

Abstract
Fabry disease is a rare, X-linked inborn error of glycosphingolipid metabolism caused by a deficiency of the lysosomal enzyme α-galactosidase A. Progressive deposition of GL-3 starts early in life, presumably as early as in fetal life. Chronic burning or provoked attacks of excruciating pain in hands and feet in Fabry disease are common in most children as well as GI-symptoms.
We describe a case of pediatric Fabry disease with gastrointestinal dysmotility symptoms as primary and most severe complaints. Colonic pseudoobstruction and necrosis developed by the age of 15 years. We hypothesize that this patient developed a gastrointestinal phenotype of pediatric Fabry disease that has not been described before.
Introduction
Fabry disease is a rare, X-linked metabolic disorder caused by a defect in the gene encoding the lysosomal enzyme α-galactosidase A (Desnick et al. 2001). Particularly globotriaosylceramide (GL-3) accumulates in various cell types, including vascular endothelial and neural cells, and in visceral tissues throughout the body Fabry disease usually presents in childhood often with symptoms resulting from damage to small nerves from the peripheral and autonomic nervous systems (Zarate and Hopkin 2008). Chronic pain and attacks of excruciating pain in the limbs result from damage to small peripheral nerve fibers. Gastrointestinal dysmotility symptoms are early manifestations of autonomic neuropathy and are commonly reported. Nevertheless, the diagnosis of Fabry disease may be delayed for years, or even decades.
Here, we report a case of severe gastrointestinal phenotype of Fabry disease.
Clinical Report
The male was born at 36 weeks gestation (birth weight 3,750 g, Apgar score 9) following an uncomplicated pregnancy. Numerous respiratory infections occurred during infancy and early childhood and growth was retarded. Weight loss and progressive abdominal pain necessitated hospital admission at the age of 9 years. The diagnosis of secondary malabsorption syndrome was suggested. Despite symptomatic treatment (gluten-free diet), the gastrointestinal symptoms (abdominal pain, diarrhea, nausea) persisted. In addition, the boy started to complain about pain in the extremities with reddish discoloration of the skin. These complaints exacerbated after meals, exercise, and rise in body temperature. Two years later, he began to experience recurrent episodes of unexplained fever. Antirheumatic medication was introduced but the pain in the lower limbs persisted.
Endoscopy, performed because of severe abdominal pain, revealed changes suggesting chronic gastritis and colitis. He was admitted to the rheumatology clinic where the diagnosis of Raynaud’s disease was suggested based on capillaroscopic findings. From age 14 onward, the pain in the stomach, lower abdomen, and hands and feet worsened. Progressive loss of weight, marked loss of appetite, vomiting, nausea, and episodic diarrhea developed. Furthermore, skin cyanosis on the feet were noted and he complained of paresthesias.
The autoimmune disorders (Crohn’s disease, polymyositis overlap syndrome) were taken into consideration and treatment with steroids and methotrexate was started. Parenteral nutrition had to be initiated because of cachexia (BMI – 11.7, below the 3rd percentile). Clinical improvement (less pain) and weight gain were noted (BMI – 15.2) but, two years later, despite treatment with steroids, abdominal pain became more severe, both in intensity and frequency. At the age of 15 years, he had to be admitted to the Children’s Memorial Health Institute. Thorough examinations revealed the following abnormalities: cachexia; dry, pale, desquamating skin; red, mottling skin lesions in the lumbosacral region; erythema palmare; red lips; and swollen, cyanosed and cold feet and hands. Parenteral nutrition was reinstituted (Fig. 1).
Fig. 1.
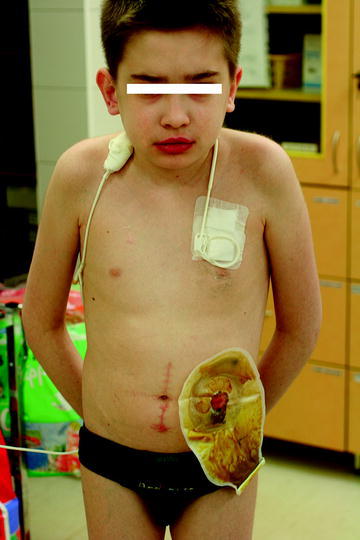
Patient at the age of 15 presented with cachexia, pale skin, red lips. Total parenteral nutrition and colostomy were performed
Necrosis of the sigmoid colon was suspected because of the deterioration of his clinical condition – cachexia (BMI – 12) and symptoms of obstruction (abdominal pain, constipation, vomiting) necessitated resection of the sigmoid colon and a colostomy procedure. Histopathological examination of bowel specimens revealed the presence of lipid deposits in autonomic ganglion cells. The causes of colitis and malnutrition i.e., infections, ischemia, collagenous colitis, vasculitis, intestinal lipodystrophies were excluded. Low levels of α-galactosidase A activity in plasma [0.4 nmol/ml per h (normal: 8.6 ± 1.5)] and in leukocytes: 0.15 nmol/mg of protein per h (normal: 10 ± 2.5) confirmed the diagnosis of Fabry disease. In addition plasma GL-3 Quantitation by LC/MS/MS was 8.8 μg/ml (normal: <7), urinary GL-3 analysis by HPLC-MS/MS was 0.858 mg/mmol of creatinine (normal: <0.03).The results of genotyping revealed hemizygous status for nonsense mutation c.71 G>A [p.Tryp24X]. There were no signs of renal or cardiac involvement (echocardiography normal, glomerular filtration rate: 130.8 ml/min per 1.73 m2, proteinuria). An auditory brainstem response study revealed mild bilateral sensorineural hearing loss. After surgery, and due to parenteral nutrition weight gain (BMI – 16.2) was noted.
