

Abstract
Akt is a serine/threonine protein kinase that is activated by a variety of growth factors or cytokines in a phosphatidylinositol 3-kinase–dependent manner. By using a conditional transgenic system in which Akt signaling can be turned on or off in the adult heart, we previously showed that short-term Akt activation induces a physiological form of cardiac hypertrophy with enhanced coronary angiogenesis and maintained contractility. Here we tested the hypothesis that induction of physiological hypertrophy by short-term Akt activation might improve contractile function in failing hearts. When Akt signaling transiently was activated in murine hearts with impaired contractility, induced by pressure overload or doxorubicin treatment, contractile dysfunction was attenuated in both cases. Importantly, improvement of contractility was observed before the development of cardiac hypertrophy, indicating that Akt activation improves contractile function independently of its growth-promoting effects. To gain mechanistic insights into Akt-mediated positive inotropic effects, transcriptional profiles in the heart were determined in a pressure overload–induced heart failure model. Biological network analysis of differentially expressed transcripts revealed significant alterations in the expression of genes associated with cell death, and these alterations were reversed by short-term Akt activation. Thus, short-term Akt activation improves contractile function in failing hearts. This beneficial effect of Akt on contractility is hypertrophy-independent and may be mediated in part by inhibition of cell death associated with heart failure.
Heart failure is a major cause of mortality and morbidity worldwide.1–4 One common feature of heart failure is hypertrophy of individual cardiac muscle cells in the myocardium.5–8 Cardiac hypertrophy observed in patients with hypertension, myocardial infarction, or valvular heart disease is considered to be an adaptive response because hypertrophy can normalize the increase in wall stress induced by mechanical overload. However, increased cardiac mass is clinically associated with increased morbidity and mortality,9 and sustained overload eventually leads to contractile dysfunction and heart failure.10–12 Thus, stress-induced or pathologic cardiac hypertrophy appears to be detrimental for the heart. Conversely, normal postnatal growth of the heart or exercise-induced cardiac growth also occurs through hypertrophy of individual cardiac muscle cells. These forms of so-called physiological cardiac hypertrophy are associated with normal or enhanced contractile function and are morphologically and molecularly distinct from stress-induced hypertrophy.13,14
Akt is a serine/threonine protein kinase that mediates cellular growth responses in multiple organisms and cell types.15,16 There are three Akt genes in mammalian genomes (Akt1/PKBα, Akt2/PKBβ, and Akt3/PKBγ). Loss of Akt1 gene in mice leads to general growth retardation and spontaneous apoptosis in restricted cell types.17,18 Akt2 gene disruption results in abnormal glucose metabolism and mild growth deficiency,19,20 whereas Akt3 knockout mice show a selective growth defect of the brain.21,22 Furthermore, these genes display a considerable degree of functional overlap, exemplified by the combined deletion of Akt1 and Akt2 genes, which results in perinatal lethality with severe growth retardation and multiple developmental defects23 and by Akt1/Akt3 double-knockout mice, which are embryonic-lethal owing to a placental defect.24 In the heart, Akt is an important positive regulator of normal postnatal cardiac growth25 and also is activated by exercise training,26–28 by pressure overload,29 and is activated in diseased human hearts.30 Overexpression of activated Akt1 or Akt3 in the heart is sufficient to induce cardiac hypertrophy and, in some cases, heart failure in transgenic (TG) mice.31–34 These data suggest that Akt-dependent signaling pathways are involved in both physiologic and pathologic cardiac growth.
By using a conditional TG system, in which the expression of the Akt transgene can be turned on or off at desired time points, we previously showed that short-term Akt activation induces physiologic hypertrophy but long-term Akt activation leads to pathologic hypertrophy and heart failure.35 In cardiac hypertrophy induced by short-term Akt activation, contractile function was preserved. Because physiologic hypertrophy can be adaptive by reducing wall stress without adverse effects on contractile function, we tested the hypothesis that induction of physiologic hypertrophy by short-term Akt activation might attenuate contractile dysfunction in failing hearts. We show that transient Akt activation improves contractile function in two different heart failure models and we explored the potential mechanisms of Akt-mediated positive inotropic effects by transcriptome analysis.
Materials and Methods
Animals
Generation, genotyping, and the initial characterization of cardiac-specific inducible Akt TG mice have been described previously.35 In brief, two lines of TG mice (tet-Akt and αMHC-tTA) were crossed to generate double-TG (DTG) mice. When DTG mice are treated with doxycycline (DOX), the expression of Akt transgene is turned off. When DOX is withdrawn, Akt transgene expression is induced specifically in the heart. αMHC-tTA single-TG mice were used as controls and treated with DOX in the same manner as DTG animals.
Heart Failure Models
For pressure overload–induced heart failure models, mice were subjected to ascending aorta constriction (AAC) at the age of 8 weeks as described previously.36 DOX was withdrawn at the age of 12 weeks (4 weeks after AAC) to induce Akt transgene expression in the heart. DOX had been given continuously since before birth. Echocardiography was performed at the age of 8 (before AAC), 12 (4 weeks after AAC), 13 (1 week after Akt activation), and 14 weeks (2 weeks after Akt activation). For Adriamycin (ADR; Sigma-Aldrich, St. Louis, MO)-induced heart failure models, ADR was administered to 12-week-old mice in 3 equal intraperitoneal injections for 2 weeks (8 mg/kg for a single injection, cumulative dose of 24 mg/kg body weight). DOX was withdrawn at the age of 16 weeks (4 weeks after initial ADR injection) to induce Akt transgene in the heart. DOX had been given continuously since before birth. Echocardiography was performed at the age of 12 (before ADR injection), 16 (4 weeks after initial ADR injection), 17 (1 week after Akt activation), and 18 weeks (2 weeks after Akt activation).
