

Abstract
In antigen-presenting cells (APC), presentation by class II-MHC molecules of the chemically-dominant peptide from the protein hen egg white lysozyme (HEL) generates different conformational isomers of the peptide-MHC II complexes (pMHC). Type B pMHCs are formed in early endosomes from exogenous peptides in the absence of H2-DM, while in contrast, type A pMHC complexes are formed from HEL protein in late vesicles after editing by H2-DM. Thus, H2-DM edits off the more unstable pMHC complexes, which are not presented from HEL. Here we show that type B pMHC complexes were presented from HEL protein only after stimulation of dendritic cells (DC) with TLR ligands or type I IFN. Type I IFN contributed to most TLR ligand-induced type B pMHC generation as presentation decreased in DC lacking the receptor for type I interferons (IFNAR1-/-). In contrast, presentation of type A pMHC from HEL and from peptide was minimally affected by TLR ligands. The relative effectiveness of CD8α+ DC or CD8α- DC in presenting type B pMHC complexes varied depending on the TLR-ligand used. The mechanisms of generation of type B pMHC from HEL protein with TLR stimulation did not involve H2-DM or release of peptides. DC from H2-DM-deficient mice in the presence of TLR-ligands presented type B pMHC. Such DC showed a slight enhancement of HEL catabolism, but peptide-release was not evident. Thus, TLR ligands and type I IFN alter the pathways of presentation by class II MHC molecules of DC such that type B pMHCs are generated from protein antigen.
INTRODUCTION
The chemically-dominant peptide from the model protein HEL binds to the MHC class II molecule, IAk, in multiple conformations (1-3). Both the more stable pMHC complexes, termed type A, and the less stable conformers, type B, bind to the same peptide, DYGILQINS, and in the same register (1-4). The type B-pMHC conformers result when APC interact with peptides in early endosomes, but are removed by H2-DM in late endosomes (4). In contrast, the type A-pMHCs are formed in late vesicles where H2-DM edits off the unstable complexes, allowing the persistence of only the type A (4). The type B-specific T cells to the HEL 52-60 are frequent (30-50% of CD4 T cells) and escape tolerance, for example in transgenic mice expressing HEL by all APCs (5). Since T cells to type B pMHC complexes are found to autologous proteins, it is critical to understand how type B responses could be protective during infection or detrimental in the context of inflammation-associated autoimmunity (5-7). We previously reported that type B pMHC were generated from HEL protein in vivo when administered along with inflammatory stimuli such as LPS, listeriolysin O (LLO), or complete Freund’s adjuvant (CFA) (8). Here, we explore the signals that regulate Toll-like receptor (TLR) -induced generation of type B pMHC in vitro
Dendritic cells (DC) play a key role in initiating the adaptive immune pathway through the presentation of antigen on MHC class II molecules to activate CD4 T cells. While resting DC are able to process and present exogenous antigen on MHC class II molecules, there are noticeable changes that occur following exposure to inflammatory stimulants (9-17). TLR signaling induces DC maturation involving the production of critical cytokines, increased migration to draining lymph nodes, and increased expression of MHC I, MHC II, and many co-stimulatory molecules (18-21). While many details of how TLR signaling affects the MHC II pathway are still to be considered, it is clear that TLR engagement leads to a transient increase in endocytosis, a transient increase in MHC II transcription, and stabilization of surface pMHC (10-12, 22, 23).
Stimulation through TLR 3, 4, 7, and 9 leads to production of type I IFN by DC (24). TLR 2 also initiates type I IFN production (25, 26). The contribution of type I IFN signaling to TLR-initiated responses has been explored primarily in vivo and shown to be important for events ranging from DC maturation to antibody production by B cells to bystander activation of T cells and NK cells (27-33). In the MHC class I pathway, type I IFN has been shown to be important for TLR-induced cross-priming (13, 33, 34) and specifically for CpG-induced cross-presentation of proteins like soluble ovalbumin (35). However, the contribution of type I IFN to TLR-initiated events in the MHC class II pathway has not been explored. Here we show that most TLR ligands and type I IFN-induced presentation of type B pMHC by DCs from HEL and that TLR-induced type I IFN contributed to induced presentation of type B pMHC.
MATERIALS AND METHODS
Mice
Mice were maintained under specific pathogen-free conditions according to institutional animal care guidelines. B10.BR mice were either from a colony maintained in our facility or purchased from Jackson Laboratory (Bar Harbor, ME.) MyD88-/- mice and the type 1 interferon receptor gene knockout (IFNAR1-/) mice were backcrossed to the B10.BR background. mHEL mice (expressing membrane-bound HEL under the MHC class II promoter) were previously described (5). H2-DMα-deficient mice were also described previously (36).
Stimulants
Stimulants used were Ultra-pure LPS-EB from E. coli 0111:B4 (Invivogen, San Diego, CA), Gardiquimod (Invivogen), Flagellin (Invivogen), CpG-B: ODN 1826 (Integrated DNA Technologies, Coralville, IA), Poly (I:C) (Sigma-Aldrich, St. Louis, MO), Zymosan A from Saccharomyces cerevisiae (Sigma-Aldrich), Resiquimod (a kind gift from Dr. Marco Colonna), recombinant IFN-α and IFN-β (PBL Interferon Source, Piscataway, NJ), and IFN-γ (kind gift from Dr. Robert Schreiber). TLR ligands used in this study are listed in Table I.
Table I.
TLR Ligands used in this study.
| TLR | TLR2/6 | TLR3 | TLR4 | TLR5 | TLR7 | TLR9 |
|---|---|---|---|---|---|---|
| Agonist(s) | Zymosan A | Poly(I:C) | LPS | Flagellin | Gardiquimod Resiquimod | ODN 1826 (CpG-B) |
|
| ||||||
| Also binds Dectin-1 | ||||||
Flow Cytometry
For cell sorting, DC were stained with CD11c (N418, eBioscience, San Diego, CA or BioLegend, San Diego, CA), CD8α (53-6.7, eBioscience) CD45RA (14.8, BD Pharmingen, San Diego, CA) Siglec H (440c, a kind gift from Dr. Marco Colonna) B220 (RA3-6B2, BD Pharmingen) and CD19 (1D3, BD Pharmingen.) For analysis of DC, the following antibodies were used: CD4 (RM4-5, eBioscences) CD40 (3/23, BD Pharmingen) CD80 (16-10A1, BD Pharmingen), CD86 (GL1, BD Pharmingen) I-Ak (40F, made in house), and H-2Kk (36-7-5, BD Pharmingen). For intracellular staining, cells were fixed and permeabilized using a Cytofix/Cytoperm kit according to manufacturer’s instructions (BD Pharmingen.) Antibodies for intracellular staining were H2-DM (2E5A, BD Pharmingen,) goat anti-Rat-Cy5 (Zymed, Carlsbad, CA), and H2-DO-Alexa647 (Mags.Ob1, a kind gift from Dr. Lisa Denzin.) Flow cytometry data were collected on a BD FACSLSR™ II or BD FACSCalibur™ and analyzed using FlowJo software (Tree Star, Ashland, OR.)
