

Abstract
Generalized arterial calcification of infancy (GACI) is a rare condition characterized by arterial calcification within the internal elastic lamina associated with intimal proliferation, leading to stenosis of great and medium-sized vessels. This disease, caused by mutations in multiple exons of ENPP1, frequently results in death in infancy. Nowadays, the most promising therapeutic compounds for this rare disease are bisphosphonates. We describe a case of GACI associated with the novel mutation c.653A>T (p.D218V) in ENPP1 on both alleles. The male infant was delivered prematurely and developed heart failure, severe hypertension, and diffuse calcifications of all arterial districts. He was treated with etidronate (18 mg/kg/day); however, the clinical condition did not improve, and a resolution of calcifications was not observed. The infant died within the 6th month of life of ischemic heart failure. We conclude that even if the diagnosis of GACI is established early and bisphosphonate treatment is started early, the prognosis can be very poor.
Keywords: Bisphosphonates, Heart failure, Myocardial infarction, Inorganic pyrophosphate
Introduction
Generalized arterial calcification of infancy (GACI, MIM #208000) is a rare condition characterized by arterial calcification within the internal elastic lamina associated with intimal proliferation leading to stenosis of great and medium-sized arteries. The disease frequently results in death in infancy due to progressive ischemic heart failure. Although some cases of prenatal diagnosis have been reported, some patients are diagnosed postmortem at the earliest (Eronem et al. 2001). Presentation after the neonatal period is also unusual. Familial occurrence has suggested a genetic basis for this disease, which has been shown to be autosomal recessive (Rutsch et al. 2001). Although most patients die within the first 6 months of life, there have been rare reports of spontaneous resolution of the calcifications and cases of successful treatment with bisphosphonates (Sholler et al. 1984, Gleason et al. 1994, Ciana et al. 2006, Rutsch et al. 2008, Ramjan et al. 2009). We describe a case of GACI associated with a novel mutation in ENPP associated with preterm delivery and death early in life despite etidronate therapy.
Case Report
The patient was the second child of consanguineous Moroccan parents. The mother is a 40-year-old woman, who had seven previous pregnancies: a 16-year-old healthy boy, two miscarriages, two voluntary interruptions, and two stillborn infants delivered at 28 and 30 weeks of gestation. Amniocentesis of the current pregnancy revealed a normal 46, XY karyotype. In the 28th week of gestation, maternal hypertension was detected and fetal ultrasound outlined a severe pulmonary stenosis, tricuspid regurgitation (TR), cardiomegaly, polyhydramnios, and poor fetal movements. The mother underwent an urgent cesarean section because of acute fetal distress. The infant had a birth weight of 1,094 g, APGAR score was 4/6/10 at 1, 5 and 10 min, respectively, and the baby was intubated and ventilated. Physical examination showed no dysmorphic features, a grade 1 systolic murmur, a heart rate of 165 bpm and poor peripheral pulses. The liver was palpable 0.5 cm below the lower costal margin. The arterial blood pressure was normal (48/23 mmHg). Echocardiography, performed on the first day of life, revealed poor ventricular function (ejection fraction 40%), severe left ventricular hypokinesia and hypertrophy, brightness and hyperreflexia of the aortic wall from the arch to the abdominal tract with a low abdominal aorta pulsatility index. Inotropic support was immediately started with dobutamine and dopamine. Laboratory evaluation on the first day of life revealed elevated inflammatory markers (leukocytes 26,2100/μl [5,000–21,000/μl]); CrP 5.97 mg/dl [<0.5 mg/dl]; platelets 32,000/μl [250,000–450,000/μl]), acute kidney (creatinine 212.2 μmol/l [53–106 μmol/l]) and hepatic injury (aspartate aminotransferase, AST 397 U/l [15–60 U/l]; alanine aminotransferase, ALT 51 U/l [5–25 U/l]) with indirect hyperbilirubinemia (252 μmol/l [< 103 μmol/l]) that required exchange transfusion. A chest X-ray showed multiple periarticular calcifications of the left elbow (Fig. 1a). Cerebral ultrasound revealed marked cerebral parenchymal hyperechogenicity. Since the first