

Abstract
Newborn screening was implemented in the 1960s with screening for phenylketonuria (PKU). In the same decade, it became possible to screen for classical galactosemia, a rare autosomal recessive inherited disorder, which is potentially life threatening if not treated. While newborn screening for PKU has become almost universal, galactosemia is included only in a minority of European newborn screening programs. The major arguments why galactosemia is excluded from newborn screening programs are that the disease can be diagnosed clinically, there is a high rate of false positives and long-term complications are common despite early diagnosis.
Here, we report how we have decreased the number of false-positive galactosemia recalls to less than 0.01%, using a two-tier test strategy. All samples are tested with the Beutler blood spot test, a method that measures galactose-1-phosphate uridyltransferase activity. Samples with less than ≤15% activity are tested for galactose with a galactose dehydrogenase test (the rapid GAL-DH test), which catalyzes the oxidation of galactose and the reduction of NAD+ to NADH that is estimated visually by fluorescence under UV-light. Both tests are semiquantitative.
With this strategy, screening for galactosemia is inexpensive, does not demand a heavy workload, and has a low false-positive re-call rate. The incidence of classical galactosemia in Sweden is 1/100,000, which is lower than the reported incidence in other European countries. Despite this, newborn screening for galactosemia has never been questioned.
Concise sentence: Screening for galactosemia using well-established methods to reduce the false-positive rate.
Introduction
Galactosemia is a rare autosomal recessive inherited disorder. Depending on which enzyme is affected the disorder can be divided into three subgroups; galactose-1-phosphate uridyltransferase deficiency (GALT; OMIM 230400), galactokinase deficiency (GALK; OMIM 230200), and galactose-4-epimerase deficiency (GALE; OMIM 230350). GALT-deficiency, also known as classical galactosemia, was first described in 1917 by Goppert (1917) and in 1956 galactose-1-phosphate uridyltransferase (EC 2.7.7.12) was identified as the enzyme that was deficient in this disease (Isselbacher et al. 1956). Galactosemia due to GALT-deficiency is the most common of the galactosemia disorders, with a frequency of approximately 1/60,000 in Caucasians.
Galactosemia became a candidate disease for newborn screening (NBS) programs, which were under development in the 1960s. A bacterial inhibition assay was introduced in 1964 followed by the Beutler enzyme spot test in 1966 (Beutler and Baluda 1966). With the bacterial inhibition assay, all galactosemias can be detected while the Beutler method only detects patients with GALT-deficiency. Today, there are other screening methods for galactosemia as well, the most common being measurement of galactose (Gal) and galactose-1-phosphate (G-1-P) in blood spots. A major disadvantage with these screening methods for galactosemia has been the high frequency of false-positive results (Jeong et al. 2007; Freer et al. 2010).
GALT-deficiency is a potentially lethal disease with symptoms occurring within the first weeks of life. Untreated the neonate will show progressive symptoms from weight loss, vomiting and diarrhea, lethargy and hypotonia to jaundice, cataracts, hepatomegaly, prolonged bleeding time, and septicemia leading to neonatal death. With the initiation of a galactose and lactose restricted diet, the acute symptoms will regress, but despite early treatment long-term complications such as mild speech and language delay, premature ovarian failure (POF), and neurological defects are not uncommon (Waggoner et al. 1990).
While newborn screening for phenylketonuria (PKU; OMIM 261600) has become almost universal, galactosemia screening is included only in a minority of European NBS programs. In 2007, nine countries in Europe had a nationwide screening program for galactosemia while Finland was the only country that did not have a nationwide screening program for PKU (www.isns-neoscreening.org). There are three major arguments why galactosemia is excluded from the NBS programs (1) the disease can be diagnosed clinically, (2) high rate of false positives, and (3) long-term complications are common in spite of early treatment.
Most newborns with classical galactosemia will show symptoms of the disease at the time of recall but by screening most deaths in Escherichia coli sepsis will be avoided. In Sweden, only four patients with classical galactosemia born before the start of screening in 1967 are alive. After 1967, one patient has died due to galactosemia. Considering these results, neonatal deaths in galactosemia decrease significantly if screening for the disease is performed. It is possible to perform screening for classical galactosemia with a low false-positive rate using well-established methods and appropriate cut-off values, which is described in this article. Long-term complications cannot be avoided with the knowledge that is available today but this may change in the near future when new methods for treatment and follow up have been developed and in use (Coman et al. 2010). Screening for galactosemia also makes it possible to offer parents genetic counselling and prenatal diagnosis in subsequent pregnancies.