Dendritic cell isolation and Flt3L treatment
DC were isolated from spleens of mice injected with 10 μg Flt3L intraperitoneally for three consecutive days. On day eight, spleens were harvested, digested with 0.14 U/mL Liberase Blendzyme 3 or 1.67 U/mL Liberase TL (Roche Applied Science, Indianapolis, IN) to make single-cell suspensions from which DC were isolated by CD11c magnetic bead (Miltenyi Biotec Inc., Auburn, CA). Enriched DC were ≥ 95% pure as determined by flow cytometry. For sorting, DC isolated as above were stained for surface markers and re-suspended in phenol-free DMEM (Invitrogen, Carlsbad, CA) supplemented with 2% FCS and 2 mM EDTA. Cells were sorted on a BD FACSAria™ II. Conventional DC (cDC) were sorted as CD11chigh CD45RA- B220- or CD11chigh Siglec H- B220-. cDC were further separated by CD8α expression. For analysis of H2-DM and H2-DO, cDC were further selected as CD19-.
Assays
For antigen presentation assays, 105 DC were incubated with HEL protein or the HEL (48-62) peptide (DGSTDYGILQINSRW), with or without stimulants for 18 hours in 100 μL volume in v-bottom 96-well plates. Stimulant concentrations used were 10 μg/mL Zymosan A, 10 μg/mL Poly (I:C), 1 μg/mL LPS, 1 μg/mL Gardiquimod, 6 μg/mL Resiquimod, 1 μM ODN 1826 (CpG-B), 100 U/mL IFN-γ, 10 U/mL IFN-α, 10 U/mL IFN-β. DC were washed three times with serum-free DMEM after incubation before adding 5×104 T cell hybridomas per well in a 200 μL volume; 24 hrs later the release of IL-2 was measured in the culture fluid using CTLL as an indicator cell. Most experiments used two previously characterized T cell hybridomas: 11A10, which only reacts with type B-pMHC, and 3A9, which recognizes type A-pMHC. Sorted DC were pre-treated with 1 μg/well anti-IFNAR1 mAb MAR1-5A3 (a kind gift of Dr. Robert Schreiber (37)), or control anti-human IFN-gamma receptor-1 GIR.208 antibody for 1 hour at 37°C. Without washing, HEL or peptide was added with or without stimulants to the wells. The remaining assay followed the protocol described above. For examination of surface markers by flow cytometry, DC isolated as described above were incubated with or without stimulants for 18 hours in 100 μL volume in 96-well v-bottom plates.
Peptide-Release Assay
DC from CB.17 mice were incubated overnight with HEL with or without 1 μM CpG-B. Supernatants were collected and spun twice to remove cells and debris. Supernatants were serially diluted and added to paraformaldehyde-fixed DC from B10.BR mice. DC were fixed in 1% paraformaldehyde for 5 min, incubated with 0.2M DL-Lysine, and washed extensively in media. T cells were added as described above.
HEL Catabolism
DC were incubated for eighteen hours with 1 μM CpG-B (ODN1826) or without stimulation. Equal numbers of DC were incubated with I125-HEL for two hours at room temperature with intermittent mixing. After the incubation, DC were washed and re-platted for indicated times. At each time point, TCA soluble and precipitable fractions were collected from the cells and the supernatant.
qRT-PCR
Sorted cDC were incubated for two, six, or eighteen hours untreated or stimulated with TLR ligands or IFN-beta. Total RNA was isolated using RNAqueous®-Micro kit according to manufacturer’s instructions (Applied Biosystems, Carlsbad, CA.) Equal amounts of RNA were used to generate cDNA using the High Capacity Reverse cDNA Transcription Kit according to manufacturer’s instructions (Applied Biosystems.) Forty nanograms of cDNA were used per reaction for qRT-PCR analysis. qRT-PCR was performed using Fast SYBR Green kit and ΔΔCΤ calculations on a StepOnePlus Instrument (Applied Biosystems.) Primers used were the following: 18SF-GTAACCCGTTGAACCCCATT, 18SR-CCATCCAATCGGTAGTAGCG; IFN-β F-ATAAGCAGCTCCAGCTCCAAG, IFN-β R-GTCTCATTCCACCCAGTGCTG; DMα F-TAGGGCTCTCGGAGACCTATG, DMα R-AGTCGAAGGAGAAGAGTTCGT; DOα F-CCGCAATGAGCTTCCTGAGTCCC, DOα R-GTGGTCGGCCTTGATGGCCCTT; H2-Aα F-TCAGTCGCAGACGGTGTTTAT, H2-Aα R-GGGGGCTGGAATCTCAGGT. 18S was used as the qRT-PCR standard.
RESULTS
Toll-like receptor ligands induce type B presentation from HEL
To assess the ability of TLR ligands to affect type B pMHC presentation, a culture system was set up involving the culture of DC with HEL in the presence of the stimulants for 18 hours, after which the stimuli were removed and the extent of presentation was assayed by adding the indicator T cell hybridomas. In agreement with the basic definition of type B presentation, un-stimulated DC presented type B pMHC only when incubated with the HEL:48-61 peptide, but not HEL (Figure 1A, C). However, all TLR ligands except flagellin (Figure 2A) allowed for the presentation of type B pMHC conformers from HEL, referred to as induced type B presentation. Notably, TLR ligands had little effect on presentation from peptide (Figure 1C, D) only affecting type B presentation from peptide at low levels (0.01 μM), and modestly augmented presentation to 3A9, a T cell that recognizes type A pMHC, only at low levels of HEL (0.01 μM) (Figure 1B). Interferon-gamma did not induce type B presentation from HEL (Figure 1A). The induced type B presentation by TLR-ligands was reproducible, testing three different type B T cell hybridomas recognizing the HEL 48-61 epitope (data not shown.)
Figure 1. TLR Ligand-Induced Presentation of Type B pMHC from HEL.
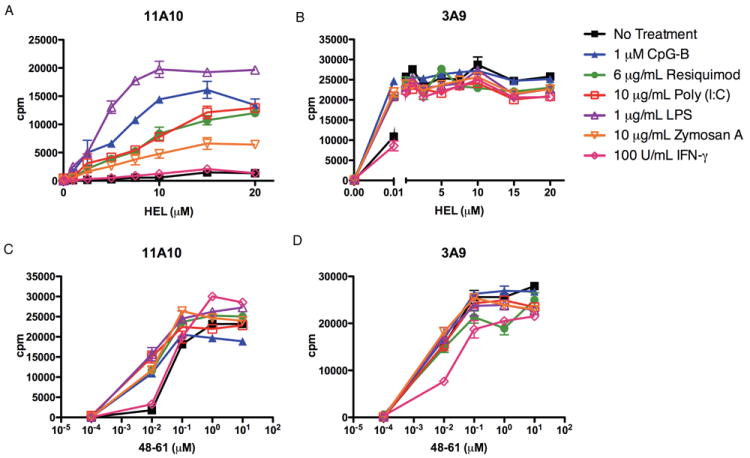
Presentation by DC incubated with HEL (A, B) or HEL:48-61 peptide (C, D) to type B 11A10 (A, C) or type A 3A9 (B, D) T cell hybridomas, in the presence or absence of TLR-ligands. Representative of at least four independent experiments. Error bars represent SD.