Echocardiography
Transthoracic echocardiography was performed with an ACUSON 256 sector scanner (Siemens, Mountain View, CA) equipped with a 13-MHz transducer as described previously.35
Microarray Analysis
Transcriptional profiles were determined in pressure overload–induced heart failure models. Mice were sacrificed at predetermined time points. Hearts were excised and snap-frozen in liquid nitrogen. RNA extraction, cDNA synthesis, biotinylated cRNA preparation, and hybridization of cRNA to microarrays (GeneChip Mouse Expression Set 430; Affymetrix, Santa Clara, CA) were performed as described.37–39 Data were obtained at three time points [before AAC, 4 weeks after AAC, and 6 weeks after AAC (2 weeks after Akt activation)], and four independent sets of hybridizations were performed at each time point. An automated analysis of related transcripts was performed using Ingenuity Pathways Analysis (Redwood City, CA), a web-delivered application that evaluates biological networks. Data analysis was performed as described.37–39 In addition, automated analysis of groups of biologically related genes was performed using GenMAPP and MAPPFinder version 2.0 (http://www.genmapp.org) as described.37 MAPPFinder is a tool that integrates the annotations of the Gene Ontology Project (http://www.geneontology.org) with the free software package Gen-MAPP. MAPPFinder identifies Gene Ontology Project terms with overrepresented numbers of gene-expression charges. GenMAPP generates graphic files within which gene expression data can be viewed in the context of the biological knowledge contained in the Gene Ontology Project database.
qPCR Analysis
Expression levels of selected transcripts were examined by quantitative real-time PCR (qPCR) as described.37 The primers in Table 1 were used for qPCR analysis.
Table 1.
qPCR Analysis. Expression levels of selected transcripts were examined by qPCR as described.37
| Transcript | Forward primer | Reverse primer |
|---|---|---|
| Actin α 1 | 5'-TGCGCGACATCAAAGAGAAG-3' | 5'-ACCGATAAAGGAAGGCTGGAA-3' |
| ATP-binding cassette, sub-family A (ABC1), member 4 | 5'-GTCCTCAGTTTGATGCAATCGA-3' | 5'-CAGGCGGTCAGCGTAGAGA-3' |
| BCL2-like 11 | 5'-GCCCCTACCTCCCTACAGACA-3' | 5'-CCGCAGCTCCTGTGCAAT-3' |
| Bone morphogenetic protein 8b | 5'-GCCCCTCGAACAGCAAGAC-3' | 5'-TGCCTGCGGCAAACTTCT-3' |
| Calsequestrin 1 | 5'-CAAGGTGGCAAAGAAGCTGACT-3' | 5'-CCTCCACGAAGCTCACAATCTC-3' |
| Carboxylesterase 3 | 5'-CAGCCTGTGGCTGTTTTCCT-3' | 5'-CCTGCCCTCCAACAGCAT-3' |
| Cartilage intermediate layer protein, nucleotide pyrophosphohydrolase | 5'-GATGCCCAAGACTAGCCTGAAGT-3' | 5'-GGTTTCCCCGTGGCTTTG-3' |
| Cullin 5 | 5'-AGACTCCAGGACAGTGCAATGA-3' | 5'-CCTCTCTGTTGAGTCCAAGTATGC-3' |
| Cyclin B2 | 5'-CACTTTAGCCAAGTACCTGATGGA-3' | 5'-GCCTGTGTAATACTGCTGCTTCA-3' |
| Cytokine receptor-like factor 1 | 5'-TCACCACCAGCTCTCAAGGATT-3' | 5'-GCACTTGGACGAAGTAAACG-3' |
| Fast skeletal myosin alkali light chain | 5'-ACAAGGACCAGGGAGGTTATGA-3' | 5'-CCAGAGTGGCGAGGACATG-3' |
| Fibroblast growth factor 4 | 5'-CTACCTGCTGGGCCTCAAAA-3' | 5'-GTAAAGAAAGGCACACCGAAGAG-3' |
| Fibrillin 1 | 5'-GCCCTGCTGGACACAAATTTAA-3' | 5'-CCGTTTGCCAGAGCTGTGTA-3' |
| Follistatin related protein | 5'-CAAGTGCCTCAACCCATCCT-3' | 5'-TCCTGTCTTCTCCTCCTCTGTGT-3' |
| Gap junction membrane channel protein α1 | 5'-CAGCCTGAGTGCGGTCTACAC-3' | 5'-AAGGACACCACCAGCATGAAG-3' |
| Glucosaminyl (N-acetyl) transferase 1, core 2 | 5'-GCTGGAGATGATCCTTACAGCAA-3' | 5'-GGCACAGTCACGGGTCATG-3' |
| Histocompatibility 2, L region | 5'-CGACGGCTGCGATTACATC-3' | 5'-CGTTCCCGTTCTTCAGGTATCT-3' |
| Insulin-like growth factor binding protein 1 | 5'-CCGCGGATGAGCTTTCTG-3' | 5'-ATTTCTTGAGGTCGGCGATCT-3' |
| Insulin-like growth factor binding protein 5 | 5'-GAAGGACCGCAGAAAGAAGCT-3' | 5'-GTTCGGATTCCTGTCTCATCTCA-3' |
| Mitogen activated protein kinase 13 | 5'-ACTGGCTCACCCCTTCTTTGA-3' | 5'-CCACGCTGAGTTTCTCATGTTC-3' |
| Myomesin 2 | 5'-CCTGCCTATGACCTGACGTTCT-3' | 5'-TGGCATTCACTGCTCGAATTC-3' |
| Myotrophin | 5'-GCTGTCTATGAGGGTCATGTTTCC-3' | 5'-GCTTTGATTGCCTGGTTGTCA-3' |
| Ninjurin 1 | 5'-GGTGGAGCAGGGCAATGA-3' | 5'-TGACCACGACGATGATGAAAAC-3' |
| Phospholipase A2, group IIC | 5'-CCTCCACCCTCAGCAGTTTCT-3' | 5'-TCACCGTCCCATTGACAATG-3' |
| Procollagen, type V, α 1 | 5'-GATGGCATCCGAGGTCTGAA-3' | 5'-CACGCCAAGCTTTCCCTTT-3' |
| Procollagen, type VIII, α 1 | 5'-CCAGGGAGAGTATCTGCCAGATA-3' | 5'-AAGGTACAGTCAGCTCGGCAGTA-3' |
| Prolactin receptor | 5'-GCCTTCCACATCCCTGAGATC-3' | 5'-CATCGGCAATGCTGTGGTAA-3' |
| Prostaglandin D2 synthase | 5'-TCCGGGAGAAGAAAGCTGTATT-3' | 5'-CTGGTTTTTCCTGAGGAAGGTAGAG-3' |
| SRY-box containing gene 4 | 5'-CAAGCGGCTAGGCAAACG-3' | 5'-GTTGCCCGACTTCACCTTCTT-3' |
| Tenascin C | 5'-ACAGCTACCGACGGGATCTTC-3' | 5'-TTGTCAACTTCCGGTTCAGCTT-3' |
| Thrombospondin 1 | 5'-CAACGTCCTTCTTACCCTTGACA-3' | 5'-CCACAGATAGCTTGGAGGTCCTT-3' |
| Thymoma viral proto-oncogene 1 | 5'-CCTTCCTTACGGCCCTCAA-3' | 5'-ACACAATCTCCGCACCATAGAA-3' |
| Zinc finger protein 352 | 5'-GCCATTGGTTTCCATTTTGG-3' | 5'-AAGTCTCCCTGGTGTCAACTCTTG-3' |
Results
Improvement of Contractile Function by Transient Akt Activation
To examine the effects of inducible Akt activation in failing hearts, mice were subjected to AAC at the age of 8 weeks and Akt transgene was induced by withdrawing DOX at the age of 12 weeks, when contractile dysfunction was apparent by echocardiography (Figure 1). At the age of 13 weeks (1 week after Akt activation), there was a significant increase in the percentage of fractional shortening in DTG animals compared with control animals. This improvement of contractility was maintained at 14 weeks of age (2 weeks after Akt activation). Of note, there was a significant increase in posterior wall thickness in DTG mice compared with control animals at 14 weeks old but not at 13 weeks old. This is consistent with our previous observation that a significant increase in heart weight starts to be observed at day 10 after Akt transgene induction.35 These findings indicate that transient Akt activation in failing hearts leads to improvement of contractile function, and this positive inotropic effect of Akt activation is independent of its growth-promoting effect.
