j.cmet.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 26.Kandolf R., Canu A., Hofschneider P.H. Coxsackie B3 virus can replicate in cultured human foetal heart cells and is inhibited by interferon. J Mol Cell Cardiol. 1985;17:167–181. doi: 10.1016/s0022-2828(85)80019-5. [DOI] [PubMed] [Google Scholar]
- 27.Yoneda S., Senda K., Hayashi K. Experimental study of virus myocarditis in culture. Jpn Circ J. 1979;43:1048–1054. doi: 10.1253/jcj.43.1048. [DOI] [PubMed] [Google Scholar]
- 28.Younes H.M., Amsden B.G. Interferon-gamma therapy: evaluation of routes of administration and delivery systems. J Pharm Sci. 2002;91:2–17. doi: 10.1002/jps.10007. [DOI] [PubMed] [Google Scholar]
- 29.Matsakas A. Molecular advances shed light on cardiac myosin heavy chain expression in health and disease. Exp Physiol. 2009;94:1161–1162. doi: 10.1113/expphysiol.2009.050211. [DOI] [PubMed] [Google Scholar]
- 30.Chizzonite R.A., Zak R. Regulation of myosin isoenzyme composition in fetal and neonatal rat ventricle by endogenous thyroid hormones. J Biol Chem. 1984;259:12628–12632. [PubMed] [Google Scholar]
- 31.Hilfiker-Kleiner D., Hilfiker A., Schieffer B., Engel D., Mann D.L., Wollert K.C., Drexler H. TNFalpha decreases alphaMHC expression by a NO mediated pathway: role of E-box transcription factors for cardiomyocyte specific gene regulation. Cardiovasc Res. 2002;53:460–469. doi: 10.1016/s0008-6363(01)00463-1. [DOI] [PubMed] [Google Scholar]
- 32.Kawano H., Okada R., Kawano Y., Sueyoshi N., Shirai T. Apoptosis in acute and chronic myocarditis. Jpn Heart J. 1994;35:745–750. doi: 10.1536/ihj.35.745. [DOI] [PubMed] [Google Scholar]
- 33.Acharyya S., Ladner K.J., Nelsen L.L., Damrauer J., Reiser P.J., Swoap S., Guttridge D.C. Cancer cachexia is regulated by selective targeting of skeletal muscle gene products. J Clin Invest. 2004;114:370–378. doi: 10.1172/JCI20174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Price S.R., England B.K., Bailey J.L., Van Vreede K., Mitch W.E. Acidosis and glucocorticoids concomitantly increase ubiquitin and proteasome subunit mRNAs in rat muscle. Am J Physiol. 1994;267:C955–C960. doi: 10.1152/ajpcell.1994.267.4.C955. [DOI] [PubMed] [Google Scholar]
- 35.Li Y.P., Lecker S.H., Chen Y., Waddell I.D., Goldberg A.L., Reid M.B. TNF-alpha increases ubiquitin-conjugating activity in skeletal muscle by up-regulating UbcH2/E220k. FASEB J. 2003;17:1048–1057. doi: 10.1096/fj.02-0759com. [DOI] [PubMed] [Google Scholar]
- 36.Clarke B.A., Drujan D., Willis M.S., Murphy L.O., Corpina R.A., Burova E., Rakhilin S.V., Stitt T.N., Patterson C., Latres E., Glass D.J. The E3 ligase MuRF1 degrades myosin heavy chain protein in dexamethasone-treated skeletal muscle. Cell Metab. 2007;6:376–385. doi: 10.1016/j.cmet.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 37.Razeghi P., Taegtmeyer H. Hypertrophy and atrophy of the heart: the other side of remodeling. Ann N Y Acad Sci. 2006;1080:110–119. doi: 10.1196/annals.1380.011. [DOI] [PubMed] [Google Scholar]
- 38.Cosper P.F., Leinwand L.A. Cancer causes cardiac atrophy and autophagy in a sexually dimorphic manner. Cancer Res. 2010;71:1710–1720. doi: 10.1158/0008-5472.CAN-10-3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Linke W.A. Sense and stretchability: the role of titin and titin-associated proteins in myocardial stress-sensing and mechanical dysfunction. Cardiovasc Res. 2008;77:637–648. doi: 10.1016/j.cardiores.2007.03.029. [DOI] [PubMed] [Google Scholar]