Figure 2. Dependence on MyD88-/- DC for TLR Ligand-Induced Presentation of Type B pMHC from HEL.
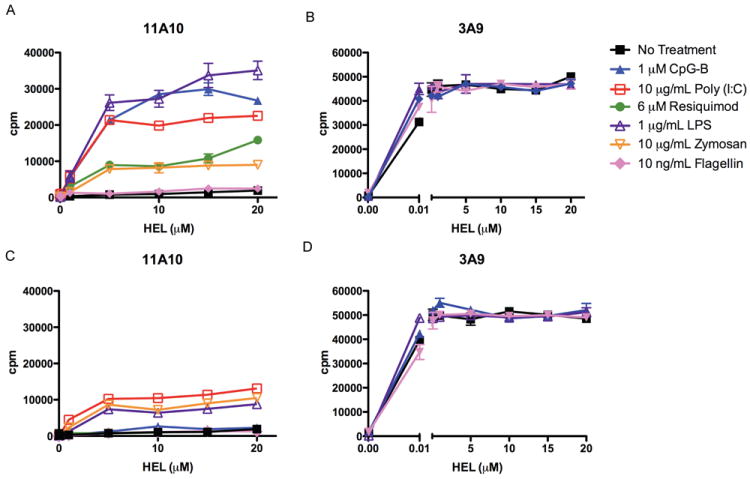
Presentation to type B 11A10 (A, C) or type A 3A9 (B, D) T cell hybridomas by DC from wild-type (A, B) or MyD88-/- mice (C, D) incubated with HEL, with or without TLR ligands. Error bars represent SD. Representative of three independent experiments.
Induced type B pMHC presentation by MyD88-deficient DC followed the expected pattern with TLR7 and 9 ligands being completely eliminated, TLR3 ligands being minimally affected, and TLR4 ligands only partially reduced, reflecting the degree to which each TLR depends on MyD88 as a signaling adapter (Figure 2) (38).
Role of DC subsets in induced presentation
Whether a subset of DC was responsible for the presentation of type B epitopes from HEL was examined. Plasymacytoid DC (pDC) comprised approximately 10 percent of the total DC population (identified as CD11cint/low Siglec H+ B220+), but were not essential for induced presentation as sorted conventional DC (cDC), identified as CD11chi Siglec H- B220- presented type B pMHC from HEL with TLR stimulation (data not shown.) To examine the role of the conventional DC subsets, sorted CD8α+ and CD8α- DC were incubated with HEL protein with or without CpG as in previous experiments. CD8α+ DC strongly presented type B pMHC from HEL upon CpG-stimulation (Figure 3B), but not in the absence of stimuli. CD8α- DC, however, had some degree of presentation of type B pMHC in the absence of stimulation at high HEL doses: it increased upon CpG stimulation, but not as strongly as the CD8α+ DC (Fig 3C). The ability of CD8α+ DC or CD8α- DC to present type B pMHC from HEL slightly differed, depending on the TLR ligands, except for gardiquimod, a ligand of TLR7/8 (Figure 3D). Gardiquimod induced a small level of presentation only by CD8α- DC, which is not surprising as TLR7 is minimally expressed in CD8α+ DC (39). There was no significant difference in presentation of HEL peptide by the DCs (Figure 3E). To note is that the degree of presentation of HEL at high doses by the unstimulated CD8α- DC varied greatly from no response whatsoever, to the small response found in panel B, reflecting most likely a degree of activation by environmental stimuli. In sum, these data indicate that both splenic DC subsets are capable of presenting type B pMHC from protein HEL in response to TLR stimulation.
Figure 3. Presentation by Sorted CD8α+ and CD8α- DC.
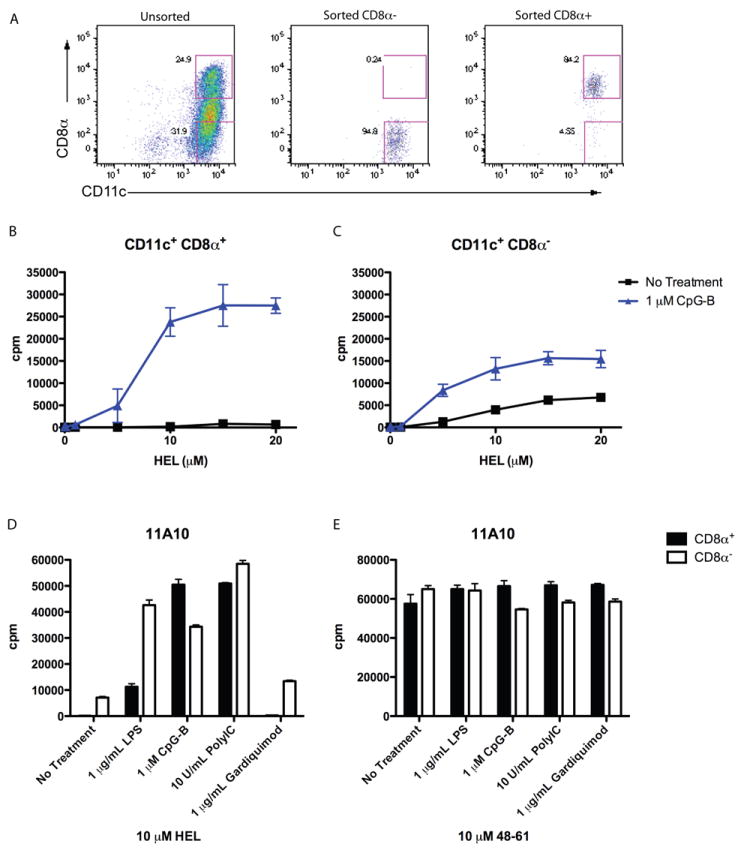
(A) Representative flow cytometry assay on sorted cells showing CD11c and CD8α expression on unsorted and sorted DC populations. (B, C). Presentation to type B 11A10 T cell hybridoma by sorted CD8α+ (B) and CD8α- (C) DC incubated with HEL protein, with or without CpG. (D, E). Presentation to type B 11A10 T cell hybridoma by sorted DC incubated with HEL protein (D) or peptide (E) with or without TLR stimulants. Error bars represent SD. B and C representative of eleven independent experiments; D and E representative of four independent experiments.