Figure 1.

Short-term Akt activation improves contractile function in pressure overload–induced heart failure. A: Temporal profile of DOX treatment, AAC operation, and echocardiography. B: Echocardiography. Top panels: representative M-mode recordings; bottom panels: posterior wall thickness (PWT), left ventricular end-diastolic dimension (LVDd), and percentage fractional shortening (%FS). C, control animals; D, DTG animals. *P < 0.01.
To examine whether this beneficial effect of Akt on contractility is observed in a different form of heart failure, Akt transiently was activated in failing hearts induced by ADR treatment. Mice received three injections of ADR once a week beginning at the age of 12 weeks. Akt transgene was induced by withdrawing DOX at the age of 16 weeks (Figure 2). At this time point, contractile dysfunction was apparent by echocardiography. As was the case with pressure overload–induced heart failure, transient Akt activation in cardiac muscle cells improved ADR-induced contractile function, and this beneficial effect of Akt was independent of its growth-promoting effect because there was a significant increase in the percentage of fractional shortening at the age of 17 weeks (1 week after Akt activation) when there was no increase in wall thickness. Taken together, Akt activation improves contractile function in two different models of heart failure independently of its growth-promoting effect on the myocardium.
Figure 2.

Short-term Akt activation improves contractile function in ADR-induced heart failure. A: Temporal profile of DOX treatment, ADR treatment, and echocardiography. B: Echocardiography. Top panels: representative M-mode recordings; bottom panels: posterior wall thickness (PWT), left ventricular end-diastolic dimension (LVDd), and percentage fractional shortening (%FS). C, control animals; D, DTG animals. *P < 0.05, **P < 0.01.
Microarray Analysis of Differentially Expressed Genes
To gain insights into Akt-mediated, positive inotropic effects, transcriptional profiles in the heart were determined in a pressure overload–induced heart failure model. RNA samples were prepared at 8 weeks (time point 1: before AAC), 12 weeks (time point 2: 4 weeks after AAC when there is apparent contractile dysfunction), and 14 weeks (time point 3: 2 weeks after Akt activation, when contractile function has been improved), and transcriptome analysis was performed. When time points 1 and 2 were compared, 381 genes differentially were regulated, of which 212 were up-regulated and 169 were down-regulated. When time points 2 and 3 were compared, 328 genes differentially were regulated, of which 162 genes were up-regulated and 166 genes were down-regulated (Figure 3A). Venn diagram analysis revealed that the expression of 619 transcripts differentially were regulated, of which 291 genes differentially were regulated only when heart failure was developing, 238 genes differentially were regulated only when heart failure was improving, and 90 genes differentially were regulated during both of these processes (Figure 3B). Differential expression of genes with a fold change more than +2.0 or less than −2.0 with a P value less than 0.01 were confirmed by qPCR and are listed in Tables 2 and 3. Categorization of differentially regulated genes using Gene Ontology Analysis revealed that genes associated with cell death and tissue development were up-regulated during heart failure progression, which was attenuated by Akt activation. Conversely, genes associated with cancer, carbohydrate metabolism, and cell cycle were up-regulated when heart failure was improving by Akt activation (Figure 3C). Biological network analysis of differentially expressed transcripts performed using Ingenuity Pathways Analysis revealed a set of genes, categorized as cell “death network,” with significant alterations in the expression levels at 12 weeks of age, 4 weeks after AAC, when there is apparent contractile dysfunction (see Supplemental Figure S1 at http://ajp.amjpathol.org). After 2 weeks of Akt activation, these alterations in the expression levels of a specific set of genes were attenuated or in some cases reversed (see Supplemental Figure S2 at http://ajp.amjpathol.org). Biological network analysis also identified a network of genes categorized as an “Akt pathway” that differentially are regulated 2 weeks after Akt activation, which support the authenticity of this type of analysis (see Supplemental Figures S3 and S4 at http://ajp.amjpathol.org). Names and symbols of the transcripts that are regulated differentially in Supplemental Figures S1 and S2 and in Supplemental Figures S3 and S4 are summarized in Supplemental Table S1 (available at http://ajp.amjpathol.org). Taken together, categorization and biological network analysis suggest that alterations in the expression levels of genes associated with cell death may be involved in the positive inotropic effects of Akt activation in the setting of heart failure.
Figure 3.
