

Abstract
Patients with severe biotinidase deficiency (BD), if untreated, may exhibit seizures, psychomotor delay, deafness, ataxia, visual pathology, conjunctivitis, alopecia, and dermatitis. Clinical features normally appear within the first months of life, between two and five. Seizures are one of the most common symptoms in these patients (55%), usually presented as generalized tonic–clonic, and improving within 24 h of biotin treatment. Treatment delay has been associated with irreversible neurological damage, mental retardation, ataxia, paraparesis, deafness, and epilepsy exceptionally.
We report the case of a girl who was admitted at 2.5 months because of vomiting, failure to thrive, flexor spasms, dermatitis, and neurological depression for 1 month. BD was identified and was treated with biotin, stopping seizures and improving symptoms. Developmental delay, paraparesis, optic atrophy, and seizures during febrile illness were observed at follow-up. At the age of 8, she suffered hemigeneralized seizures despite appropriate biotin treatment, so levetiracetam was administered, and epilepsy was controlled. Organic acid measurement was performed to determine whether the child was receiving enough or no biotin.
Even though BD is a rare condition, because the biotinidase screening is a reliable procedure and the disorder is readily treatable, the implementation of extended biotinidase screening will effectively help to prevent any acute and long-term neurological problems as well as the significant morbidity associated with untreated disease. In addition, neonatal screening and early treatment with biotin prevents severe neurological sequelae, such as epilepsy, which has not been thoroughly described in the literature.
Introduction
Biotinidase (BTD; EC 3.5.1.12) deficiency (BD; OMIM 253260) is an autosomal recessively inherited metabolic disorder with an estimated worldwide incidence of 1:60,000 newborns (Wolf 1991). It can be present at any time from 1 week to 10 years of age, but usually between 2nd and 5th month (Wolf 2010). Individuals with profound BD have less than 10% of mean normal biotinidase activity in serum. Untreated individuals with profound deficiency may exhibit neurological and cutaneous features, including seizures, hypotonia, skin rash or alopecia, developmental delay, conjunctivitis, visual problems, such as optic atrophy, and hearing (Wolf et al. 1983).
Clinical expressivity is variable even within the same family (Pindolia et al. 2010). Laboratory studies reveal elevated CSF and blood lactate and pyruvate levels, organic acidaemia, and urinary excretion of 3-hydroxyisovaleric acid (Schürmann et al. 1997; Baumgartner and Suormala 1997).
Early detection by newborn screening is possible and prompt introduction of therapy prevents the clinical expression of the disease. In patients diagnosed and treated after symptoms have appeared (symptomatic children), irreversible deficits such as hearing loss, visual problems, or developmental delay including impaired cognitive functions have been described (Salbert et al. 1993).
Many of the patients with BD initially present epilepsy but its persistence after treatment is exceptionally mentioned in the literature (Salbert et al. 1993; Bunch and Singh 2011). We report a case with BD, who presented epilepsy after some years with biotin treatment.
Case Report
A 10-year-old girl at present, without any family or perinatal history was admitted at the age of 2.5 months because of abnormal movements, dermatitis, vomiting, food refusal, failure to thrive and neurological depression over the prior month. Video-EEG showed a burst-suppression pattern (Fig. 1) and we recorded electroclinical seizures, consisting in flexor spasms. Biochemical analyses showed an elevated 3-hydroxypropionic acids, 3-hydroxyvaleric and methylcitrate as well as 3- methylcrotonyl glycine urinary excretion, which was suggestive of BD. It was confirmed by a biotinidase activity analysis: 0.73 nmolPABA/min per ml serum (normal values: 7.96–13.30 nmolPABA/min per ml). Brain MRI demonstrated diffuse supratentorial white matter abnormalities with decreased brain volume. Molecular testing of BTD gene demonstrated C.98 104del7ins3 homozygous mutation.
Fig. 1.
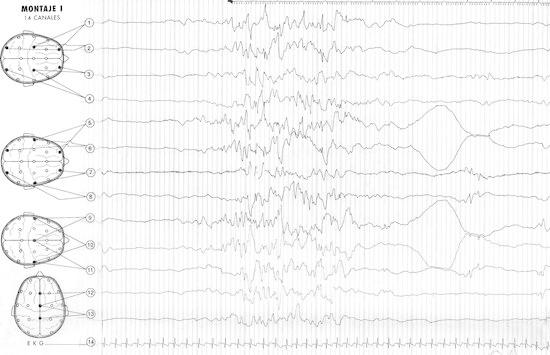
Video-EEG at 2.5 months showed a burst-suppression pattern
She was treated with biotin (20 mg/day), and she became seizure free, with resolution of skin symptoms and somatic growth. However, microcephaly, cognitive impairment, behavioral disorder, sensorineural hearing loss, optic atrophy, and spastic paraparesis persisted. Throughout her second year of life, she had three seizures during febrile illness (two were generalized, one hemigeneralized), EEG was abnormal (multifocal paroxysmal pattern), and she received phenobarbital. Then biotin was increased (30 mg/day) and neuroimaging revealed progressive improvement in the cerebral volume. At the age of 8, she suffered two afebrile hemigeneralized seizures, with no other clinical or biochemical parameters that suggested biotin deficiency. Organic acid measurement (blood and urine) was performed and organic aciduria was ruled out. Biotin plasma and urine levels were normal. EEG revealed multifocal paroxysmal pattern. Epilepsy improved after levetiracetam treatment, and administration of phenobarbital was withdrawn. Obviously, biotin treatment was maintained.