Role of type I IFN in induced type B presentation
In addition to TLR ligands, recombinant IFN-alpha and IFN-beta induced type B pMHC presentation from HEL (Figure 4A), but only affected type A presentation marginally at low levels of protein (Figure 4B). Type I IFNs had minimal effects on presentation from peptide, as with TLR ligands, only affecting type B pMHC presentation at low peptide doses (0.01 μM) (Figure 4C, D).
Figure 4. Type I IFN-Induced Presentation of Type B pMHC from HEL and Production of Type I IFN by TLR Stimulation.
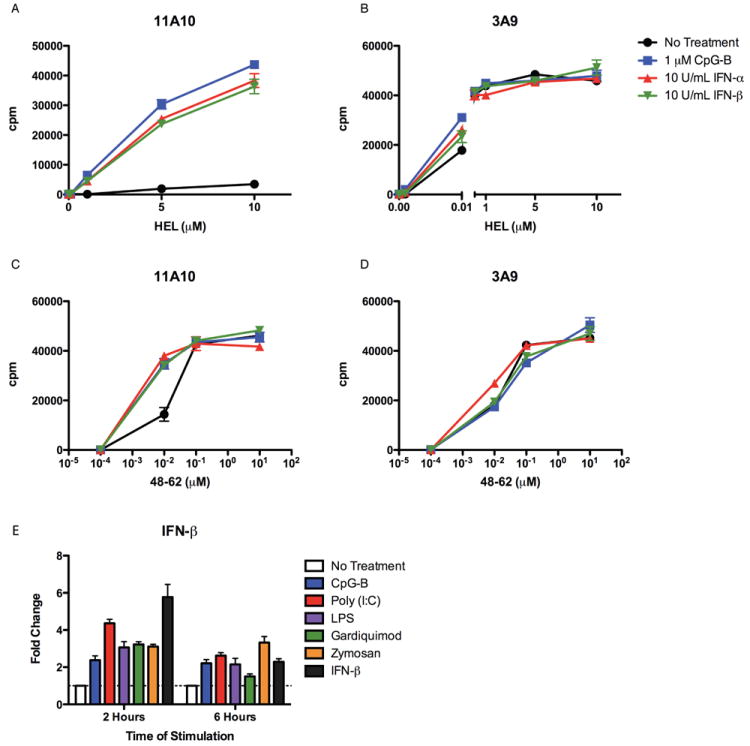
(A-D) Presentation by DC incubated with (A, B) HEL or (C, D) HEL peptide 48-62 with or without CpG or recombinant IFN-α or IFN-β to (A, C) type B 11A10 or (B, D) type A 3A9 T cell hybridomas. Representative of twelve independent experiments. (E) Sorted cDC were incubated with or without stimulants for 2 or 6 hours before RNA was isolated. qRT-PCR was performed to assess transcript levels of IFN-beta. ΔΔCΤ calculations were performed using 18S as the standard.
Quantitative RT-PCR was used to assess expression of IFN-beta by sorted cDC. Stimulation of cDC with CpG, Poly(I:C), LPS, Gardiquimod, and Zymosan induced expression of IFN-beta as indicated by increased mRNA levels after two hours, decreasing by six hours after stimulation (Figure 4E.)
To test the role of TLR-induced type I IFN on presentation, MAR1-5A3 antibody was used to block signaling through the type I IFNα/β receptor (37). DC were pretreated with control or MAR1-5A3 antibody before adding HEL with or without stimulants. MAR1-5A3 completely blocked IFN beta-induced type B presentation by CD8α+ DC (99.7% at half-max, Figure 5A). It reduced CpG-induced presentation by 57% (at half-max) by CD8α+ DC. Blocking the IFNα/β receptor had little effect on presentation from peptide (data not shown), and followed a similar trend with CD8α- DC (Figure 5B). Of note, MAR1-5A3 treatment reduced the level of type B presentation by un-stimulated CD8α- DC, indicating that cytokine production may explain why CD8α- DC were able to present low levels of type B pMHC from protein in the absence of stimulation.
Figure 5. Role of Type I IFN Signaling in CpG-Induced pMHC Presentation from HEL.
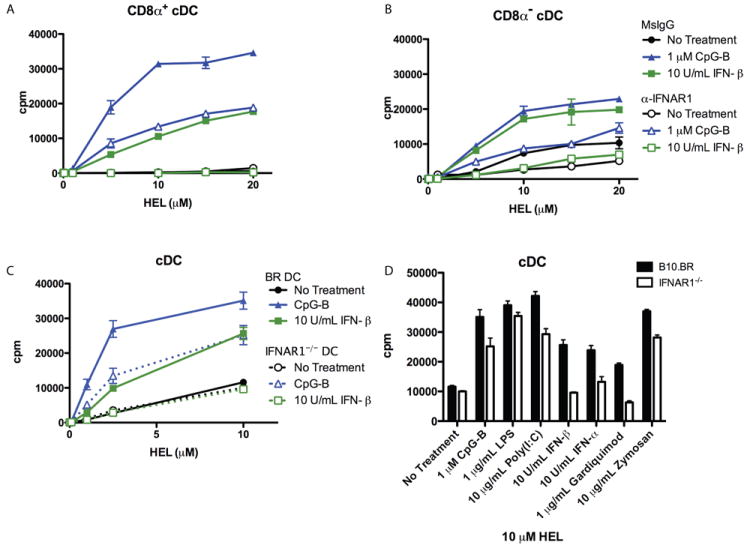
(A, B) Presentation to type B 11A10 T cell hybridoma by sorted CD11chiCD8a+ (A) versus CD11chiCD8a- (B) cells pre-treated with MAR1-5A3 (anti-IFNAR1) or control mouse IgG before adding antigen and stimulants. (C) Presentation to type B 11A10 T cell hybridoma by sorted cDC from B10.BR or IFNAR1-/- mice incubated with HEL protein with or without stimulants. (D) Presentation to 11A10 by cDC treated as in B. Results shown for 10 μM HEL protein dose. Complete antigen titrations are shown in Figure S1. Error bars represent SD. All graphs are representative of two independent experiments.
To more critically address the role of TLR-stimulated type I IFN in induced presentation, DC from IFNAR1-deficient mice were examined. While recombinant IFN-beta and IFN-alpha had no effect on presentation by IFNAR1-deficient DC (Figure 5C, D and Figure S1A), CpG-induced presentation was decreased by about 60% (at half-max) in IFNAR1-deficient DC compared to wild-type DC (Figure 5C, Figure S1A). The role of TLR-induced type I IFN was explored with the remaining TLR ligands shown to induce type B pMHC presentation from protein HEL using IFNAR1-deficient DC. Poly (I:C)- and Zymosan A-induced type B presentation were reduced by 25% and 36% respectively in IFNAR1-deficient DC (Figure 5D and Figure S1A.) Gardiquimod-induced presentation was completely inhibited and LPS-induced presentation was unaffected.