Screening in Sweden
The first method for galactosemia screening in Sweden was based on a bacterial inhibition assay (collaboration with Thalhammar, Vienna) in line with the Guthrie method for the screening of PKU (Guthrie and Susi 1963). The method was included in the NBS program in 1967 and used until 1985 when the Beutler spot test was employed (Fig. 1). During the period 1967–1985, 22 cases of GALT-deficiency and 3 cases of GALK-deficiency were identified but no cases of GALE-deficiency. In 1983, the Beutler spot test was introduced as a second tier test and in 1985, we decided to change to the Beutler spot test as the first step (Beutler and Baluda 1966). From then on we have only been screening for GALT-deficiency. Throughout the years a quantitative determination of Gal and G-1-P has been performed on all positive cases, but different cut-off values have been employed.
Fig. 1.

Determination of GALT-activity according to Beutler. GALT galactose-1-phosphate uridyltransferase, PGM phosphoglucomutase, G6P-DH glucose-6-phosphate dehydrogenase
The Beutler Spot Test
The Beutler spot test is a semiquantitative, fluorometric method, which measures GALT-activity. With the Beutler spot test, patients with GALT-deficiency are identified. In addition, severe deficiencies of glucose-6-phosphate dehydrogenase (EC 1.1.1.49) and phosphoglucomutase (EC 5.4.2.2) can be picked up, since these enzymes are required for the test to function (Fig. 1).
A disadvantage with the Beutler spot test is that it can show a false-positive result (absent enzyme activity) if the sample contains EDTA. This is common in samples taken from older children, i.e. adoptive and immigrants up to the age of 18 years, which are offered the screening test on arrival in Sweden. It is not uncommon that samples containing EDTA that are positive in the Beutler screening also show false-positive results in the screening for congenital adrenal hyperplasia (OMIM 201910) when using the Auto DELFIA method, a fact that is assisting in the evaluation of the Beutler results. Samples that have been exposed to heat are another source of false-positive results.
Two-Tier Testing
When a case of galactosemia is suspected due to a positive Beutler spot test it is of importance to verify the result rapidly. Quantitative analysis of Gal and Gal-1-P in our laboratory takes 4 h, which we consider to be too long. We therefore introduced a rapid and simple method to determine total Gal semiquantitatively in samples with low activity in the Beutler test – “the rapid galactose dehydrogenase (EC 1.1.1.120) test” (rapid GAL-DH test). This method has been in use for the past 20 years. The Beutler spot test is ready in the same day as the screening sample arrives in the laboratory. If a sample shows an activity of 15% or less in the Beutler spot test, the rapid GAL-DH test is performed. The rapid GAL-DH test is a semiquantitative, fluorometric spot test where total galactose is approximated visually within an hour. With this two-tier approach, the diagnosis of galactosemia can be confirmed on the same day the sample arrived in the laboratory. In addition, a quantitative determination of Gal and G-1-P is always performed the following day.
Method
Gal in filter paper spots is determined enzymatically with the enzyme GAL-DH, which catalyzes the oxidation of galactose. At the same time nicotinamide adenine dinucleotide (NAD+) is reduced to NADH, which is estimated visually by fluorescence under UV-light (Fig. 2).
Fig. 2.
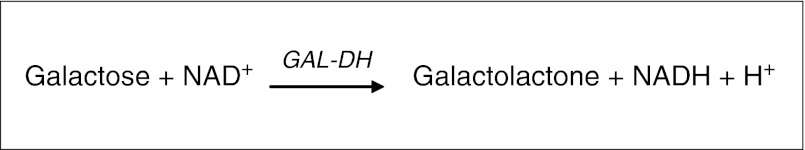
Principle for rapid GAL-DH. GAL-DH galactose dehydrogenase
The analysis has five controls consisting of one blank filter paper spot, blood filter paper samples containing none, 2, 4, and 8 mM Gal, respectively. Blood spots from a healthy child are used as a negative control. Two dried blood spots (diameter 3.0 mm) from each control and the samples to be analyzed are incubated at 37 °C in glass tubes for 30 min in a reaction mixture of 100 μl containing 50 μl Tris-buffer (170 mM, pH 8.0) and 50 μl G-1-P-mix containing: 19.61 μl Tris-buffer (50 mM pH 8.6); 24.51 μl NAD (0.013 M); 0.98 μl Gal-DH (2.5 mg/ml) and 4.9 μl alkaline phosphatase (300U/ml). An aliquot of 10 μl from each incubation mixture is spotted on a filter paper and dried in room temperature. The fluorescence is visually interpreted under long-wave ultraviolet light. The intensity of the fluorescence corresponds to the level of total Gal. Samples showing fluorescence greater than 2 mM total Gal are considered positive (Fig. 3).