hours of life the infant was treated with intravenous antibiotics, immunoglobulins, and cortisone for suspected sepsis. This was associated with a general clinical improvement and an increase of the cardiac ejection fraction, allowing for extubation on the 6th day of life. Treatment with phenobarbital and captopril was begun on the 10th day of life when the infant developed arterial hypertension (mean arterial pressure between 70–80 mmHg) and tonic-clonic seizures. The electroencephalogram (EEG) showed synchronous and asynchronous bursts, delta brush, and long interburst inactivity. During treatment the seizures resolved, while the hypertension persisted. Further echocardiography showed myocardial hyperechogenic foci on the apical portion of the left ventricle, hyperechogenicity, and brightness of both the pulmonary and the aortic valve (Fig. 2). Similar findings were also detected in the pulmonary, celiac, and renal arteries. Total body X-ray revealed more periarticular calcifications (left radiocarpal and both tarsal joints) and calcific spots in the left brachial and both iliac and femoral arteries (Fig. 1b). Total-body computed tomography (CT) confirmed widespread arterial calcifications and calcifications of the left lobe of the liver and the right kidney (Fig. 1c). At the age of 4 weeks, GACI was suspected after having excluded other potential causes of hyperechogenicity of the great vessels and hypertension. Because of congestive heart failure, which did not allow further fluid overload and because of limited venous access, an oral therapy with etidronate was started at the age of 1 month at a dose of 18 mg/kg body weight per day. Within the 2nd month of life, cerebral ultrasonography and Doppler studies revealed multicystic encephalomalacia with reduced arterial blood flow in the anterior and middle cerebral arteries bilaterally. At the age of 5 months, the infant was discharged on oral medication consisting of etidronate (18 mg/kg/day), phenobarbital (2.2 mg/kg × 2/day), captopril (0.3 mg/kg × 3/day), digoxine (0.016 mg/day), and vitamin D (900 U/day). Fifteen days later, the patient was readmitted for persistent vomiting and feeding refusal. On physical examination, the infant was tachypneic, tachycardic, and cyanotic. He started to have frequent episodes of desaturation with bradycardia requiring intubation. Echocardiography revealed a severely dilated cardiomyopathy and reduced ventricular function (EF less than 20%). Few hours later, he developed severe hypotension and died after a failed resuscitation attempt. Permission for autopsy was denied.
Fig. 1.

Radiographic manifestations of generalized arterial calcification of infancy. Panel A: X-ray scan of the patient’s left elbow. Note the calcification of the elbow joint. Panel B: Abdominal X-ray of the patient at 20 days of life. Note the calcification of the abdominal aorta (A) and of the bifurcation of the iliac arteries (B). Panel C: Chest CT of the patient at the age of 2 months. Note a ring-like aortic calcification (A) and spread calcifications over the left lobe of the liver (B) and in the right kidney (C)
Fig. 2.

Echocardiographic manifestations of generalized arterial calcification of infancy. Panel A: Echocardiogram at 1 month of life (uppersternal axis view). Note the calcification of the aortic arch, descending aorta (D) and of the supraaortic vessels: brachiocephalic trunk (A), left carotid artery (B), left subclavian artery (C). Panel B: Echocardiogram at 1 month of life (parasternal short axis view). Note the hyperechogenicity of the pulmonary (P) and aortic (A) anulus and of the coronary arteries (C)
Genetic Analysis
With a set of 25 primer pairs, all 25 exons and their flanking splice sites of the ENPP1 gene were amplified from genomic DNA by polymerase chain reaction (PCR). The PCR products were directly sequenced bidirectionally using an ABI 3730 DNA Analyzer and a BigDye Terminator v1.1 Cycle Sequencing Kit according to the manufacturer’s protocol (Applied Biosystems). All primer sequences are available on request. Mutations were compared with the ENSEMBL polymorphism database. We detected the novel mutation c.653A>T (p.D218V) on exon 6 in ENPP1 on both alleles in the patient’s DNA. By analyzing the parental DNA, it was demonstrated that both parents carried the same mutation on one allele (Fig. 3).
Fig. 3.