These data show that type I IFN signaling contributed differently to each TLR pathway of induced type B pMHC presentation, augmented presentation through TLR3 and TLR9, was the sole mediator through TLR7, and had no effect on induction through TLR4. Of note, presentation from peptide and type A pMHC presentation from protein was unaltered in IFNAR1-deficient DC (Figure S1B, C). Together these results suggest that TLR and type I IFN signaling most likely initiate the same sub-cellular events and that TLR-induced type I IFN can amplify the signal, allowing for augmented type B pMHC presentation.
Role of type I IFN on expression of co-stimulatory molecules
Because co-stimulatory molecule expression is a component of DC maturation, the contribution of type I IFN to expression of co-stimulatory molecules was examined to see if it mirrored the contribution to induced type B pMHC presentation. DC from B10.BR wild-type or IFNAR1-/- mice were analyzed for surface expression of MHC II and co-stimulatory molecules after 18 hours in culture with or without stimulation. While MHC class II levels changed little after stimulation, CD40, CD80, and CD86 were all significantly increased on B10.BR DC after exposure to TLR ligands and IFN-beta (Figure 6 and Figure S2). Co-stimulatory molecule expression after stimulation was lower on IFNAR1-/- DC than B10.BR DC for all stimulants tested, indicating that type I IFN signaling augmented up-regulation of co-stimulatory molecule expression by all TLR ligands tested. As expected, increased surface expression of co-stimulatory molecules by IFN-beta was completely dependent on type I IFN signaling, as IFNAR1-/- DC were unable to respond (Figure 6F, G). Zymosan A-stimulated co-stimulatory molecule expression was minimally dependent on type I IFN signaling (Figure 6A). Poly(I:C)- and CpG-stimulated co-stimulatory molecule expression was more significantly dependent on type I IFN signaling (Figure 6B, E). Gardiquimod-stimulated co-stimulatory molecule expression was almost completely dependent on type I IFN signaling, mirroring the complete requirement for type I IFN signaling for induced type B pMHC presentation (Figure 6D, 5E). The stimulation of co-stimulatory molecule expression by LPS was dependent partially on type I IFN signaling (Figure 6C), which was not the case for induced type B presentation (Figure 5E, Figure S1A).
Figure 6. Role of Type I IFN Signaling in TLR-Induced Changes in Costimulatory Molecule Expression.
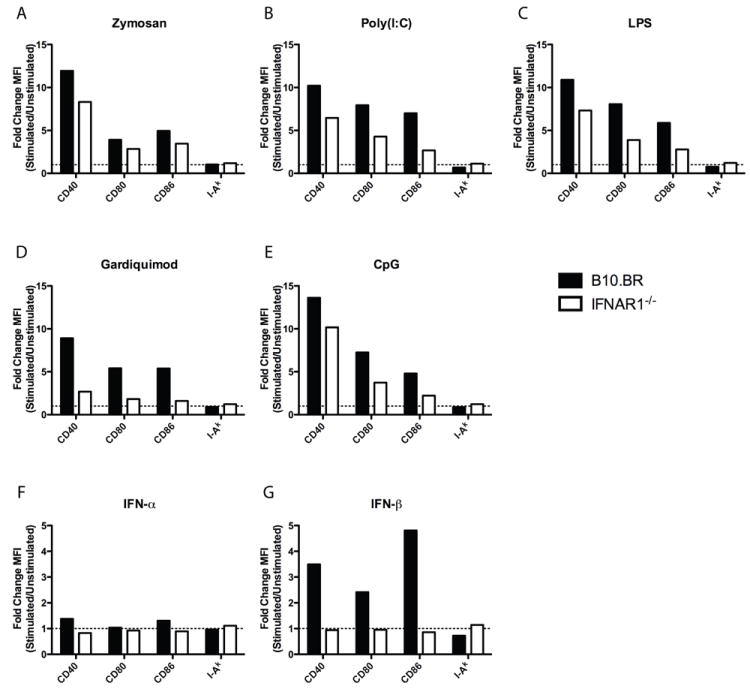
DC from wild type B10.BR or IFNAR1-/- mice were incubated alone or with (A) Zymosan, (B) Poly (I:C), (C) LPS, (D) Gardiquimod, (E) CpG, (F) IFN-alpha, or (G) IFN-beta for 18 hours. DC were stained for CD40, CD80, CD86, or I-Ak and analyzed by flow cytometry. Data represented as fold change MFI of stimulated over un-stimulated DC. Data gated on live CD11chigh Siglec H- cells. Data are representative of two independent experiments. Full histograms are shown in Figure S2.
Role of H2-DM and HEL catabolism in induced presentation
Since H2-DM prevents type B pMHC from being generated (4), regulation of H2-DM was considered as an explanation for the effects of TLR ligands. The function of H2-DM can be modulated by H2-DO, so it was necessary to examine expression of both molecules. To determine if expression of H2-DM or H2-DO was altered by TLR ligand stimulation, sorted cDC were incubated for 2, 6, or 18 hours with or without CpG-B, LPS, or IFN-beta stimulation and H2-DM, H2-DO, and H2-Aalpha (MHC II) were examined by qRT-PCR. Their expression increased two hours after stimulation dropping by six and eighteen hours (Figure 7A). Protein levels of H2-DM and H2-DO were examined by intracellular staining after eighteen hours of stimulation. CpG-B did not significantly alter levels of H2-DM protein, but did modestly increase expression of H2-DO (Figure 7B). LPS has previously been shown to have a similar effect on splenic DC, having no detectable effect on H2-DM protein levels, however they observed a modest decrease in H2-DO protein levels (40)
Figure 7. Role of H2-DM Regulation in TLR-Induced Type B pMHC Presentation from HEL and Accelerated Catabolism in CpG-B-Activated DC.