The patient’s family history included the patient’s 10-year-old brother and 11-year-old cousin who had a history of pain in the limbs, bouts of fever, and fatigue with the similar but less intense gastrointestinal symptoms. Fabry disease was enzymatically confirmed in both. All three patients began the enzyme replacement therapy just after proper diagnosis.
Discussion
Here we describe severe gastrointestinal phenotype of Fabry disease in a pediatric patient. In our patient, cramping abdominal pain was the first Fabry symptom to appear at age 9. The gastrointestinal symptoms are usually preceded (not in our patient) by the leading symptom of childhood Fabry disease, chronic burning pain or provoked attacks of excruciating pain in hands and feet, by several years.
Our patient’s abdominal pain and other reported gastrointestinal symptoms (i.e., loss of appetite, nausea, vomiting, diarrhea) were most probably consequences of autonomic Fabry neuropathy. This type of neuropathy results from GL-3 deposition in autonomic ganglia (confirmed in our patient), neurons of the myenteric plexuses, and smooth muscle cells and vascular endothelium of epineural and endoneural small blood vessels, and causes delayed gastric emptying and reduced peristaltic activity in (or throughout) the gastrointestinal tract with subsequent bacterial overgrowth (Hoffmann et al. 2007; Argoff et al. 1998). A range of corresponding histological and radiological abnormalities of the gastrointestinal tract have been described in the literature including thickening of the intestinal walls, dilatation, diverticula, and smoothing or loss of colonic haustration.
Endoscopy performed for the first time at age 11 revealed inflammatory bowel changes in the patient’s colon. At age 15, sigmoid colon resection surgery revealed ischemic intestinal necrosis. The pathogenetic mechanism linking lipid accumulation to ischemic tissue damage in Fabry disease is unclear.
To date, only a few reports have described small or large bowel infarction in adult patients with advanced Fabry disease (Sheth et al. 1981; O’Brien et al. 1982; Jardine et al. 1994; Bryan et al. 1977; Breunig et al. 2006).
Growth retardation, observed during our patient’s infancy, has been described to occur in many affected males. Older children may have difficulty gaining weight due to the variety of bothersome gastrointestinal complaints (Desnick et al. 2001; Hoffmann et al. 2007).
It is known that the accumulation of GL-3 may stimulate vascular remodeling. Deacylated GL-3, globotriaosylsphingosine (lyso-GL-3) can affect smooth muscle proliferation via interference in signal transduction and cause the local migration of inflammatory mediators (Aerts et al. 2008).
Deposition of glycosphingolipids leads also to the dysfunction of the autonomous nervous system and symptoms of the intestinal dysmotility and pseudoobstruction.
Severe gastrointestinal symptoms (i.e., loss of appetite, nausea, vomiting, diarrhea) in our patient were secondary to Fabry disease, as well as in patient’s brother and cousin, who presented the similar symptoms but less severe because of their younger age at the time of diagnosis. The harmful impact of steroid and infliximab therapy can be rather excluded. GL-3 accumulation injuring peripheral nerves may progressively induce a variety of peripheral and central pain mechanisms as well may result from impairment of autonomic sudomotor nerve fibers and sweat gland function (Hilz 2002). Another typical clinical consequence of GL-3 accumulation is the cutaneous vascular lesions (“angiokeratomata”) (Zarate and Hopkin 2008). Most patients develop proteinuria in late adolescence that gradually progresses to chronic kidney disease and ultimately to end-stage renal disease by third to fifth decades of life (Schiffmann et al. 2009). Common cardiac complications arising during adulthood include conduction abnormalities and potentially lethal arrhythmias, left ventricular hypertrophy, valvular disease, and myocardial infarction. Early stroke, thrombosis and transient ischemic attacks are among the cerebrovascular complications of Fabry disease. As a consequence of the above-mentioned complications, life expectancy is substantially diminished (Waldek et al. 2009).
Individuals with Fabry disease often come to medical attention in childhood but diagnosis of this rare disorder (estimates of its incidence range from 1:3,100 to 1:117,000 (Spada et al. 2006; Meikle et al. 1999) can be delayed for many years. Medical specialists are generally unfamiliar with the initial symptoms (e.g., peripheral and abdominal pain) pointing to this multisystem heritable disorder. Over the years, several diagnoses were considered in our patient, including secondary malabsorption syndrome, Raynaud’s disease, autoimmune disorder, and Crohn’s disease. Although malabsorption has been reported before in patients with Fabry disease, it is not a recognized feature of this disease (Desnick et al. 2001). It was not until age 15 that the correct diagnosis could be made by examining bowel tissue collected during colectomy and subsequent analysis of α-galactosidase A activity.