- 40.Barth S., Glick D., Macleod K.F. Autophagy: assays and artifacts. J Pathol. 2010;221:117–124. doi: 10.1002/path.2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seglen P.O., Gordon P.B. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci U S A. 1982;79:1889–1892. doi: 10.1073/pnas.79.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Willis M.S., Patterson C. Into the heart: the emerging role of the ubiquitin-proteasome system. J Mol Cell Cardiol. 2006;41:567–579. doi: 10.1016/j.yjmcc.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 43.Liu Z., Miers W.R., Wei L., Barrett E.J. The ubiquitin-proteasome proteolytic pathway in heart vs skeletal muscle: effects of acute diabetes. Biochem Biophys Res Commun. 2000;276:1255–1260. doi: 10.1006/bbrc.2000.3609. [DOI] [PubMed] [Google Scholar]
- 44.Meiners S., Dreger H., Fechner M., Bieler S., Rother W., Gunther C., Baumann G., Stangl V., Stangl K. Suppression of cardiomyocyte hypertrophy by inhibition of the ubiquitin-proteasome system. Hypertension. 2008;51:302–308. doi: 10.1161/HYPERTENSIONAHA.107.097816. [DOI] [PubMed] [Google Scholar]
- 45.Nowis D., Maczewski M., Mackiewicz U., Kujawa M., Ratajska A., Wieckowski M.R., Wilczynski G.M., Malinowska M., Bil J., Salwa P., Bugajski M., Wojcik C., Sinski M., Abramczyk P., Winiarska M., Dabrowska-Iwanicka A., Duszynski J., Jakobisiak M., Golab J. Cardiotoxicity of the anticancer therapeutic agent bortezomib. Am J Pathol. 2010;176:2658–2668. doi: 10.2353/ajpath.2010.090690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Khan S., van den Broek M., Schwarz K., de Giuli R., Diener P.A., Groettrup M. Immunoproteasomes largely replace constitutive proteasomes during an antiviral and antibacterial immune response in the liver. J Immunol. 2001;167:6859–6868. doi: 10.4049/jimmunol.167.12.6859. [DOI] [PubMed] [Google Scholar]
- 47.Zu L., Bedja D., Fox-Talbot K., Gabrielson K.L., Van Kaer L., Becker L.C., Cai Z.P. Evidence for a role of immunoproteasomes in regulating cardiac muscle mass in diabetic mice. J Mol Cell Cardiol. 2010;49:5–15. doi: 10.1016/j.yjmcc.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Groettrup M., Khan S., Schwarz K., Schmidtke G. Interferon-gamma inducible exchanges of 20S proteasome active site subunits: why? Biochimie. 2001;83:367–372. doi: 10.1016/s0300-9084(01)01251-2. [DOI] [PubMed] [Google Scholar]
- 49.Szalay G., Meiners S., Voigt A., Lauber J., Spieth C., Speer N., Sauter M., Kuckelkorn U., Zell A., Klingel K., Stangl K., Kandolf R. Ongoing coxsackievirus myocarditis is associated with increased formation and activity of myocardial immunoproteasomes. Am J Pathol. 2006;168:1542–1552. doi: 10.2353/ajpath.2006.050865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lecker S.H., Jagoe R.T., Gilbert A., Gomes M., Baracos V., Bailey J., Price S.R., Mitch W.E., Goldberg A.L. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004;18:39–51. doi: 10.1096/fj.03-0610com. [DOI] [PubMed] [Google Scholar]
- 51.Esfandiarei M., McManus B.M. Molecular biology and pathogenesis of viral myocarditis. Annu Rev Pathol. 2008;3:127–155. doi: 10.1146/annurev.pathmechdis.3.121806.151534. [DOI] [PubMed] [Google Scholar]
- 52.Xiong D., Yajima T., Lim B.K., Stenbit A., Dublin A., Dalton N.D., Summers-Torres D., Molkentin J.D., Duplain H., Wessely R., Chen J., Knowlton K.U. Inducible cardiac-restricted expression of enteroviral protease 2A is sufficient to induce dilated cardiomyopathy. Circulation. 2007;115:94–102. doi: 10.1161/CIRCULATIONAHA.106.631093. [DOI] [PubMed] [Google Scholar]