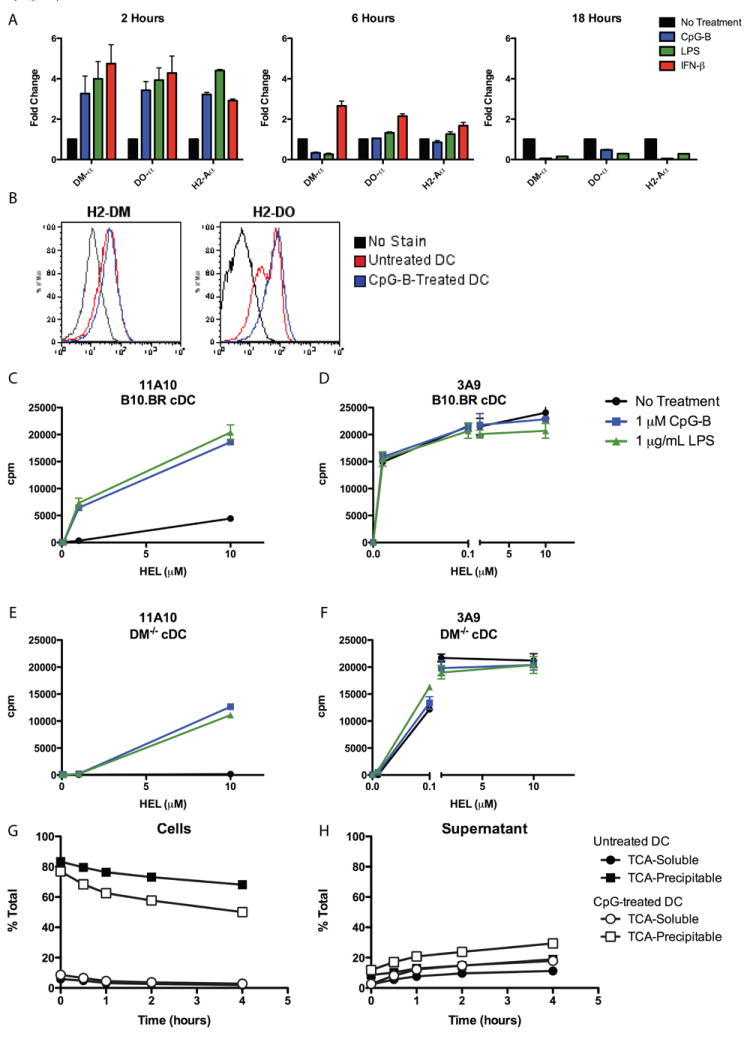
(A) qRT-PCR analysis of RNA isolated from sorted cDC (CD11chighSiglecH-CD19-) were incubated with stimulants for 2, 6, or 18 hours. (B) Intracellular flow-cytometry analysis of H2-DM and H2-DO protein levels in sorted cDC after 18 hours of stimulation. (C-F) Presentation to (A, E) type B 11A10 or (B, D) type A 3A9 by sorted (C, D) wild-type B10.BR or (E, F) H2-DM-/- cDC incubated with HEL protein with or without stimulants. (G, H) DC were incubated for 18 hours with or without CpG-B, washed, incubated with I125-HEL for two hours at room temperature, washed extensively, then chased for indicated times at 37°C. TCA-Soluble and TCA-Precipitable fractions were collected from the (G) intracellular and (H) supernatant fractions. Data plotted as percentage of total cpm at each time point.
To examine whether regulation of H2-DM contributed to induced type B pMHC presentation, cDC from B10.BR wild-type or H2-DM-deficient mice were incubated with HEL with or without stimulants as previously described. Consistent with previous reports (36), presentation of the type A pMHC was decreased by about ten fold in the H2-DM-deficient cDC, an indication that H2-DM is important for the assembly of HEL pMHC complexes. Importantly, H2-DM-deficient cDC presented type B pMHC from HEL protein in response to CpG and LPS, albeit at reduced levels compared to wild-type cDC. These data indicate that while regulation of H2-DM may have some contribution to TLR and type I IFN-induced type B presentation from HEL, it is not the key mechanism controlling the event.
Finally, we examined if CpG-B changed the rate of catabolism of I125-HEL. DC were incubated for eighteen hours alone or with CpG-B, then incubated with I125-HEL for two hours at room temperature followed by one, two, three, or four hour chases in media free of HEL. TCA soluble and precipitable fractions were collected from intracellular (Figure 7G) and supernatant (Figure 7H) fractions at each time point. CpG-B-activated DC showed slight acceleration of HEL catabolism compared to resting DC. We did not find evidence that CpG-B treated DC released peptides that would contribute to the type B presentation. Fig S3 describes the experiment.
DISCUSSION
The present report adds not only to the understanding of presentation of type B pMHC conformers of HEL, but also to the general effects of TLR ligand and type I IFN on the class II- MHC processing and presentation pathway. In the present study, exposure of DC to TLR ligands and type I IFN changed HEL protein handling, allowing strong processing and presentation of type B pMHC complexes. Notably, in the absence of TLR or type I IFN stimulation, as reported before, DC did not present type B pMHC complexes from HEL unless at very high concentrations. Both major DC subsets participated in induced type B pMHC presentation of HEL, although the degree to which each participated varied depending on the inflammatory signal.
Type I IFN signaling was important for type B pMHC presentation initiated by CpG, Poly(I:C), Gardiquimod, and Zymosan A, but not for LPS-initiated events. Type I IFN has been argued to be important for both cross-priming of CD8 T cells and for direct priming of CD8 T cells (13, 34, 41) and for cross-presentation induced by CpG (35). Here, with class II MHC, we found a strong effect not on presentation of the conventional type A epitopes, rather on the type B pMHCs. Induction of CD40 by LPS, Poly (I:C), and CpG on GM-CSF bone marrow-derived DC has been shown to be at least partially dependent on type I IFN signaling (28-31). Similarly, the increase in CD40, CD80, and CD86 expression after stimulation with Zymosan A, Poly (I:C), LPS, and CpG was partially dependent on type I IFN signaling. Gardiquimod-induced expression of these co-stimulatory molecules absolutely required type I IFN signaling. In our experiments we used T cell hybridomas that do not require costimulation, allowing examination exclusively of changes in levels of pMHC.
Previous studies showed effects of TLR ligands on the generation of class II pMHC (15-17) as well as on the modulation of co-stimulatory molecules (28-31). These findings have varied in the kind of assays the APC used for examination, the amounts and forms of ligands so a general consensus has not emerged on the key mechanisms of action. An issue thus is to separate processing and generation of pMHC from other components of the presentation pathway. We and others have shown subsequently, comparing results with T cell hybridomas and primary T cells, that both resting and TLR-activated DC were capable of processing and presenting antigen about equally (9, 10).
Although this report does not identify the way by which the type B pMHC epitopes were presented, some issues are pointing to possible mechanisms of action. The initial data indicates that H2-DM, a logical molecule to examine, may not be key in the effects seen here with TLR ligands. Pointedly, H2-DM-deficient DC presented type B pMHC from HEL protein after TLR ligands. While TLR stimulation regulated expression of H2-DO, which controls H2-DM activity, modulation of H2-DM function was ultimately not the key mechanism of type B presentation, as shown with the H2-DM gene knockout DC. We have ruled out the release of peptides from the treated APC, while the effects on catabolism by CpG were modest. The observation that TLR and type I IFN stimulation minimally increased type B pMHC presentation from peptide at low doses could be due to increased total MHC II expression and/or surface pMHC half-life. However, these effects were modest, while the effects on type B pMHC presentation from protein were robust; it is unlikely that they are the sole mechanism of induced type B pMHC presentation. We posit that presentation may be explained by changes in the dynamics of vesicular traffic leading to a flow of peptides from late vesicles into endosomes lacking H2-DM. Such a mechanism of action is now the subject of on-going examination. This could happen through changes in acidification of, or recruitment of proteases to, an early or recycling compartment. Interestingly, when bone marrow-derived DC were treated with CpG prior to antigen exposure, MHC II processing and presentation occurred in early and recycling compartments (14), a finding compatible with our hypothesis on how TLR ligands could promote presentation of type B pMHC of HEL.