Fig. 3.
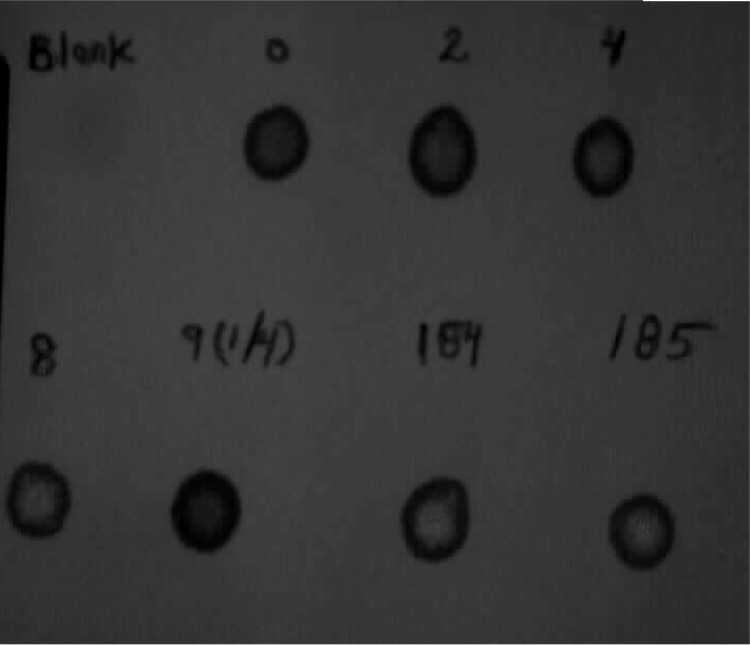
Rapid GAL-DH test. First line: samples containing 0, 2, 4 mM galactose. Second line: sample containing 8 mM galactose, number 9 healthy child, number 184 and 185 twins with classical galactosemia. Number 184 had 7.3 mmol/l galactose and number 185 had 5.3 mmol/l galactose when determined quantitatively
Results and Discussion
During 1985–2009 when the Beutler spot test has been performed, patients have been diagnosed at an average age of 6.5 days (range 4–14 days). In 2007, as a preparation for expanded screening with tandem mass technique, the recommended time of sampling was changed from as soon as possible after 72 h to after 48 h of age. The samples are taken at an average of 2.9 days after birth and reach the laboratory at an average time of 5.5 days after birth. This implies that a child with galactosemia born in Sweden will have a diagnosis and be on treatment within the first week of life.
Two cut-off levels are in use for the Beutler spot test, one for samples with an activity of 10% or less and one for samples with activity over 10% and up to 15%, see algorithm (Fig. 4). Infants with ≤10% activity are re-called promptly.
Fig. 4.

Algorithm for newborn screening for galactosemia using the two-tier approach with Beutler screening and the rapid GAL-DH on the initial screening sample. Positive Beutler test ≤15% activity. Positive rapid Gal-DH test approximately ≥2 mM galactose
The false-positive rate has dropped to less than one newborn a year after lowering the cut-off value for the Beutler spot test to ≤15%, and increasing the cut-off for G-1-P to ≥1.5 mmol/l (Table 1). After including the rapid GAL-DH test in 1991 as a second tier test all patients with classical galactosemia have had a verified diagnosis at the time of recall.
Table 1.