In summary, we describe a case of Fabry disease reporting gastrointestinal dysmotility symptoms as first and most prominent complaints.
Acknowledgment
The authors acknowledge Dr. Hans Ebels for his help in manuscript revision.
Synopsis
The study presents a case of Fabry disease with predominant severe gastrointestinal symptoms, pseudoobstruction syndrome, and cachexia, which suggests gastrointestinal phenotype of Fabry disease.
Footnotes
Competing interests: None declared.
References
- Aerts JM, Groener JE, Kuiper S, Donker-Koopman SA, Ottenhoff R, Roomen C, Mirzaian M, Wijburg FA, Linthorst GE, Vedder AC, Rombach SM, Cox-Brinkman J, Somerharju P, Boot RG, Hollak CE, Brady RO, Poorthuis BJ. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc Natl Acad Sci USA. 2008;105(8):2812–2817. doi: 10.1073/pnas.0712309105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argoff CE, Barton NW, Brady RO, Ziessman HA. Gastrointestinal symptoms and delayed gastric emptying in Fabry’s disease: response to metoclopramide. Nucl Med Commun. 1998;19:887–891. doi: 10.1097/00006231-199809000-00009. [DOI] [PubMed] [Google Scholar]
- Breunig F, Weidemann F, Strotmann J, Knoll A, Wanner C. Clinical benefit of enzyme replacement therapy in Fabry disease. 2006. Kidney Int. 2006;69:1216–1221. doi: 10.1038/sj.ki.5000208. [DOI] [PubMed] [Google Scholar]
- Bryan A, Knauft RF, Burns WA. Small bowel perforation in Fabry’s disease. Ann Intern Med. 1977;86:315–316. doi: 10.7326/0003-4819-86-3-315. [DOI] [PubMed] [Google Scholar]
- Desnick R, Ioannou Y, Eng C. Alpha-galactosidase A deficiency: Fabry disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. 8. New York: McGraw-Hill; 2001. pp. 3733–3774. [Google Scholar]
- Hilz MJ. Evaluation of peripheral and autonomic nerve function in Fabry disease. Acta Paediatr Suppl. 2002;91:38–42. doi: 10.1111/j.1651-2227.2002.tb03108.x. [DOI] [PubMed] [Google Scholar]
- Hoffmann B, Schwarz M, Mehta A, Keshav S. Gastrointestinal symptoms in 342 patients with Fabry disease: prevalence and response to enzyme replacement therapy. Clin Gastroenterol Hepatol. 2007;5:1447–1453. doi: 10.1016/j.cgh.2007.08.012. [DOI] [PubMed] [Google Scholar]
- Jardine DL, Fitzpatrick MA, Troughton WD, Tie AB. Small bowel ischaemia in Fabry’s disease. J Gastroenterol Hepatol. 1994;9:201–204. doi: 10.1111/j.1440-1746.1994.tb01244.x. [DOI] [PubMed] [Google Scholar]
- Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999;281:249–254. doi: 10.1001/jama.281.3.249. [DOI] [PubMed] [Google Scholar]
- O’Brien BD, Shnitka TK, McDougall R, Walker K, Costopoulos L, Lentle B, Anholt L, Freeman H, Thomson AB. Pathophysiologic and ultrastructural basis for intestinal symptoms in Fabry’s disease. Gastroenterology. 1982;82:957–962. [PubMed] [Google Scholar]
- Schiffmann R, Warnock D, Banikazemi M, Bultas J, Linthorst G, Packman S, Sorensen S, Wilcox W, Desnick R. Fabry disease: progression of nephropathy, and prevalence of cardiac and cerebrovascular events before enzyme replacement therapy. Nephrol Dial Transplant. 2009;24:2102–2111. doi: 10.1093/ndt/gfp031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth KJ, Werlin SL, Freeman ME, Hodach AE. Gastrointestinal structure and function in Fabry’s disease. Am J Gastroenterol. 1981;76:246–251. [PubMed] [Google Scholar]
- Spada M, Pagliardini S, Yasuda M, Tukel T, Thiagarajan G, Sakuraba H, Ponzone A, Desnick RJ. High incidence of later-onset Fabry disease revealed by newborn screening. Am J Hum Genet. 2006;79:31–40. doi: 10.1086/504601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldek S, Patel MR, Banikazemi M, Lemay R, Lee P. Life expectancy and cause of death in males and females with Fabry disease: findings from the Fabry Registry. Genet Med. 2009;11:790–796. doi: 10.1097/GIM.0b013e3181bb05bb. [DOI] [PubMed] [Google Scholar]
- Zarate YA, Hopkin RJ. Fabry’s disease. Lancet. 2008;372:1427–1435. doi: 10.1016/S0140-6736(08)61589-5. [DOI] [PubMed] [Google Scholar]