A longstanding issue has been whether T cells recognizing type B epitopes from self-antigens could be involved in autoimmunity. This study indicates that TLR- and type I IFN-activated DC present type B pMHC from soluble HEL. It will be critical to determine whether type B pMHC from self-proteins are presented by TLR- and/or type I IFN-activated DC, allowing priming of auto-reactive T cells in vivo. We have previously shown that naturally arising insulin-reactive T cells against type B pMHC can transfer diabetes in the NOD model (7).
Supplementary Material
Acknowledgments
We thank Kathy Frederick for help with mice husbandry, Shirley Petzold and Jeremy Herzog for technical assistance, Susanne Schloemann and Olga Malkova for assistance with flow cytometry-assisted cell sorting, and Drs. Javier Carrero and Danielle Atibalentja for critical discussions and reading of the manuscript.
This work is supported by National Institutes of Health grants AI024742 and AI022033.
Footnotes
The authors have no financial conflicts of interest.
References
- 1.Pu Z, Carrero JA, Unanue ER. Distinct recognition by two subsets of T cells of an MHC class II-peptide complex. Proc Natl Acad Sci U S A. 2002;99:8844–8849. doi: 10.1073/pnas.092260499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Viner NJ, Nelson CA, Deck B, Unanue ER. Complexes generated by the binding of free peptides to class II MHC molecules are antigenically diverse compared with those generated by intracellular processing. J Immunol. 1996;156:2365–2368. [PubMed] [Google Scholar]
- 3.Lovitch SB, Unanue ER. Conformational isomers of a peptide-class II major histocompatibility complex. Immunol Rev. 2005;207:293–313. doi: 10.1111/j.0105-2896.2005.00298.x. [DOI] [PubMed] [Google Scholar]
- 4.Pu Z, Lovitch SB, Bikoff EK, Unanue ER. T cells distinguish MHC-peptide complexes formed in separate vesicles and edited by H2-DM. Immunity. 2004;20:467–476. doi: 10.1016/s1074-7613(04)00073-1. [DOI] [PubMed] [Google Scholar]
- 5.Peterson DA, DiPaolo RJ, Kanagawa O, Unanue ER. Quantitative analysis of the T cell repertoire that escapes negative selection. Immunity. 1999;11:453–462. doi: 10.1016/s1074-7613(00)80120-x. [DOI] [PubMed] [Google Scholar]
- 6.Lovitch SB, Walters JJ, Gross ML, Unanue ER. APCs present A beta(k)-derived peptides that are autoantigenic to type B T cells. J Immunol. 2003;170:4155–4160. doi: 10.4049/jimmunol.170.8.4155. [DOI] [PubMed] [Google Scholar]
- 7.Mohan JF, Levisetti MG, Calderon B, Herzog JW, Petzold SJ, Unanue ER. Unique autoreactive T cells recognize insulin peptides generated within the islets of Langerhans in autoimmune diabetes. Nat Immunol. 2010;11:350–354. doi: 10.1038/ni.1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lovitch SB, Esparza TJ, Schweitzer G, Herzog J, Unanue ER. Activation of type B T cells after protein immunization reveals novel pathways of in vivo presentation of peptides. J Immunol. 2007;178:122–133. doi: 10.4049/jimmunol.178.1.122. [DOI] [PubMed] [Google Scholar]
- 9.Veeraswamy RK, Cella M, Colonna M, Unanue ER. Dendritic cells process and present antigens across a range of maturation states. J Immunol. 2003;170:5367–5372. doi: 10.4049/jimmunol.170.11.5367. [DOI] [PubMed] [Google Scholar]
- 10.Wilson N, El-Sukkari D, Villadangos J. Dendritic cells constitutively present self antigens in their immature state in vivo and regulate antigen presentation by controlling the rates of MHC class II synthesis and endocytosis. Blood. 2004;103:2187–2195. doi: 10.1182/blood-2003-08-2729. [DOI] [PubMed] [Google Scholar]
- 11.Cella M, Engering A, Pinet V, Pieters J, Lanzavecchia A. Inflammatory stimuli induce accumulation of MHC class II complexes on dendritic cells. Nature. 1997;388:782–787. doi: 10.1038/42030. [DOI] [PubMed] [Google Scholar]
- 12.West M, Wallin R, Matthews S, Svensson H, Zaru R, Ljunggren H, Prescott A, Watts C. Enhanced dendritic cell antigen capture via toll-like receptor-induced actin remodeling. Science. 2004;305:1153–1157. doi: 10.1126/science.1099153. [DOI] [PubMed] [Google Scholar]
- 13.Durand V, Wong SYC, Tough DF, Le Bon A. Shaping of adaptive immune responses to soluble proteins by TLR agonists: A role for IFN-alpha/beta. Immunol & Cell Biol. 2004;82:596–602. doi: 10.1111/j.0818-9641.2004.01285.x. [DOI] [PubMed] [Google Scholar]
- 14.Askew D, Chu RS, Krieg AM, Harding CV. CpG DNA induces maturation of dendritic cells with distinct effects on nascent and recycling MHC-II antigen-processing mechanisms. J Immunol. 2000;165:6889–6895. doi: 10.4049/jimmunol.165.12.6889. [DOI] [PubMed] [Google Scholar]
- 15.Inaba K, Turley S, Iyoda T, Yamaide F, Shimoyama S, Reis e Sousa C, Germain R, Mellman I, Steinman R. The formation of immunogenic major histocompatibility complex class II-peptide ligands in lysosomal compartments of dendritic cells is regulated by inflammatory stimuli. J Exp Med. 2000;191:927–936. doi: 10.1084/jem.191.6.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Turley S, Inaba K, Garrett W, Ebersold M, Unternaehrer J, Steinman R, Mellman I. Transport of peptide-MHC class II complexes in developing dendritic cells. Science. 2000;288:522–527. doi: 10.1126/science.288.5465.522. [DOI] [PubMed] [Google Scholar]
- 17.Blander J, Medzhitov R. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature. 2006;440:808–812. doi: 10.1038/nature04596. [DOI] [PubMed] [Google Scholar]
- 18.Inaba K, Witmer-Pack M, Inaba M, Hathcock K, Sakuta H, Azuma M, Yagita H, Okumura K, Linsley P, Ikehara S, Muramatsu S, Hodes R, Steinman R. The tissue distribution of the B7-2 costimulator in mice: abundant expression on dendritic cells in situ and during maturation in vitro. J Exp Med. 1994;180:1849–1860. doi: 10.1084/jem.180.5.1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reis e Sousa C, Hieny S, Scharton-Kersten T, Jankovic D, Charest H, Germain R, Sher A. In vivo microbial stimulation induces rapid CD40 ligand-independent production of interleukin 12 by dendritic cells and their redistribution to T cell areas. J Exp Med. 1997;186:1819–1829. doi: 10.1084/jem.186.11.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.MacPherson G, Jenkins C, Stein M, Edwards C. Endotoxin-mediated dendritic cell release from the intestine. Characterization of released dendritic cells and TNF dependence. J Immunol. 1995;154:1317–1322. [PubMed] [Google Scholar]
