

Abstract
Phylogenomics has revealed the existence of fast-evolving animal phyla in which the amino acid substitution rate, averaged across many proteins, is consistently higher than in other lineages. The reasons for such differences in proteome-wide evolutionary rates are still unknown, largely because only a handful of species offer within-species genomic data from which molecular evolutionary processes can be deduced. In this study, we use next-generation sequencing technologies and individual whole-transcriptome sequencing to gather extensive polymorphism sequence data sets from Ciona intestinalis. Ciona is probably the best-characterized member of the fast-evolving Urochordata group (tunicates), which was recently identified as the sister group of the slow-evolving vertebrates. We introduce and validate a maximum-likelihood framework for single-nucleotide polymorphism and genotype calling, based on high-throughput short-read typing. We report that the C. intestinalis proteome is characterized by a high level of within-species diversity, efficient purifying selection, and a substantial percentage of adaptive amino acid substitutions. We conclude that the increased rate of amino acid sequence evolution in tunicates, when compared with vertebrates, is the consequence of both a 2–6 times higher per-year mutation rate and prevalent adaptive evolution.
Keywords: substitution rate, population size, mutation rate, next-generation sequencing, transcriptome
Introduction
Phylogenomic data have changed our view of metazoan diversity and evolution (Delsuc et al. 2005). The joint phylogenetic analysis of multiple genes has uncovered a number of unexpected relationships among animal phyla, modifying classical interpretations of body plan evolution and opening new perspectives in the field of Evo–Devo research (Philippe et al. 2005; Bourlat et al. 2006; Delsuc et al. 2006; Dunn et al. 2008; Philippe et al. 2011). Besides organismal evolution, phylogenomic data have also been of great relevance for the study of molecular evolutionary processes. Molecular phylogenies based on large protein data sets have revealed that the rate of amino acid substitution, averaged over many genes, varies by several orders of magnitude across certain metazoan groups. Nematodes, platyhelminthes, and tunicates, for instance, evolve much faster than cnidarians or vertebrates as far as protein sequences are concerned (Delsuc et al. 2006; Lartillot et al. 2007). In this large body of literature, rate variation across lineages has only been considered as a methodological issue. Fast-evolving lineages are highly problematic for phylogenetic tree inference because of the multiple substitutions and saturation—resulting in the so-called long-branch attraction effect. To this respect, important methodological developments have taken place, including taxon sampling optimization (Wiens 2005; Paps et al. 2009) and improved modeling of substitution rate heterogeneity (Lartillot and Philippe 2004; Lartillot et al. 2007; Wang et al. 2008).
However, the pattern is in itself intriguing: why do metazoan proteins evolve quickly in some groups and slowly in others? The question is a difficult one, because multiple evolutionary forces affect the amino acid substitution rate (Bromham 2009). The most obvious one is mutation. A higher per-year mutation rate, perhaps due to a shorter generation time, could explain the high amino acid substitution rate in some groups (Nabholz et al. 2008; Thomas et al. 2010). Differences in selective regimes could also be involved. The fraction of amino acid substitutions corresponding to adaptive events was found quite variable between species (Smith and Eyre-Walker 2002; Boyko et al. 2008; Gossmann et al. 2010; Halligan et al. 2010)—for yet unclear reasons. Therefore, a high rate of amino acid substitution in specific taxa could be explained by a stronger contribution of adaptive processes. Conversely, protein evolution could be accelerated in some lineages by a less efficient purifying selection. This is specifically expected in species of reduced population size, in which an enhanced genetic drift increases the probability of fixation of the slightly deleterious mutations (Ohta 2000; Nikolaev et al. 2007; Popadin et al. 2007).
All these hypotheses are not mutually exclusive: the amino acid substitution rate is presumably determined by a complex combination of mutation rate, the distribution of selection coefficients, and population size, each of these parameters likely varying in time and between lineages. Within-species variation data in a genome-wide scale are required to disentangle these many influences, through the comparison of nonsynonymous (selected) versus synonymous (neutral) patterns of polymorphism and divergence, according to the McDonald and Kreitman (1991) approach (Keightley and Eyre-Walker 2010). In Metazoa, most of the published population genomic data sets concern the relatively slow-evolving vertebrates (Boyko et al. 2008; Axelsson and Ellegren 2009; Halligan et al. 2010) and just one genus of insects (i.e., Drosophila, Bierne and Eyre-Walker 2004; Begun et al. 2007), providing only limited opportunity for comparison. A genome-wide survey of within-species molecular variations in a typical fast-evolving animal taxon is still lacking.