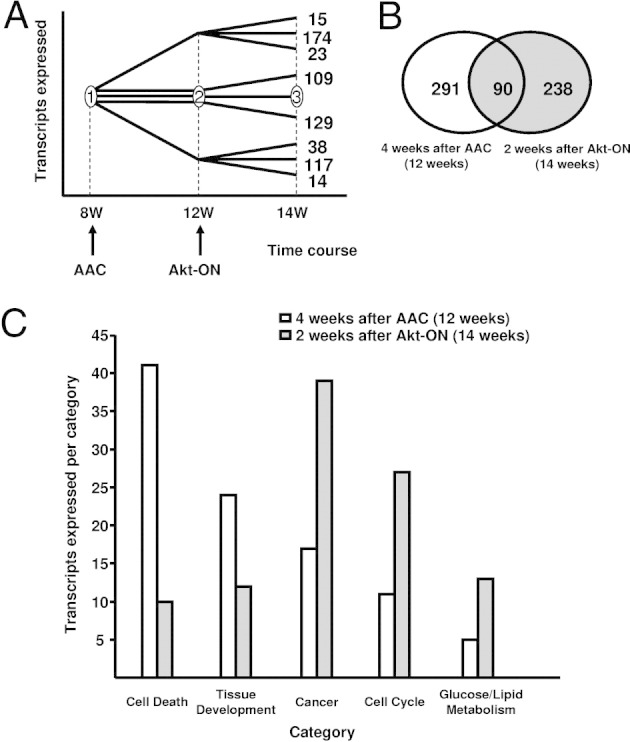
Transcriptome analysis of DTG hearts after AAC and Akt activation. A: Temporal profile evaluation of transcripts that are expressed differentially at time points 4 weeks after AAC (12 weeks) and 2 weeks after Akt-ON (14 weeks). B: Venn diagram representing the sets of transcripts that are regulated differentially 4 weeks after AAC (12 weeks) and 2 weeks after Akt-ON (14 weeks). C: Categorization of differentially expressed transcripts 4 weeks after AAC (12 weeks) and 2 weeks after Akt-ON (14 weeks) revealed by Ingenuity.
Table 2.
Summary of Transcripts Identified by Microarray and Confirmed by qPCR as Differentially Regulated Between 8 Weeks (Before AAC) and 12 Weeks (4 Weeks After AAC)
| Transcript name | Fold change (microarrays), P < 0.01 | Fold change (qPCR), P < 0.01 | Accession code | Transcript symbol |
|---|---|---|---|---|
| Histocompatibility 2, L region | +16.2 | +12.2 | BC023409 | H2-L |
| Glucosaminyl (N-acetyl) transferase 1, core 2 | +4.7 | +3.7 | NM173442 | Gcnt1 |
| Ninjurin 1 | +3.7 | +2.0 | NM013610 | Ninj1 |
| Actin α1 | +2.4 | +2.0 | NM009606 | Acta1 |
| Cullin 5 | +2.1 | +2.1 | NM027807 | Cul5 |
| Phospholipase A2, group IIC | −2.1 | −4.7 | NM008868 | Pla2g2c |
| BCL2-like 11 | −2.2 | −2.4 | NM009754 | Bcl2l11 |
| Carboxylesterase 3 | −2.5 | −3.0 | NM053200 | Ces3 |
| Fibroblast growth factor 4 | −2.6 | −3.1 | NM010202 | Fgf4 |
| Zinc finger protein 352 | −3.1 | −2.5 | NM153102 | Zfp352 |
| Bone morphogenetic protein 8b | −3.7 | −4.4 | NM007559 | Bmp8b |
| Insulin-like growth factor binding protein 1 | −4.3 | −5.9 | NM008341 | Igfbp1 |
| SRY-box containing gene 4 | −4.9 | −4.0 | NM009238 | Sox4 |
| ATP-binding cassette, subfamily A (ABC1), member 4 | −28.8 | −15.8 | NM007378 | Abca4 |
Table 3.
Summary of Transcripts Identified by Microarray and Confirmed by qPCR as Differentially Regulated Between 12 Weeks (4 Weeks After AAC) and 14 Weeks (2 Weeks After Akt-ON)
| Transcript name | Fold change (microarrays), P < 0.01 | Fold change (qPCR), P < 0.01 | Accession code | Transcript symbol |
|---|---|---|---|---|
| ATP-binding cassette, subfamily A (ABC1), member 4 | +13.8 | +11.4 | NM007378 | Abca4 |
| Mitogen-activated protein kinase 13 | +9.7 | +10.8 | NM011950 | Mapk13 |
| Cyclin B2 | +7.1 | +5.5 | NM007630 | Ccnb2 |
| Tenascin C | +6.5 | +4.8 | NM011607 | Tnc |
| Cartilage intermediate layer protein, nucleotide pyrophosphohydrolase | +6.1 | +6.5 | NM173385 | Cilp |
| Thymoma viral proto-oncogene 1 | +4.2 | +6.6 | NM009652 | Akt1 |
| Procollagen, type V, α1 | +3.5 | +4.6 | NM015734 | Col5a1 |
| Insulin-like growth factor binding protein 5 | +3.0 | +2.1 | NM010518 | Igfbp5 |
| Follistatin-related protein | +2.9 | +1.9 | NM008047 | Fstl |
| Bone morphogenetic protein 8b | +2.8 | +3.1 | NM007559 | Bmp8b |
| Thrombospondin 1 | +2.7 | +3.0 | NM011580 | Thbs1 |
| Cytokine receptor-like factor 1 | +2.7 | +3.1 | NM018827 | Crlf1 |
| Calsequestrin 1 | +2.5 | +3.5 | NM009813 | Casq1 |
| Zinc finger protein 352 | +2.3 | +2.9 | NM153102 | Zfp352 |
| Fast skeletal myosin alkali light chain | +2.2 | +2.4 | NM021285 | Myl1 |
| Procollagen, type VIII, alpha 1 | +2.2 | +2.8 | NM007739 | Col8a1 |
| Fibrillin 1 | +2.1 | +2.6 | NM007993 | Fbn1 |
| Myotrophin | +2.1 | +2.0 | NM008098 | Mtpn |
| Gap junction membrane channel protein α 1 | −2.4 | −5.5 | NM010288 | Gja1 |
| Prostaglandin D2 synthase | −3.2 | −5.2 | NM008963 | Ptgds |
| Carboxylesterase 3 | −5.2 | −3.2 | NM053200 | Ces3 |
| Glucosaminyl (N-acetyl) transferase 1, core 2 | −5.6 | −6.6 | NM173442 | Gcnt1 |
| Myomesin 2 | −10.8 | −17.0 | NM008664 | Myom2 |
Discussion
Unlike pathologic cardiac hypertrophy observed in patients with hypertension or valvular heart diseases, physiologic cardiac hypertrophy as observed in trained athletes or during normal postnatal growth is associated with maintained or enhanced contractile function.