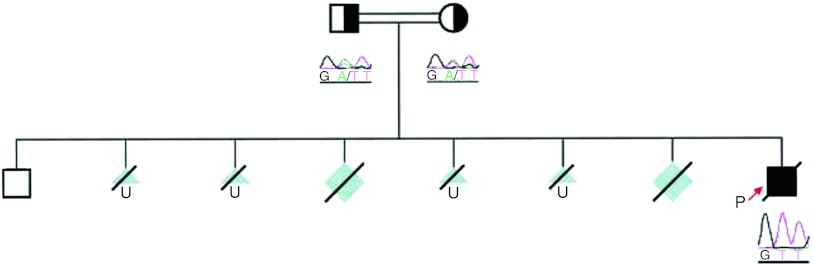
Pedigree tree of the family showing known genotypes and clinical phenotypes. The affected allele is shaded black, whereas wild type alleles are white, unknown affected is shaded gray
Discussion
GACI is a rare autosomal recessive disorder due to systemic deficiency of nucleotide pyrophosphatase/phosphodiesterase 1 (NPP1) activity caused by mutations in multiple exons of ENPP1 (MIM #173335), a gene located on chromosome 6q22–q23 (Rutsch et al. 2003). This gene encodes NPP1, a class II (intracellular NH2 terminus) transmembrane glycoprotein ectoenzyme of 130 kDa with an extracellular domain containing two somatomedin B-like regions, a conserved calcium-binding EF hand, and a conserved phosphodiesterase/pyrophosphatase catalytic site (Terkeltaub 2001). The mutation c.653A>T (p.D218V) on exon 6 present on both alleles in our patient leads to a change of a conserved aspartate to a valine residue at position 218 in the catalytic side of NPP1.
The NPP1 cell surface enzyme regulates soft tissue calcification and joint cartilage mineralization by generating extracellular inorganic pyrophosphate (PPi) (Rutsch et al. 2001). NPP1 is also involved both in the physiological inhibition of hydroxyapatite crystal growth and in chondrogenesis (Johnson et al. 2005). Deficiency of this enzyme therefore results in deposition of calcium hydroxyapatite crystals at the level of the lamina elastica interna of muscular arteries. Myointimal proliferation is found to be associated with calcified spots, but may also occur in areas lacking calcification and vice versa. This suggests a separate pathological process involved in the development of GACI (Dlamini et al. 2009). The aorta, renal, mesenteric, and carotid arteries are usually involved, while the cerebral vessels are commonly spared by the calcification process (Rutsch et al. 2008). Extravascular foci of periarticular calcification are often present. The first cases of GACI described were mostly recognized postmortem (Moran 1975). However, along with improving imaging technology in recent years, the diagnosis has frequently been established in early life and antenatal diagnosis has already been documented. Fetal ultrasound may reveal dilated cardiac ventricles, hydrops fetalis, and hyperechogenic large vessels (Levine et al. 2001). The most common clinical presentation, however, occurs within the neonatal period. Respiratory distress is the presenting feature in more than 50% of cases, followed by feeding intolerance, poor weight gain, tachypnea, tachycardia, lethargy, pallor, and cyanosis (Glatz et al. 2006, Rutsch et al. 2008). Patients may present with failure to thrive, hypertension, myocardial infarctions, and convulsions (Van der Sluis et al. 2006). Arterial biopsy is the gold standard for diagnosis, but this rather invasive technique can be replaced by combining standard imaging techniques (conventional radiographs, CT, and ultrasound). Recently, MRI and MR angiography (MRA) have been successfully used in the evaluation of patients with suspected GACI avoiding radiation exposure and potential renal toxicity associated with contrast enhanced CT examination (Pao et al. 1998; Tran and Boechat 2006). Widespread arterial calcifications can be related to numerous causes; thus, renal, osseous, parathyroid, and metabolic disorders should be ruled out before suspecting GACI (Patel et al. 2004). The differential diagnosis includes metastatic calcification secondary to advanced renal disease, arterial calcifications associated with anomalies of the heart and great vessels, hypervitaminosis D, primary or secondary hyperparathyroidism, Williams syndrome, syphilis, osteogenesis imperfecta, pseudoxanthoma elasticum, sepsis with myocarditis, endocarditis, coronary arteritis, rheumatic fever, polyarteritis nodosa, and also maternal conditions such as diabetes mellitus, allergy with asthma, and bronchiectasis (Glatz et al. 2006).