Within chordates, tunicates (or urochordates) are a large phylum of morphologically simplified but highly diversified marine filter-feeding animals (Satoh 2003; Lambert 2005). Phylogenomics has recently identified tunicates as the closest living relatives of vertebrates (Delsuc et al. 2006; Dunn et al. 2008; Delsuc et al. 2008; Putnam et al. 2008). However, unlike vertebrates, tunicates show a very high rate of genome evolution (Delsuc et al. 2006; Singh et al. 2009; Denoeud et al. 2010; Tsagkogeorga et al. 2010), offering promising comparative perspectives with respect to their slow-evolving sister group. In this study, we investigate the population genomics of the tunicate Ciona intestinalis. Ciona is a popular model species in Evo–Devo studies (Holland and Gibson-Brown 2003), and its complete genome sequence has been previously characterized (Dehal et al. 2002). Furthermore, C. intestinalis is one of the very few tunicate species for which species boundaries have been delineated, with recent phylogenetic and population genetic data suggesting that it represents a species complex (Zhan et al. 2010). Specifically, several lines of evidence from crosses (Caputi et al. 2007), mitochondrial data (Iannelli et al. 2007), microsatellites, and five nuclear genes (Nydam and Harrison 2010) currently corroborate the existence of two cryptic species, called C. intestinalis A and B.
In this study, we use the Illumina next-generation sequencing (NGS) technology to approach the transcriptome of eight individuals of C. intestinalis B sampled in the wild. We introduce a novel probabilistic approach for single-nucleotide polymorphism (SNP) and genotype calling from transcriptome-based data. Using the fully sequenced C. intestinalis A as an outgroup, we investigate the patterns of polymorphism and divergence in species B based on >1,500 cDNA sequences and >30,000 SNPs. Finally, comparing Ciona versus human, we show that the increased rate of amino acid substitution in tunicates is explained by both a high per-year mutation rate and prevalent adaptive evolution.
Materials and Methods
Sampling and Sequencing
Adult specimens of Ciona intestinalis B were collected from natural populations encompassing two geographic localities: 1) Northern Europe, Norway, and 2) the East Coast of the Northern Atlantic Ocean, Canada. Of these, we sampled five individuals from Norway collected at local sites near Bergen and three individuals from Canada originated from three locations along the coast of Nova Scotia (table 1). Gonads and muscles were dissected from fresh adults for each population, rapidly stabilized in RNAlater®, and stored at −80°C until use. Total RNA extractions were performed using the RNeasy® Plus Kit (Qiagen, Chatsworth, CA, USA). Tissues were pooled, homogenized, lysed together, and subsequently passed over spin columns for purification according to the manufacturer’s instructions, following Gayral et al. (2011). The quantity and quality of the RNA extracts for each sample were assessed using spectrophotometry (NanoDrop), agarose gel electrophoresis, and capillary electrophoresis (Agilent). cDNA library construction and transcriptome sequencing were performed by the GATC Biotech according to standard Illumina protocols. A 3'-primed non-normalized cDNA library was created from 5 μg of total RNA extract for each sample. Individuals were tagged, pooled, and sequenced in reads of 100 bp length on a Genome Analyzer II (Illumina, Inc.).
Table 1.
Illumina sequencing and read mapping statistics
| Sample | Locality | No. of reads | Total length (Mb) | Mapped reads (%) | Genes ≥ 5X | Genes ≥ 10X |
|---|---|---|---|---|---|---|
| GA02G | Grimstad, Norway | 3,909,166 | 293 | 32.8 | 2,834 | 1,920 |
| GA02I | South Askøy, Norway | 3,474,750 | 261 | 30.2 | 2,248 | 1,557 |
| GA02J | South Søtra, Norway | 3,415,890 | 256 | 39.2 | 3,078 | 2,077 |
| GA12M | South Askøy, Norway | 4,941,551 | 371 | 29.3 | 1,473 | 797 |
| GA12N | South Søtra, Norway | 2,440,102 | 183 | 31.9 | 1,543 | 970 |
| GA02L | Chester, Canada | 5,048,582 | 379 | 29.6 | 2,727 | 1,702 |
| GA02M | Port La Tour, Canada | 5,493,035 | 412 | 21.5 | 1,864 | 1,182 |
| GA02N | Petit-de-Grat, Canada | 7,115,473 | 534 | 35.6 | 3,624 | 2,565 |
| Total | 35,838,549 | 2,688 | 31.0 | 4,744 | 3,261 |
Read Mapping
All the reads of each individual were independently aligned to a single reference database, consisting of all 20,225 Ciona intestinalis A cDNA sequences (including untranslated regions, or UTRs) downloaded from Ensembl release 58-59 (http://www.ensembl.org/). The read alignment against the Ciona type A transcriptome was performed using the Burrows–Wheeler Alignment (BWA) tool (Li and Durbin 2009). By default, BWA aligns read sequences with a low error rate (<3%). Therefore—and because of the relatively high level of sequence divergence between the two species Ciona A and B (Nydam and Harrison 2010)—mapping analyses were repeated varying the options for the match stringency (data not shown). Final mapping analyses were conducted specifying the maximum edit distance n = 10−3 and the maximum edit distance in the seed k = 5 in BWA. When multiple mapping positions were obtained for the same read, a single hit was considered (the most significant one). Reads introducing gaps in the reference sequence were discarded. For genes with multiple transcripts, a single cDNA was selected (the longest). The average coverage of a cDNA was calculated by multiplying the number of matching reads by their mean length and dividing the result by the cDNA length. The cDNA coverage was calculated separately for each individual.