By using an inducible TG system in which cardiac Akt signaling can be turned on or off at desired time points, we previously showed that short-term Akt activation in the adult heart induces a physiologic form of cardiac hypertrophy.35 Because cardiac hypertrophy can be beneficial for the heart through reduction of wall stress, we hypothesized that physiologic hypertrophy induced by short-term Akt activation might improve contractile function in failing hearts. To test this hypothesis, we used two different murine heart failure models (pressure overload– or ADR-induced heart failure models) and showed that transient Akt activation after the establishment of heart failure improved contractility in both cases. This experiment is relevant because it mimics the clinical situation in which therapeutic intervention is started after the onset of heart failure, and has been difficult to perform with conventional knockout or TG mice in which deletion or overexpression of a specific gene is initiated during early embryogenesis. It was rather unexpected, however, that the transient Akt activation attenuated contractile dysfunction at earlier time points when no apparent hypertrophy was observed. This clearly indicates that the beneficial effect of Akt on contractility is independent of its effect to promote physiologic cardiac hypertrophy. Thus, although transient Akt activation does improve contractile function in the failing heart, this improvement is not caused by a reduction in wall stress by Akt-mediated physiologic hypertrophy. Of note, short-term Akt activation does not affect contractile function when Akt is turned on in normal hearts,35 indicating that the beneficial effect of Akt is specific to failing hearts. This raises the question as to the mechanism by which transient Akt activation has positive inotropic effects in the setting of heart failure.
Previous studies have associated Akt activation with pathologic cardiac hypertrophy. It recently was shown in the canine congestive heart failure/cardiac recovery model that phosphatidylinositol 3Kγ and phosphatidylinositol 3Kα expressions increase during the congestive heart failure and cardiac recovery phases, respectively.40 The study went on to extrapolate that, based on the measured increase of phosphatidylinositol 3Kα/Akt during the experimental recovery phase, the phosphatidylinositol 3Kα/Akt signaling pathway potentially plays a role in compensatory cardiac hypertrophy. However, this and other studies linking Akt and pathologic cardiac hypertrophy generally were based on constitutively active forms of the Akt pathway. In contrast, our model indicates that contractile improvements after heart failure may not be completely linked with hypertrophic growth, as evidenced by the lack of hypertrophy (heart rate/body weight) seen in our model. We do recognize, however, that any direct comparison of experimental results must be viewed with caution because of differences in the experimental models. Therefore, echocardiography was chosen as an outcome marker because it is a well-established technique for measuring cardiac function and thus allows our work to be compared directly with previous works in which cardiac recovery was associated with hypertrophy.
In our previous study, we showed that short-term Akt activation enhances vascular endothelial growth factor secretion from the myocardium and promotes coronary angiogenesis, which is required for the maintenance of cardiac function during Akt-induced physiologic heart growth: blocking the vascular endothelial growth factor signal during short-term Akt activation results in early transition from physiologic to pathologic hypertrophy and contractile dysfunction.35 It also has been shown that myocardial capillary density is decreased in hearts subjected to pressure overload.41 Therefore, one possible explanation for the Akt-mediated positive inotropic effect is that Akt induces vascular endothelial growth factor expression in cardiac muscle cells, thereby increasing the otherwise reduced capillary density in failing hearts. A second possibility is that Akt modulates Ca2+ handling in cardiac myocytes and enhances contractility. It has been shown that Akt increases the protein expression levels of sarcoplasmic reticulum calcium ATPase 2 (SERCA2) in TG mice overexpressing constitutively active Akt42 or in rat hearts infected with Akt adenoviruses.43 Because overexpression of SERCA2 has been shown to improve contractile function in heart failure models,44 Akt may show positive inotropic effects by increasing the expression levels of SERCA2. Previous investigations,45 among others, have shown a link between activation of the phosphatidylinositol 3K/Akt pathway and SERCA2a regulation. Although the scope of our investigations did not specifically include measurements of SERCA2a or other calcium-regulating proteins, it seems more than likely that in light of the aforementioned investigations, Akt-dependent regulation of SERCA2a is a potential mechanism for affecting myocyte contractility. Moreover, the potential mechanisms responsible for our model's improved cardiac contractility will be the subject of our next series of investigations. A third possibility suggested by our transcriptome analysis is that Akt improves contractile function by suppressing cell death in the failing heart. Several lines of evidence support the notion of a causal role of apoptosis in heart failure. It was shown that even low levels of cardiomyocyte apoptosis, induced by procaspase-8, led to lethal dilated cardiomyopathy in TG mice, and that this phenotype was prevented by treatment with caspase inhibitor.46 Caspase inhibitor treatment also ameliorated the development of heart failure in Gαq TG mice.47 Moreover, loss of gp130-mediated cell survival signals led to early transition from adaptive cardiac growth to heart failure in response to pressure overload.48 Given that Akt strongly promotes cell survival through phosphorylation of multiple downstream targets including forkhead transcription factor or BCL-2 antagonist of cell death,49 improved contractile function by inducible expression of Akt in the heart may be owing in part to its prosurvival effects. Collectively, transient activation of Akt signaling in cardiac muscle cells attenuates contractile dysfunction in the failing heart, presumably through multiple mechanisms including modulation of coronary angiogenesis, Ca2+ handling, and cell survival.
In the clinical setting, Akt is not a suitable therapeutic target for patients with heart failure because sustained activation of Akt is deleterious for the heart, presumably through induction of extensive cardiac hypertrophy.31,35 Because the positive inotropic effects of Akt can be separated from its growth-promoting effects, downstream substrates of Akt kinase that mediate its cardioprotective effects may be promising therapeutic targets for heart failure.
Footnotes
Supported by NIH grants HL102874, AG34972, AG15052, and HL68758 (K.W.), and Landesforschungsschwerpunkt Baden-Württemberg (S.S.).
I.S. and S.S. contributed equally to this work.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at http://dx.doi.org/10.1016/j.ajpath.2012.08.020.
Supplementary data
Biological network analysis. Network of functionally related genes constructed from differentially regulated transcripts between 8 weeks (before AAC) and 12 weeks (4 weeks after AAC). This set of genes represents the cell death network according to the assignment by Ingenuity software version 4 (Ingenuity Systems, Redwood City, CA). Red shading indicates up-regulation at 12 weeks, and green shading indicates down-regulation at 12 weeks. The extent of shading is indicative of the magnitude of regulation according to assignment by Ingenuity software. Node shapes indicate function. Diamond, enzyme; square, growth factor; triangle, kinase; circle, other; —‖, physical interactions (eg, formation of complexes); arrow, functional interaction (activation); — , inhibition.
Biological network analysis. Genes that are regulated differentially between 12 weeks (4 weeks after AAC) and 16 weeks (2 weeks after Akt-ON) superimposed on the network constructed as described in Supplemental Figure S1. Names and symbols of the transcripts that are differentially regulated are summarized in Supplemental Table S1 (available at http://ajp.amjpathol.org).
Biological network analysis. Genes that are regulated differentially between 8 weeks (before AAC) and 12 weeks (4 weeks after AAC) superimposed on the network constructed as described in Supplemental Figure S4 (available at http://ajp.amjpathol.org).