Newborn screening for galactosemia in Sweden 1967–2010
| Years | Newborns | True positives | Incidence | False positive | Incidence | Method | Cut off% | Quantitative determination | |
|---|---|---|---|---|---|---|---|---|---|
| Gal mmol/l | G-1-P mmol/l | ||||||||
| 1967–1985 | 1,780,600 | 22 | 1/81,000 | 336 | 1/5,300 | Bacterial inhibition assay | >0.6 | >0.6 | |
| 1986–1991 | 677,900 | 3 | 1/226,000 | 80 | 1/8,500 | Beutler | 30 | >0.6 | >0.6 |
| 1992–2010 | 1,973,400 | 18 | 1/108,000 | 10 | 1/194,000 | Beutler | 15 | >0.5 | >1.5 |
| Total | 4,401,900 | 43 | 1/103,000 | 424 | 1/10,000 | ||||
The two most important changes to reduce the false-positive rate in the galactosemia screening were change of method from bacterial inhibition assay to the Beutler spot test in 1985 and lowering the cut-off value for the Beutler spot test in 1992. When the Beutler spot test was introduced as the test method for galactosemia screening in 1985, the cut-off value was set at an activity of 30% or less. With this cut-off value, newborns with Duarte galactosemia (DG) (newborns carrying one Duarte allele and one classic galactosemia allele) who have approximately 25% rest activity and also some patients heterozygous for one severe mutation were recalled giving a high false-positive incidence (1/8,500 newborns). When patients with DG were caught in the NBS for galactosemia, our program chose to recommend treatment and follow up of the newborns with half of the breast milk exchanged for galactose free formula for 4–6 months. This was routine in 1983–1989. Already at this time it was uncertain if newborns with DG needed treatment with a galactose restricted diet (Gitzelmann and Bosshard 1995). Our experience was that at the end of the follow-up at 6 months of age the patients had normal galactose metabolites. A decision was made in 1992 that the goal of the galactosemia screening was to detect patients with classical galactosemia excluding newborns with DG. Evaluation of the true positive cases showed that all cases with classical galactosemia have an enzyme activity of 8% or less in the Beutler test. Hence, the criteria for recall were changed to an activity of ≤15% and a positive rapid Gal-DH test. With this approach, almost all newborns with DG are unrecognized. There is still an uncertainty of whether DG patients need treatment or not but several publications indicate that infants with DG have a favorable outcome on galactose unrestricted diet (Ficicioglu et al. 2008, 2010; Powell et al. 2009; Fernhoff 2010).
Some laboratories combine metabolite screening and enzymatic testing, either performing both or as a two-tier test. If the Beutler spot test is the second test in a two-tier test combination where Gal and Gal-1-P are measured first, galactosemia due to GALK-deficiency and GALE-deficiency can also be identified. The latter conditions are, however, extremely rare. When metabolite screening is the approach, ingestion of galactose/lactose containing formula is necessary. Another disadvantage when using the galactose metabolites Gal and G-1-P to detect galactosemia is a high rate of false-positive cases, due to an increase of Gal and G-1-P seen in newborns with DG and in some newborns who are heterozygous for one classical galactosemia allele. There are also newborns who have a transient increase of galactose metabolites. With our two-tier approach the false-positive rate has decreased to almost null and furthermore no missed cases of galactosemia have been reported to the laboratory.
A drawback with the present lower age at sampling is an increased risk of the child not having been fed lactose when the screening sample is taken. In this case the rapid GAL-DH test will be negative. This has been verified in two cases of samples taken from younger siblings with galactosemia having not been given any Gal containing food. In these cases, we have found only a slight elevation of Gal-1-P in the screening sample, 0.53 and 0.90 mmol/l respectively (reference value <0.1 mmol/l) but no increase in Gal when measured quantitatively. To assure that we do not miss any cases, all samples with less than 10% activity and negative on the rapid GAL-DH tests are evaluated considering the age of the child at sampling and an assurance that the child has been fed galactose.
Screening for galactosemia using the Beutler method is inexpensive. The total cost per child for working time, reagents, and machines is less than 0.25 Euro.
Conclusion
We find it surprising that galactosemia is not included in more screening programs, whilst organic acidurias and urea cycle disorders are. Several of the disorders included in the expanded screening by tandem mass result in severe sequelae in spite of early treatment. Neonatal screening cannot be governed by methodology when cheap and well-performing methods are available.
The incidence of classical galactosemia in Sweden is 1/100 000, which is lower than the reported incidence in other European countries. Despite this, newborn screening for galactosemia has never been questioned. One reason could be that we have had a well-working strategy for the screening procedure with a low false-positive rate especially after 1992. The screening methods we use are well established, inexpensive, and do not demand a heavy workload.