Few attempts to treat patients with GACI have been made with corticosteroids, estrogens, thyroid extract, and prostaglandins (Ciana et al. 1997). The most promising therapeutic compounds are bisphosphonates. Bisphosphonates are synthetic analogs of inorganic pyrophosphate, which block the conversion of calcium phosphate to hydroxyapatite and therefore may reduce ectopic calcification. So far, a standardized treatment approach for this rare disease could not be established. However, a recent multicenter genetic study and observational analysis has grouped 55 subjects affected by this disease. Seventeen patients have been treated with bisphosphonates, namely etidronate (10–20 mg/kg body weight per day p.o), pamidronate (0.1 mg/kg per week up to 5 mg/kg per day i.v.), clodronate or risedronate. Survival of these subjects was 65%, while 69% of patients not treated with bisphosphonates died during infancy (Rutsch et al. 2008). Treatment with aminobisphosphonates such as pamidronate disodium followed by oral risedronate was associated with a complete resolution of the calcifications and a good outcome (Ramjan et al. 2009). However, long-term survival has been reported also in patients receiving no specific anti-mineralisation therapy, suggesting that spontaneous resolution of calcification may also occur (Glatz et al. 2006). Etidronate, a non-nitrogen-containing compound among the bisphosphonates, has a stronger effect in inhibiting mineralization compared to the newer aminobisphosphonates and showed no adverse effect on growth (Van der Sluis et al. 2006). It is reasonable that the reduction of vascular calcifications increases vascular compliance and thereby reduces cardiac afterload. However, high-dose etidronate injections have been shown to induce vitamin D resistant rickets in rats (Atkin et al. 1988). Therefore, parameters of bone metabolism have to be closely monitored under etidronate therapy.
It has been hypothesized that hypophosphatemia may inhibit potential effects of defective NPP1-mediated PPi generation, as PPi and inorganic phosphate seem to have mutually antagonistic roles in tissue mineralization. In this respect, an interesting link has recently been reported (Lorenz-Depiereux et al. 2010, Levy-Litan et al. 2010) between inactivating mutations of ENPP1 and hypophosphatemic rickets characterized by phosphaturia and elevated FGF23 levels. Moreover, previous studies on human subjects with ENPP1 mutations have found a variable and incomplete reciprocal association between aberrant calcification and hypophosphatemic rickets (Ciana et al. 2006, Rutsch et al. 2008). Thus, inducing phosphaturia may have a potential benefit for GACI patients. This, however, has not become common practice. Nevertheless, bisphosphonates promote resolution of calcifications, but have no influence on the myointimal proliferation, which plays an important role in vascular stenoses (Rutsch et al. 2008).
In this particular case, the extreme prematurity of the infant, the severity and variability of clinical conditions since birth and the lack of diagnostic certainty may have delayed the diagnosis and thus the start of bisphosphonate therapy. Furthermore, during 4 months of therapy, a resolution of calcifications has not been observed. It may well be that the initiation of therapy might have been too late to resolve preexisting calcifications. Resistance to bisphosphonates may also be due to the particular mutation p.D218V in ENPP1 affecting a conserved residue in the catalytic domain of the protein. Our experience again stresses the fact that GACI can present with a very rapid and fatal course. More data should be thus collected to better define prognosis and therapeutic possibilities.
Acknowledgment
Y.N. and F.R are supported by a grant from the Interdisciplinary Center for Clinical Research, Münster University.
References
- Atkin I, Ornoy A, Pita JC, et al. EHDP-induced rachitic syndrome in rats is not reversed by vitamin D metabolites. Anat Rec. 1988;220(1):22–30. doi: 10.1002/ar.1092200104. [DOI] [PubMed] [Google Scholar]
- Ciana G, Colonna F, Forleo V, et al. Idiopathic arterial calcification in infancy: effectiveness of prostaglandin infusion for treatment of secondary hypertension refractory to conventional therapy: a case report. Pediatr Cardiol. 1997;18:67–71. doi: 10.1007/s002469900114. [DOI] [PubMed] [Google Scholar]
- Ciana G, Trappan A, Bembi B, et al. Generalized arterial calcification in infancy: two siblings with prolonged survival. Eur J Pediatr. 2006;165:258–263. doi: 10.1007/s00431-005-0035-6. [DOI] [PubMed] [Google Scholar]