Discussion
Biotin is an imidazole derivative found in many natural foods, which serves as a cofactor in its unbound state for pyruvate carboxylase, propionyl-CoA carboxylase, beta-methylcrotonyl-CoA carboxylase, and two isoenzymes of acetyl-CoA carboxylase. These enzymes catalyze key reactions in gluconeogenesis, fatty acid synthesis, and amino acid catabolism (Wolf 1991, 1983). The carboxyl group of biotin is covalently attached to an ε-amino group of specific lysine residues of the apocarboxylases. Following the proteolytic degradation of carboxylases, the resulting biocytin or small biotinylpeptides are cleaved by biotinidase at the amide bond, releasing lysine or lysyl peptides and free biotin, which can then be recycled. Biotinidase may also play a role in the processing of dietary protein-bound biotin (Wolf 2010).
To date there have been described more than 140 mutations in the BTD gene (OMIM 609019), which is located in the 3p25 locus (Pindolia et al. 2010). Most symptomatic children have serum biotinidase activity between undetectable and 10% of mean normal serum activity; these children are considered to have profound BD. However, there have been many children ascertained by newborn screening who were found to have activity in the 10–30% of mean normal serum activity; this is considered to be partial BD. Our patient was homozygous for the C.98-104del7ins3 mutation, which was described by Pomponio et al. in 1997. It seems to be one of the most common profound BD-related mutations (51% in mentioned series).
The pathogenesis of the central nervous system involvement is unknown. Brain and cerebrospinal fluid biotinidase activity is very low relative to that in serum and other tissues. Therefore, the carboxylase deficiencies and the resulting accumulation of lactate and organic acids such as 3-hydroxyisovalerate, 2-hydroxybutyrate, and 3-hydroxypropionate may develop in the brain before it occurs in other organs (Baumgartner and Suormala 1997; Schürmann et al. 1997).
Moreover, seizures are a common feature in profound BD (55% in Salbert series) and very usual as initial symptom (38%) as well. The seizures are usually generalized tonic–clonic (56%), but there have also been described some patients with myoclonic seizures (Bunch and Singh 2011), infantile spasms (Desai et al. 2008; Lafuente-Hidalgo et al. 2010), or even Ohtahara syndrome (Singhi and Ray 2011). In mentioned series with 43 patients with BD and epilepsy, seizures were controlled with antiepileptic drugs in 51% of cases, and were removed after starting treatment with biotin. Biotin therapy stopped the seizures within 24 h in 75% of the children whose seizures were uncontrolled by antiepileptic drugs. The metabolic and cutaneous abnormalities were corrected by biotin in the four infants (0.09%), who continued to have seizures and residual neurologic deficits. This small group of patients, who experienced severe or multiple episodes of metabolic compromise, may develop irreversible neurologic damage (epilepsy, neuropathy, ataxia, mental delay), which subsequently may not respond to biotin therapy. Early initiation of treatment has been shown to prevent further manifestations of the disease with many clinical findings that turned out to be reversible. (Mc Sweeney et al. 2010, Couce Pico et al. 1999).
We would like to emphasize that valproic acid treatment impairs the liver mitochondrial function, resulting in a low biotinidase activity and/or biotin deficiency (Schulpis et al. 2001), so we recommend avoiding it in patients with BD.
Most frequent neuroimaging findings include diffuse cerebral edema, delayed myelination, basal ganglia calcification, and diffuse supratentorial white matter abnormalities with decreased brain volume, as it was seen in our patient (Schürmann et al. 1997). These findings were reversible after biotin treatment, as it has been described (Mc Sweeney et al. 2010).