Biological network analysis. Network of functionally related genes constructed from differentially regulated transcripts between 12 weeks (4 weeks after AAC) and 16 weeks (2 weeks after Akt-ON). This set of genes represents the Akt pathway according to the assignment by Ingenuity software. Red shading indicates up-regulation at 16 weeks, and green shading indicates down-regulation at 16 weeks. The extent of shading is indicative of the magnitude of regulation according to assignment by Ingenuity software. Node shapes indicate function. Diamond, enzyme; square, growth factor; triangle, kinase; circle, other; —‖, physical interactions (eg, formation of complexes); arrow, functional interaction (activation); — , inhibition. Names and symbols of the transcripts that are regulated differentially are summarized in Supplemental Table S1 (available at http://ajp.amjpathol.org).
References
- 1.Thom T., Haase N., Rosamond W., Howard V.J., Rumsfeld J., Manolio T., Zheng Z.J., Flegal K., O'Donnell C., Kittner S., Lloyd-Jones D., Goff D.C., Jr, Hong Y., Adams R., Friday G., Furie K., Gorelick P., Kissela B., Marler J., Meigs J., Roger V., Sidney S., Sorlie P., Steinberger J., Wasserthiel-Smoller S., Wilson M., Wolf P. Heart disease and stroke statistics–2006 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2006;113:e85–e151. doi: 10.1161/CIRCULATIONAHA.105.171600. [DOI] [PubMed] [Google Scholar]
- 2.Braunwald E., Colucci W., Grossman W. Clinical aspects of heart failure: high-output heart failure; pulmonary edema. In: Braunwald E., editor. Heart disease: a textbook of cardiovascular medicine. W.B. Sanders; Philadelphia: 1997. pp. 445–470. [Google Scholar]
- 3.Harjola V.P., Follath F., Nieminen M.S., Brutsaert D., Dickstein K., Drexler H., Hochadel M., Komajda M., Lopez-Sendon J.L., Ponikowski P., Tavazzi L. Characteristics, outcomes, and predictors of mortality at 3 months and 1 year in patients hospitalized for acute heart failure. Eur J Heart Fail. 2010;12:239–248. doi: 10.1093/eurjhf/hfq002. [DOI] [PubMed] [Google Scholar]
- 4.Lam C.S., Donal E., Kraigher-Krainer E., Vasan R.S. Epidemiology and clinical course of heart failure with preserved ejection fraction. Eur J Heart Fail. 2011;13:18–28. doi: 10.1093/eurjhf/hfq121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Olivetti G., Capasso J.M., Meggs L.G., Sonnenblick E.H., Anversa P. Cellular basis of chronic ventricular remodeling after myocardial infarction in rats. Circ Res. 1991;68:856–869. doi: 10.1161/01.res.68.3.856. [DOI] [PubMed] [Google Scholar]
- 6.Gerdes A.M., Kellerman S.E., Moore J.A., Muffly K.E., Clark L.C., Reaves P.Y., Malec K.B., McKeown P.P., Schocken D.D. Structural remodeling of cardiac myocytes in patients with ischemic cardiomyopathy. Circulation. 1992;86:426–430. doi: 10.1161/01.cir.86.2.426. [DOI] [PubMed] [Google Scholar]
- 7.Barry S.P., Davidson S.M., Townsend P.A. Molecular regulation of cardiac hypertrophy. Int J Biochem Cell Biol. 2008;40:2023–2039. doi: 10.1016/j.biocel.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 8.Toischer K., Rokita A.G., Unsold B., Zhu W., Kararigas G., Sossalla S., Reuter S.P., Becker A., Teucher N., Seidler T., Grebe C., Preuss L., Gupta S.N., Schmidt K., Lehnart S.E., Kruger M., Linke W.A., Backs J., Regitz-Zagrosek V., Schafer K., Field L.J., Maier L.S., Hasenfuss G. Differential cardiac remodeling in preload versus afterload. Circulation. 2010;122:993–1003. doi: 10.1161/CIRCULATIONAHA.110.943431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levy D., Garrison R.J., Savage D.D., Kannel W.B., Castelli W.P. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med. 1990;322:1561–1566. doi: 10.1056/NEJM199005313222203. [DOI] [PubMed] [Google Scholar]
- 10.MacLellan W.R., Schneider M.D. Genetic dissection of cardiac growth control pathways. Annu Rev Physiol. 2000;62:289–319. doi: 10.1146/annurev.physiol.62.1.289. [DOI] [PubMed] [Google Scholar]
- 11.Molkentin J., Dorn I.G. Cytoplasmic signaling pathways that regulate cardiac hypertrophy. Annu Rev Physiol. 2001;63:391–426. doi: 10.1146/annurev.physiol.63.1.391. [DOI] [PubMed] [Google Scholar]
- 12.Frey N., Olson E.N. Cardiac hypertrophy: the good, the bad, and the ugly. Annu Rev Physiol. 2003;65:45–79. doi: 10.1146/annurev.physiol.65.092101.142243. [DOI] [PubMed] [Google Scholar]
- 13.Hudlicka O., Brown M.D. Postnatal growth of the heart and its blood vessels. J Vasc Res. 1996;33:266–287. doi: 10.1159/000159155. [DOI] [PubMed] [Google Scholar]
- 14.Richey P.A., Brown S.P. Pathological versus physiological left ventricular hypertrophy: a review. J Sports Sci. 1998;16:129–141. doi: 10.1080/026404198366849. [DOI] [PubMed] [Google Scholar]
- 15.Shiojima I., Walsh K. Role of Akt signaling in vascular homeostasis and angiogenesis. Circ Res. 2002;90:1243–1250. doi: 10.1161/01.res.0000022200.71892.9f. [DOI] [PubMed] [Google Scholar]
- 16.Shiojima I., Walsh K. Regulation of cardiac growth and coronary angiogenesis by the Akt/PKB signaling pathway. Genes Dev. 2006;20:3347–3365. doi: 10.1101/gad.1492806. [DOI] [PubMed] [Google Scholar]
- 17.Chen W.S., Xu P.Z., Gottlob K., Chen M.L., Sokol K., Shiyanova T., Roninson I., Weng W., Suzuki R., Tobe K., Kadowaki T., Hay N. Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev. 2001;15:2203–2208. doi: 10.1101/gad.913901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cho H., Thorvaldsen J.L., Chu Q., Feng F., Birnbaum M.J. Akt1/pkbalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem. 2001;276:38349–38352. doi: 10.1074/jbc.C100462200. [DOI] [PubMed] [Google Scholar]