Acknowledgements
This work was supported by grants from the Karolinska Institute Research Foundation.
Abbreviations
- DG
Duarte galactosemia
- G-1-P
Galactose-1-phosphate
- Gal
Galactose
- GAL-DH
Galactose dehydrogenase
- GALE
Galactose epimerase
- GALK
Galactokinase
- GALT
Galactose-1-phosphate uridyltransferase
- NBS
Newborn screening
- PKU
Phenylketonuria
References to Electronic Databases
OMIM disorder: http://www.ncbi.nlm.nih.gov/omim
Enzyme Commission (EC) number: http://www.chem.qmul.ac.uk/iubmb/enzyme
Footnotes
Competing interests: None declared.
References
- Beutler E, Baluda MC. A simple spot screening test for galactosemia. J Lab Clin Med. 1966;68:137–141. [PubMed] [Google Scholar]
- Coman DJ, Murray DW, Byrne JC, Rudd PM, Bagaglia PM, Doran PD, Treacy EP. Galactosemia, a single gene disorder with epigenetic consequences. Pediatr Res. 2010;67:286–292. doi: 10.1203/PDR.0b013e3181cbd542. [DOI] [PubMed] [Google Scholar]
- Fernhoff PM. Duarte galactosemia: how sweet is it? Clin Chem. 2010;56:1045–1046. doi: 10.1373/clinchem.2010.147371. [DOI] [PubMed] [Google Scholar]
- Ficicioglu C, Thomas N, Yager C, Gallagher PR, Hussa C, Mattie A, Day-Salvatore DL, Forbes BJ. Duarte (DG) galactosemia: a pilot study of biochemical and neurodevelopmental assessment in children detected by newborn screening. Mol Genet Metab. 2008;95:206–212. doi: 10.1016/j.ymgme.2008.09.005. [DOI] [PubMed] [Google Scholar]
- Ficicioglu C, Hussa C, Gallagher PR, Thomas N, Yager C. Monitoring of biochemical status in children with Duarte galactosemia: utility of galactose, galactitol, galactonate, and galactose 1-phosphate. Clin Chem. 2010;56:1177–1182. doi: 10.1373/clinchem.2010.144097. [DOI] [PubMed] [Google Scholar]
- Freer DE, Ficicioglu C, Finegold D. Newborn screening for galactosemia: a review of 5 years of data and audit of a revised reporting approach. Clin Chem. 2010;56:437–444. doi: 10.1373/clinchem.2009.135947. [DOI] [PubMed] [Google Scholar]
- Gitzelmann R, Bosshard NU. Partial deficiency of galactose-1-phosphate uridyltransferase. Eur J Pediatr. 1995;154:S40–S44. doi: 10.1007/BF02143802. [DOI] [PubMed] [Google Scholar]
- Goppert F (1917) Galaktosurie nach Milchzuckergabe bei angeborenem, familiaerem chronishen Leberleiden Klin Wschr 54:473–477
- Guthrie R, Susi A. A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants. Pediatrics. 1963;32:338–343. [PubMed] [Google Scholar]
- Isselbacher KJ, Andersson EP, Kurahashi K, Kalckar HM. Congenital galactosemia, a single enzymatic block in galactose metabolism. Science. 1956;123:635–636. doi: 10.1126/science.123.3198.635. [DOI] [PubMed] [Google Scholar]
- Jeong JS, Yoon HR, Hong SP. Development of a new diagnostic method for galactosemia by high-performance anion-exchange chromatography with pulsed amperometric detection. J Chromatogr A. 2007;1140:157–162. doi: 10.1016/j.chroma.2006.11.085. [DOI] [PubMed] [Google Scholar]
- Powell KK, Van Naarden BK, Singh RH, Shapira SK, Olney RS, Yeargin-Allsopp M. Long-term speech and language developmental issues among children with Duarte galactosemia. Genet Med. 2009;11:874–879. doi: 10.1097/GIM.0b013e3181c0c38d. [DOI] [PubMed] [Google Scholar]
- Waggoner DD, Buist NR, Donnell GN. Long-term prognosis in galactosaemia: results of a survey of 350 cases. J Inherit Metab Dis. 1990;13:802–818. doi: 10.1007/BF01800204. [DOI] [PubMed] [Google Scholar]