- 53.Pruznak A.M., Hong-Brown L., Lantry R., She P., Frost R.A., Vary T.C., Lang C.H. Skeletal and cardiac myopathy in HIV-1 transgenic rats. Am J Physiol Endocrinol Metab. 2008;295:E964–E973. doi: 10.1152/ajpendo.90482.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scaglione J., Puyo A.M., Dupuy H.A., Postan M., Fernandez B.E. Behavior of atrial natriuretic factor in an experimental model of Trypanosoma cruzi infection in rats. J Parasitol. 2001;87:923–926. doi: 10.1645/0022-3395(2001)087[0923:BOANFI]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 55.Torzewski M., Wenzel P., Kleinert H., Becker C., El-Masri J., Wiese E., Brandt M., Pautz A., Twardowski L., Schmitt E., Munzel T., Reifenberg K. Chronic inflammatory cardiomyopathy of interferon gamma-overexpressing transgenic mice is mediated by tumor necrosis factor-alpha. Am J Pathol. 2012;180:73–81. doi: 10.1016/j.ajpath.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 56.Weekes J., Morrison K., Mullen A., Wait R., Barton P., Dunn M.J. Hyperubiquitination of proteins in dilated cardiomyopathy. Proteomics. 2003;3:208–216. doi: 10.1002/pmic.200390029. [DOI] [PubMed] [Google Scholar]
- 57.Kobayashi S., Mao K., Zheng H., Wang X., Patterson C., O'Connell T.D., Liang Q. Diminished GATA4 protein levels contribute to hyperglycemia-induced cardiomyocyte injury. J Biol Chem. 2007;282:21945–21952. doi: 10.1074/jbc.M703048200. [DOI] [PubMed] [Google Scholar]
- 58.Cohen S., Brault J.J., Gygi S.P., Glass D.J., Valenzuela D.M., Gartner C., Latres E., Goldberg A.L. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation. J Cell Biol. 2009;185:1083–1095. doi: 10.1083/jcb.200901052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Campos Y., Qiu X., Zanoteli E., Moshiach S., Vergani N., Bongiovanni A., Harris A.J., d'Azzo A. Ozz-E3 ubiquitin ligase targets sarcomeric embryonic myosin heavy chain during muscle development. PLoS One. 2010;5:e9866. doi: 10.1371/journal.pone.0009866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yewdell J.W. The seven dirty little secrets of major histocompatibility complex class I antigen processing. Immunol Rev. 2005;207:8–18. doi: 10.1111/j.0105-2896.2005.00309.x. [DOI] [PubMed] [Google Scholar]
- 61.Fairweather D., Kaya Z., Shellam G.R., Lawson C.M., Rose N.R. From infection to autoimmunity. J Autoimmun. 2001;16:175–186. doi: 10.1006/jaut.2000.0492. [DOI] [PubMed] [Google Scholar]
- 62.Afanasyeva M., Wang Y., Kaya Z., Park S., Zilliox M.J., Schofield B.H., Hill S.L., Rose N.R. Experimental autoimmune myocarditis in A/J mice is an interleukin-4-dependent disease with a Th2 phenotype. Am J Pathol. 2001;159:193–203. doi: 10.1016/S0002-9440(10)61685-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Caforio A.L., Mahon N.J., Tona F., McKenna W.J. Circulating cardiac autoantibodies in dilated cardiomyopathy and myocarditis: pathogenetic and clinical significance. Eur J Heart Fail. 2002;4:411–417. doi: 10.1016/s1388-9842(02)00010-7. [DOI] [PubMed] [Google Scholar]
- 64.Du J., Wang X., Miereles C., Bailey J.L., Debigare R., Zheng B., Price S.R., Mitch W.E. Activation of caspase-3 is an initial step triggering accelerated muscle proteolysis in catabolic conditions. J Clin Invest. 2004;113:115–123. doi: 10.1172/JCI200418330. [DOI] [PMC free article] [PubMed] [Google Scholar]