As mentioned above, the most frequent permanent neurological sequelae are mental retardation, spastic paraparesis, ataxia, deafness, and visual impairment, meanwhile epilepsy remains a rare finding in previous literature, which must be taken into account for prognostic and therapeutic implications. We strongly believe that epilepsy should be considered as a neurological sequelae, as they are mental retardation, the paraparesis and the hearing and visual impairment suffered by the child. We also believe that these serious consequences are due to the delay in consultation and, therefore, in the diagnosis and initiation of treatment. We advocate for introducing BD in the neonatal metabolic screening because although this disease has a low incidence, the response to early treatment is excellent.
Acknowledgments
To Mr. Jose Jimenez and Dr. Paloma Alonso for their big effort with translation.
Take-Home Message
Epilepsy is a possible neurological sequelae in biotinidase deficiency, due to delay in the diagnosis and initiation of treatment.
Footnotes
Competing interests: None declared.
References
- Baumgartner ER, Suormala T. Multiple carboxylase deficiency: inherited and acquired disorders of biotin metabolism. Int J Vitam Nutr Res. 1997;67(5):377–384. [PubMed] [Google Scholar]
- Bunch M, Singh A. Peculiar neuroimaging and electrophysiological findings in a patient with biotinidase deficiency. Seizure. 2011;20(1):83–86. doi: 10.1016/j.seizure.2010.10.001. [DOI] [PubMed] [Google Scholar]
- Couce Pico ML, Martinón-Torres F, Castiñeiras DE, et al. Biotinidase deficiency: importance of its neonatal diagnosis and early treatment. An Esp Pediatr. 1999;50:504–506. [PubMed] [Google Scholar]
- Desai S, Ganesan K, Hegde A. Biotinidase deficiency: a reversible metabolic encephalopathy. Neuroimaging and MR spectroscopic findings in a series of four patients. Pediatr Radiol. 2008;38(8):848–856. doi: 10.1007/s00247-008-0904-z. [DOI] [PubMed] [Google Scholar]
- Lafuente-Hidalgo M, Ranz Angulo R, López Pisón J, et al. Epileptic encephalopathy due to partial biotinidase deficiency. An Pediatr (Barc) 2010;72(3):227–228. doi: 10.1016/j.anpedi.2009.10.015. [DOI] [PubMed] [Google Scholar]
- Mc Sweeney N, Grunewald S, Bhate S, et al. Two unusual clinical and radiological presentations of biotinidase deficiency. Eur J Paediatr Neurol. 2010;14(6):535–538. doi: 10.1016/j.ejpn.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Pindolia K, Jordan M, Wolf B. Analysis of mutations causing biotinidase deficiency. Hum Mutat. 2010;31(9):983–991. doi: 10.1002/humu.21303. [DOI] [PubMed] [Google Scholar]
- Pomponio RJ, Hymes J, Reynolds TR, et al. Mutations in the human biotinidase gene that cause profound biotinidase deficiency in symptomatic children: molecular, biochemical, and clinical analysis. Pediatr Res. 1997;42(6):840–848. doi: 10.1203/00006450-199712000-00020. [DOI] [PubMed] [Google Scholar]
- Salbert BA, Pellock JM, Wolf B. Characterization of seizures associated with biotinidase deficiency. Neurology. 1993;43(7):1351–1355. doi: 10.1212/WNL.43.7.1351. [DOI] [PubMed] [Google Scholar]
- Schulpis KH, Karikas GA, Tjamouranis J, et al. Low serum biotinidase activity in children with valproic acid monotherapy. Epilepsia. 2001;42(10):1359–1362. doi: 10.1046/j.1528-1157.2001.47000.x. [DOI] [PubMed] [Google Scholar]
- Schürmann M, Engelbrecht V, Lohmeier K, et al. Cerebral metabolic changes in biotinidase deficiency. J Inherit Metab Dis. 1997;20(6):755–760. doi: 10.1023/A:1005307415289. [DOI] [PubMed] [Google Scholar]
- Singhi P, Ray M. Ohtahara syndrome with biotinidase deficiency. J Child Neurol. 2011;26(4):507–509. doi: 10.1177/0883073810383018. [DOI] [PubMed] [Google Scholar]
- Wolf B. Worldwide survey of neonatal screening of biotinidase deficiency. J Inherit Metab Dis. 1991;14:923–937. doi: 10.1007/BF01800475. [DOI] [PubMed] [Google Scholar]
- Wolf B. Clinical issues and frequent questions about biotinidase deficiency. Mol Genet Metab. 2010;100(1):6–13. doi: 10.1016/j.ymgme.2010.01.003. [DOI] [PubMed] [Google Scholar]
- Wolf B, Grier RE, Allen RJ, et al. Biotinidase deficiency: the enzymatic defect in late-onset multiple carboxylase deficiency. Clin Chim Acta. 1983;131(3):273–281. doi: 10.1016/0009-8981(83)90096-7. [DOI] [PubMed] [Google Scholar]