- 19.Cho H., Mu J., Kim J.K., Thorvaldsen J.L., Chu Q., Crenshaw E.B., 3rd, Kaestner K.H., Bartolomei M.S., Shulman G.I., Birnbaum M.J. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta) Science. 2001;292:1728–1731. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- 20.Garofalo R.S., Orena S.J., Rafidi K., Torchia A.J., Stock J.L., Hildebrandt A.L., Coskran T., Black S.C., Brees D.J., Wicks J.R., McNeish J.D., Coleman K.G. Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKB beta. J Clin Invest. 2003;112:197–208. doi: 10.1172/JCI16885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Easton R.M., Cho H., Roovers K., Shineman D.W., Mizrahi M., Forman M.S., Lee V.M., Szabolcs M., de Jong R., Oltersdorf T., Ludwig T., Efstratiadis A., Birnbaum M.J. Role for Akt3/protein kinase Bgamma in attainment of normal brain size. Mol Cell Biol. 2005;25:1869–1878. doi: 10.1128/MCB.25.5.1869-1878.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tschopp O., Yang Z.Z., Brodbeck D., Dummler B.A., Hemmings-Mieszczak M., Watanabe T., Michaelis T., Frahm J., Hemmings B.A. Essential role of protein kinase B gamma (PKB gamma/Akt3) in postnatal brain development but not in glucose homeostasis. Development. 2005;132:2943–2954. doi: 10.1242/dev.01864. [DOI] [PubMed] [Google Scholar]
- 23.Peng X.D., Xu P.Z., Chen M.L., Hahn-Windgassen A., Skeen J., Jacobs J., Sundararajan D., Chen W.S., Crawford S.E., Coleman K.G., Hay N. Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes Dev. 2003;17:1352–1365. doi: 10.1101/gad.1089403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang Z.Z., Tschopp O., Di-Poi N., Bruder E., Baudry A., Dummler B., Wahli W., Hemmings B.A. Dosage-dependent effects of Akt1/protein kinase Balpha (PKBalpha) and Akt3/PKBgamma on thymus, skin, and cardiovascular and nervous system development in mice. Mol Cell Biol. 2005;25:10407–10418. doi: 10.1128/MCB.25.23.10407-10418.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shiojima I., Yefremashvili M., Luo Z., Kureishi Y., Takahashi A., Tao J., Rosenzweig A., Kahn C.R., Abel E.D., Walsh K. Akt signaling mediates postnatal heart growth in response to insulin and nutritional status. J Biol Chem. 2002;277:37670–37677. doi: 10.1074/jbc.M204572200. [DOI] [PubMed] [Google Scholar]
- 26.Wilkins B.J., Dai Y.S., Bueno O.F., Parsons S.A., Xu J., Plank D.M., Jones F., Kimball T.R., Molkentin J.D. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res. 2004;94:110–118. doi: 10.1161/01.RES.0000109415.17511.18. [DOI] [PubMed] [Google Scholar]
- 27.Konhilas J.P., Maass A.H., Luckey S.W., Stauffer B.L., Olson E.N., Leinwand L.A. Sex modifies exercise and cardiac adaptation in mice. Am J Physiol Heart Circ Physiol. 2004;287:H2768–H2776. doi: 10.1152/ajpheart.00292.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Konhilas J.P., Widegren U., Allen D.L., Paul A.C., Cleary A., Leinwand L.A. Loaded wheel running and muscle adaptation in the mouse. Am J Physiol Heart Circ Physiol. 2005;289:H455–H465. doi: 10.1152/ajpheart.00085.2005. [DOI] [PubMed] [Google Scholar]
- 29.Naga Prasad S.V., Esposito G., Mao L., Koch W.J., Rockman H.A. Gbetagamma-dependent phosphoinositide 3-kinase activation in hearts with in vivo pressure overload hypertrophy. J Biol Chem. 2000;275:4693–4698. doi: 10.1074/jbc.275.7.4693. [DOI] [PubMed] [Google Scholar]
- 30.Haq S., Choukroun G., Lim H., Tymitz K.M., del Monte F., Gwathmey J., Grazette L., Michael A., Hajjar R., Force T., Molkentin J.D. Differential activation of signal transduction pathways in human hearts with hypertrophy versus advanced heart failure. Circulation. 2001;103:670–677. doi: 10.1161/01.cir.103.5.670. [DOI] [PubMed] [Google Scholar]
- 31.Shioi T., McMullen J.R., Kang P.M., Douglas P.S., Obata T., Franke T.F., Cantley L.C., Izumo S. Akt/protein kinase B promotes organ growth in transgenic mice. Mol Cell Biol. 2002;22:2799–2809. doi: 10.1128/MCB.22.8.2799-2809.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matsui T., Li L., Wu J.C., Cook S.A., Nagoshi T., Picard M.H., Liao R., Rosenzweig A. Phenotypic spectrum caused by transgenic overexpression of activated Akt in the heart. J Biol Chem. 2002;277:22896–22901. doi: 10.1074/jbc.M200347200. [DOI] [PubMed] [Google Scholar]
- 33.Condorelli G., Drusco A., Stassi G., Bellacosa A., Roncarati R., Iaccarino G., Russo M.A., Gu Y., Dalton N., Chung C., Latronico M.V., Napoli C., Sadoshima J., Croce C.M., Ross J., Jr Akt induces enhanced myocardial contractility and cell size in vivo in transgenic mice. Proc Natl Acad Sci U S A. 2002;99:12333–12338. doi: 10.1073/pnas.172376399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taniyama Y., Ito M., Sato K., Kuester C., Veit K., Tremp G., Liao R., Colucci W.S., Ivashchenko Y., Walsh K., Shiojima I. Akt3 overexpression in the heart results in progression from adaptive to maladaptive hypertrophy. J Mol Cell Cardiol. 2005;38:375–385. doi: 10.1016/j.yjmcc.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 35.Shiojima I., Sato K., Izumiya Y., Schiekofer S., Ito M., Liao R., Colucci W.S., Walsh K. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. 2005;115:2108–2118. doi: 10.1172/JCI24682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liao R., Jain M., Cui L., D'Agostino J., Aiello F., Luptak I., Ngoy S., Mortensen R.M., Tian R. Cardiac-specific overexpression of GLUT1 prevents the development of heart failure attributable to pressure overload in mice. Circulation. 2002;106:2125–2131. doi: 10.1161/01.cir.0000034049.61181.f3. [DOI] [PubMed] [Google Scholar]
- 37.Schiekofer S., Galasso G., Sato K., Kraus B.J., Walsh K. Impaired revascularization in a mouse model of type 2 diabetes is associated with dysregulation of a complex angiogenic-regulatory network. Arterioscler Thromb Vasc Biol. 2005;25:1603–1609. doi: 10.1161/01.ATV.0000171994.89106.ca. [DOI] [PubMed] [Google Scholar]