- 21.Jakubzick C, Bogunovic M, Bonito A, Kuan E, Merad M, Randolph G. Lymph-migrating, tissue-derived dendritic cells are minor constituents within steady-state lymph nodes. J Exp Med. 2008;205:2839–2850. doi: 10.1084/jem.20081430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Niel G, Wubbolts R, Ten Broeke T, Buschow S, Ossendorp F, Melief C, Raposo G, van Balkom B, Stoorvogel W. Dendritic cells regulate exposure of MHC class II at their plasma membrane by oligoubiquitination. Immunity. 2006;25:885–894. doi: 10.1016/j.immuni.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 23.Shin J, Ebersold M, Pypaert M, Delamarre L, Hartley A, Mellman I. Surface expression of MHC class II in dendritic cells is controlled by regulated ubiquitination. Nature. 2006;444:115–118. doi: 10.1038/nature05261. [DOI] [PubMed] [Google Scholar]
- 24.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 25.Dietrich N, Lienenklaus S, Weiss S, Gekara N. Murine toll-like receptor 2 activation induces type I interferon responses from endolysosomal compartments. PLoS One. 2010;5:e10250. doi: 10.1371/journal.pone.0010250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barbalat R, Lau L, Locksley R, Barton G. Toll-like receptor 2 on inflammatory monocytes induces type I interferon in response to viral but not bacterial ligands. Nat Immunol. 2009;10:1200–1207. doi: 10.1038/ni.1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Swanson C, Wilson T, Strauch P, Colonna M, Pelanda R, Torres R. Type I IFN enhances follicular B cell contribution to the T cell-independent antibody response. J Exp Med. 2010;207:1485–1500. doi: 10.1084/jem.20092695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Honda K, Sakaguchi S, Nakajima C, Watanabe A, Yanai H, Matsumoto M, Ohteki T, Kaisho T, Takaoka A, Akira S, Seya T, Taniguchi T. Selective contribution of IFN-alpha/beta signaling to the maturation of dendritic cells induced by double-stranded RNA or viral infection. Proc Natl Acad Sci USA. 2003;100:10872–10877. doi: 10.1073/pnas.1934678100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoshino K, Kaisho T, Iwabe T, Takeuchi O, Akira S. Differential involvement of IFN-beta in Toll-like receptor-stimulated dendritic cell activation. Int Immunol. 2002;14:1225–1231. doi: 10.1093/intimm/dxf089. [DOI] [PubMed] [Google Scholar]
- 30.Longhi M, Trumpfheller C, Idoyaga J, Caskey M, Matos I, Kluger C, Salazar A, Colonna M, Steinman R. Dendritic cells require a systemic type I interferon response to mature and induce CD4+ Th1 immunity with poly IC as adjuvant. J Exp Med. 2009;206:1589–1602. doi: 10.1084/jem.20090247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoebe K, Janssen E, Kim S, Alexopoulou L, Flavell R, Han J, Beutler B. Upregulation of costimulatory molecules induced by lipopolysaccharide and double-stranded RNA occurs by Trif-dependent and Trif-independent pathways. Nat Immunol. 2003;4:1223–1229. doi: 10.1038/ni1010. [DOI] [PubMed] [Google Scholar]
- 32.Kamath A, Sheasby C, Tough D. Dendritic cells and NK cells stimulate bystander T cell activation in response to TLR agonists through secretion of IFN-alpha beta and IFN-gamma. J Immunol. 2005;174:767–776. doi: 10.4049/jimmunol.174.2.767. [DOI] [PubMed] [Google Scholar]
- 33.Van Uden JH, Tran CH, Carson DA, Raz E. Type I interferon is required to mount an adaptive response to immunostimulatory DNA. Eur J Immunol. 2001;31:3281–3290. doi: 10.1002/1521-4141(200111)31:11<3281::aid-immu3281>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 34.Le Bon A, Etchart N, Rossmann C, Ashton M, Hou S, Gewert D, Borrow P, Tough DF. Cross-priming of CD8(+) T cells stimulated by virus-induced type I interferon. Nat Immunol. 2003;4:1009–1015. doi: 10.1038/ni978. [DOI] [PubMed] [Google Scholar]
- 35.Kuchtey J, Chefalo PJ, Gray RC, Ramachandra L, Harding CV. Enhancement of dendritic cell antigen cross-presentation by CpG DNA involves type IIFN and stabilization of class I MHC mRNA. J Immunol. 2005;175:2244–2251. doi: 10.4049/jimmunol.175.4.2244. [DOI] [PubMed] [Google Scholar]
- 36.Koonce CH, Wutz G, Robertson EJ, Vogt AB, Kropshofer H, Bikoff EK. DM loss in k haplotype mice reveals isotype-specific chaperone requirements. J Immunol. 2003;170:3751–3761. doi: 10.4049/jimmunol.170.7.3751. [DOI] [PubMed] [Google Scholar]
- 37.Sheehan KCF, Lai KS, Dunn GP, Bruce AT, Diamond MS, Heutel JD, Dungo-Arthur C, Carrero JA, White JM, Hertzog PJ, Schreiber RD. Blocking monoclonal antibodies specific for mouse IFN-alpha/beta receptor subunit 1 (IFNAR-1) from mice immunized by in vivo hydrodynamic transfection. J Interferon Cytokine Res. 2006;26:804–819. doi: 10.1089/jir.2006.26.804. [DOI] [PubMed] [Google Scholar]
- 38.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 39.Edwards A, Diebold S, Slack E, Tomizawa H, Hemmi H, Kaisho T, Akira S, Reis e Sousa C. Toll-like receptor expression in murine DC subsets: lack of TLR7 expression by CD8 alpha+ DC correlates with unresponsiveness to imidazoquinolines. Eur J Immunol. 2003;33:827–833. doi: 10.1002/eji.200323797. [DOI] [PubMed] [Google Scholar]
- 40.Chen X, Reed-Loisel LM, Karlsson L, Jensen PE. H2-O expression in primary dendritic cells. J Immunol. 2006;176:3548–3556. doi: 10.4049/jimmunol.176.6.3548. [DOI] [PubMed] [Google Scholar]
- 41.Ahonen CL, Doxsee CL, McGurran SM, Riter TR, Wade WF, Barth RJ, Vasilakos JP, Noelle RJ, Kedl RM. Combined TLR and CD40 triggering induces potent CD8(+) T cell expansion with variable dependence on type I IFN. J Exp Med. 2004;199:775–784. doi: 10.1084/jem.20031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.