- Dlamini N, Splitt M, Durkan A et al (2009) Generalized arterial calcification of infancy: phenotypic spectrum among three siblings including one case without obvious arterial calcifications. Am J Med Genet A 149A:456–460 [DOI] [PubMed]
- Eronem M, Pohjavuori M, Heikkila P. Fatal outcome of two siblings with idiopathic arterial calcification of infancy diagnosed in utero. Pediatr Cardiol. 2001;22:167–169. doi: 10.1007/s002460010189. [DOI] [PubMed] [Google Scholar]
- Glatz AC, Pawel BR, Hsu DT, et al. Idiopathic Infantile arterial calcification: two case reports, a review of the literature and a role for cardiac transplantation. Pediatr Transplant. 2006;10:225–233. doi: 10.1111/j.1399-3046.2005.00414.x. [DOI] [PubMed] [Google Scholar]
- Gleason MM, Weber HS, Curan SE, et al. Idiopathic infantile arterial calcinosis: intermediate-term survival and cardiac sequelae. Am Heart J. 1994;127:691–695. doi: 10.1016/0002-8703(94)90683-1. [DOI] [PubMed] [Google Scholar]
- Johnson K, Polewski M, van Etten D, et al. Chondrogenesis mediated by PPi depletion promotes spontaneous aortic calcification in NPP1-/- mice. Arterioscler Thromb Vasc Biol. 2005;25(4):686–691. doi: 10.1161/01.ATV.0000154774.71187.f0. [DOI] [PubMed] [Google Scholar]
- Levine JC, Campbell J, Nadel A. Prenatal diagnosis of idiopathic infantile arterial calcification. Circulation. 2001;103:325–326. doi: 10.1161/01.CIR.103.2.325. [DOI] [PubMed] [Google Scholar]
- Levy-Litan V, Hershkovitz E, Avizov L et al (2010) Autosomal-recessive hypophosphatemic rickets is associated with an inactivation mutation in the ENPP1 gene. Am J Hum Genet 86:273–278 [DOI] [PMC free article] [PubMed]
- Lorenz-Depiereux B, Schnabel D et al (2010) Loss-of-function ENPP1 mutations cause both generalized arterial calcification of infancy and autosomal-recessive hypophosphatemic rickets. Am J Hum Genet 86:267–272 [DOI] [PMC free article] [PubMed]
- Moran JJ. Idiopathic arterial calcification of infancy: a clinicopathologic study. Pathol Annu. 1975;10:393–417. [PubMed] [Google Scholar]
- Pao DG, DeAngelis GA, Lovell MA. Idiopathic arterial calcification in infancy: sonographic and magnetic resonance findings with pathologic correlation. Pediatr Radiol. 1998;28:256–259. doi: 10.1007/s002470050344. [DOI] [PubMed] [Google Scholar]
- Patel M, Savvas A, Rustum S, et al. Idiopathic arterial calcification in childhood. Pediatr Radiol. 2004;34:652–655. doi: 10.1007/s00247-004-1166-z. [DOI] [PubMed] [Google Scholar]
- Ramjan KA, Roscioli T, Rutsch F, et al. Generalized arterial calcification of infancy: treatment with bisphosphonates. Nat Clin Pract Endocrinol Metab. 2009;5:167–172. doi: 10.1038/ncpendmet1067. [DOI] [PubMed] [Google Scholar]
- Rutsch F, Vaingankar S, Johnson K, et al. PC-1 nucleoside triphosphate pyrophosphohydrolase deficiency in idiopathic infantile arterial calcification. Am J Pathol. 2001;158:543–554. doi: 10.1016/S0002-9440(10)63996-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutsch F, Ruf N, Vaingankar S, et al. Mutations in ENPP1 are associated with “idiopathic” infantile arterial calcification. Nat Genet. 2003;34(4):379–381. doi: 10.1038/ng1221. [DOI] [PubMed] [Google Scholar]
- Rutsch F, Böyer P, Nitschke Y, et al. Hypophosphatemia, hyperphosphaturia, and bisphosphonate treatment are associated with survival beyond infancy in generalized arterial calcification of infancy. Circ Cardiovasc Genet. 2008;1:133–140. doi: 10.1161/CIRCGENETICS.108.797704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sholler GF, Yu JS, et al. Generalized arterial calcification in infancy: three case reports, including spontaneous regression with long-term survival. J Pediatr. 1984;105:257–260. doi: 10.1016/S0022-3476(84)80123-7. [DOI] [PubMed] [Google Scholar]
- Terkeltaub RA. Inorganic pyrophosphate generation and disposition in pathophysiology. Am J Physiol Cell Physiol. 2001;281:C1–C11. doi: 10.1152/ajpcell.2001.281.1.C1. [DOI] [PubMed] [Google Scholar]
- Tran KH, Boechat MI. Idiopathic infantile arterial calcification: imaging evaluation and the usefulness of MR angiography. Pediatr Radiol. 2006;36:247–253. doi: 10.1007/s00247-005-0044-7. [DOI] [PubMed] [Google Scholar]
- Van Der Sluis IM, Boot AM, Vernooij M, et al. Idiopathic infantile arterial calcification: clinical presentation, therapy and long-term follow-up. Eur J Pediatr. 2006;165:590–593. doi: 10.1007/s00431-006-0146-8. [DOI] [PubMed] [Google Scholar]