- 38.Schiekofer S., Belisle K., Galasso G., Schneider J.G., Boehm B.O., Burster T., Schmitz G., Walsh K. Angiogenic-regulatory network revealed by molecular profiling heart tissue following Akt1 induction in endothelial cells. Angiogenesis. 2008;11:289–299. doi: 10.1007/s10456-008-9112-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schiekofer S., Shiojima I., Sato K., Galasso G., Oshima Y., Walsh K. Microarray analysis of Akt1 activation in transgenic mouse hearts reveals transcript expression profiles associated with compensatory hypertrophy and failure. Physiol Genomics. 2006;27:156–170. doi: 10.1152/physiolgenomics.00234.2005. [DOI] [PubMed] [Google Scholar]
- 40.Braz J.C., Gill R.M., Corbly A.K., Jones B.D., Jin N., Vlahos C.J., Wu Q., Shen W. Selective activation of PI3Kalpha/Akt/GSK-3beta signalling and cardiac compensatory hypertrophy during recovery from heart failure. Eur J Heart Fail. 2009;11:739–748. doi: 10.1093/eurjhf/hfp094. [DOI] [PubMed] [Google Scholar]
- 41.Izumiya Y., Shiojima I., Sato K., Sawyer D.B., Colucci W.S., Walsh K. VEGF blockade promotes the transition from compensated cardiac hypertrophy to failure in response to pressure overload. Hypertension. 2006;47:887–893. doi: 10.1161/01.HYP.0000215207.54689.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim Y.K., Kim S.J., Yatani A., Huang Y., Castelli G., Vatner D.E., Liu J., Zhang Q., Diaz G., Zieba R., Thaisz J., Drusco A., Croce C., Sadoshima J., Condorelli G., Vatner S.F. Mechanism of enhanced cardiac function in mice with hypertrophy induced by overexpressed Akt. J Biol Chem. 2003;278:47622–47628. doi: 10.1074/jbc.M305909200. [DOI] [PubMed] [Google Scholar]
- 43.Cittadini A., Monti M.G., Iaccarino G., Di Rella F., Tsichlis P.N., Di Gianni A., Stromer H., Sorriento D., Peschle C., Trimarco B., Sacca L., Condorelli G. Adenoviral gene transfer of Akt enhances myocardial contractility and intracellular calcium handling. Gene Ther. 2006;13:8–19. doi: 10.1038/sj.gt.3302589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.del Monte F., Hajjar R.J. Targeting calcium cycling proteins in heart failure through gene transfer. J Physiol. 2003;546:49–61. doi: 10.1113/jphysiol.2002.026732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim S.J., Abdellatif M., Koul S., Crystal G.J. Chronic treatment with insulin-like growth factor I enhances myocyte contraction by upregulation of Akt-SERCA2a signaling pathway. Am J Physiol Heart Circ Physiol. 2008;295:H130–H135. doi: 10.1152/ajpheart.00298.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wencker D., Chandra M., Nguyen K., Miao W., Garantziotis S., Factor S.M., Shirani J., Armstrong R.C., Kitsis R.N. A mechanistic role for cardiac myocyte apoptosis in heart failure. J Clin Invest. 2003;111:1497–1504. doi: 10.1172/JCI17664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hayakawa Y., Chandra M., Miao W., Shirani J., Brown J.H., Dorn G.W., 2nd, Armstrong R.C., Kitsis R.N. Inhibition of cardiac myocyte apoptosis improves cardiac function and abolishes mortality in the peripartum cardiomyopathy of Galpha (q) transgenic mice. Circulation. 2003;108:3036–3041. doi: 10.1161/01.CIR.0000101920.72665.58. [DOI] [PubMed] [Google Scholar]
- 48.Hirota H., Chen J., Betz U.A., Rajewsky K., Gu Y., Ross J., Jr, Muller W., Chien K.R. Loss of a gp130 cardiac muscle cell survival pathway is a critical event in the onset of heart failure during biomechanical stress. Cell. 1999;97:189–198. doi: 10.1016/s0092-8674(00)80729-1. [DOI] [PubMed] [Google Scholar]
- 49.Datta S.R., Brunet A., Greenberg M.E. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Biological network analysis. Network of functionally related genes constructed from differentially regulated transcripts between 8 weeks (before AAC) and 12 weeks (4 weeks after AAC). This set of genes represents the cell death network according to the assignment by Ingenuity software version 4 (Ingenuity Systems, Redwood City, CA). Red shading indicates up-regulation at 12 weeks, and green shading indicates down-regulation at 12 weeks. The extent of shading is indicative of the magnitude of regulation according to assignment by Ingenuity software. Node shapes indicate function. Diamond, enzyme; square, growth factor; triangle, kinase; circle, other; —‖, physical interactions (eg, formation of complexes); arrow, functional interaction (activation); — , inhibition.
Biological network analysis. Genes that are regulated differentially between 12 weeks (4 weeks after AAC) and 16 weeks (2 weeks after Akt-ON) superimposed on the network constructed as described in Supplemental Figure S1. Names and symbols of the transcripts that are differentially regulated are summarized in Supplemental Table S1 (available at http://ajp.amjpathol.org).
Biological network analysis. Genes that are regulated differentially between 8 weeks (before AAC) and 12 weeks (4 weeks after AAC) superimposed on the network constructed as described in Supplemental Figure S4 (available at http://ajp.amjpathol.org).
Biological network analysis. Network of functionally related genes constructed from differentially regulated transcripts between 12 weeks (4 weeks after AAC) and 16 weeks (2 weeks after Akt-ON). This set of genes represents the Akt pathway according to the assignment by Ingenuity software. Red shading indicates up-regulation at 16 weeks, and green shading indicates down-regulation at 16 weeks. The extent of shading is indicative of the magnitude of regulation according to assignment by Ingenuity software. Node shapes indicate function. Diamond, enzyme; square, growth factor; triangle, kinase; circle, other; —‖, physical interactions (eg, formation of complexes); arrow, functional interaction (activation); — , inhibition. Names and symbols of the transcripts that are regulated differentially are summarized in Supplemental Table S1 (available at http://ajp.amjpathol.org).