Data Cleaning
cDNA sequences were trimmed at both 5′- and 3′-ends to discard the noncoding UTRs (ENSEMBL annotations). This was done after the read-mapping step, to avoid a drop in coverage close to the start and stop positions of the coding sequence. Then the first and last five bases of each aligned read were removed. Ambiguously aligned sites were then eliminated using Gblocks (Castresana 2000) set to the following parameters: type of sequence = codons, minimum number of sequences for a conserved position ≈ 0.7 × n (where n = number of sequences), minimum number of sequences for a flanking position ≈ 0.7 × n, Maximum number of contiguous nonconserved positions = 1, minimum length of a block = 6, and allowed gap positions = with half. Alignments less than 100 bp long were discarded.
SNP and Genotype Calling
A novel maximum-likelihood framework, adapted to transcriptome-based high-throughput short sequence read data, was here introduced for SNP and genotype calling. This method is based on the assumption of a multinomial distribution of read numbers at each position, the multinomial probabilities being functions of the putative genotype, and error rate. Two error models were used. The M1 model has a single parameter, ε, which stands for the probability of misreading a nucleotide. The transcriptome-specific M2 model has two parameters, ε (same as in M1) and γ. Parameter γ measures the amount of allele-specific expression bias. When γ equals zero, the two alleles of the considered locus are equally expressed, so that the two have the same probability of being sequenced. When γ equals one, only one of the two alleles is expressed, as in imprinted genes. Intermediate values of γ represent intermediate levels of allelic expression bias. The ε and γ parameters are assumed to be shared by all the positions of a given gene. For each gene, parameters were estimated by maximum likelihood (ML) under each M1 and M2, and the two models were compared through likelihood-ratio tests, assuming that twice the log-likelihood ratio follows a chi-squared distribution with one degree of freedom under M1. Then the posterior probabilities of the 16 possible genotypes were calculated for each position of each gene in each individual using the empirical Bayes method. When one of the 16 possible genotypes had a posterior probability above 0.95, it was validated. Otherwise, the genotype was considered as unknown. Positions at which more than one allele was inferred across the eight individuals were called SNPs. Details about the method, which was programmed in C++ using the Bio++ library (Dutheil et al. 2006), are given in the Supplementary Material online. The source code is freely available upon request to the authors.
Polymorphism and Divergence Analyses
The proportion of missing data (individual positions at which genotype is unknown) in the data set was reduced by removing the most gappy positions and individuals. For each gene, codon sites showing a proportion of missing data above the arbitrary threshold site_p were discarded. Several values of site_p were tried. Then individuals showing more than a half the positions with undetermined genotypes were removed. For each gene of the data set, the following summary statistics were calculated using the Bio++ library: synonymous (πS) and nonsynonymous (πN) diversity in C. intestinalis B, number of synonymous (pS) and nonsynonymous (pN) segregating sites in C. intestinalis B, number of synonymous (dS) and nonsynonymous (dN) fixed differences between C. intestinalis A and B, neutrality index (NI) = (pN/pS)/(dN/dS) (Rand and Kann 1996), and NI calculated after removing SNPs for which the minor allele frequency was below 0.2 (NI0.2).
The proportion of adaptive amino acid substitutions was estimated as α = 1 − NI (naïve estimate, Fay et al. 2001), α0.2 = 1 − NI0.2 (estimate tentatively accounting for slightly deleterious nonsynonymous mutations segregating at a low frequency), and αEWK (estimate based on the full site frequency spectra, Eyre-Walker and Keightley 2009). The method of Eyre-Walker and Keightley (2009) was also used to estimate the proportion P of effectively neutral nonsynonymous mutations. The πS and πN statistics were calculated using complete sites only, i.e., codon sites for which all individuals in the sample were genotyped. We also calculated a multi-SNP FIS, defined as 1 − (Hobs/Hexp), where Hexp is the expected number of heterozygotes and Hobs the observed number, summed across all SNPs of a gene. The genomic averages of πS, πN, and FIS were calculated, weighting each gene by its length. The genomic proportion of adaptive amino acid substitutions was calculated by first summing the pS, pN, dS, and dN values across genes, then calculating the collective NI and α. Confidence intervals around estimates were obtained by bootstrapping genes, following Smith and Eyre-Walker (2002). The number of bootstrap replicates was 100 for αEWK and 1,000 for the other statistics.
These results were compared with equivalent calculations performed in Drosophila simulans (Begun et al. 2007) and Homo sapiens (Bustamante et al. 2005). In these two previously published data sets, we identified the subsets of genes orthologous to the C. intestinalis genes analyzed in this study, to maximize comparability and assess the bias in gene sampling of the transcriptome-based studies. This was achieved thanks to the Ensembl orthology annotations.
Results and Discussion
Sequencing Layout, Mapping, and Data Set Assembly
The Illumina high-throughput sequencing of the C. intestinalis B eight individuals yielded approximately 2.7 Gb of raw data, corresponding to a total of 35,838,549 single-end sequence reads with an average length of 91 bp. The number of reads obtained per sample ranged from 2,440,102 to 7,115,473 (table 1). Reads were aligned to a collection of 20,225 cDNA sequences corresponding to all the transcripts of the 14,547 C. intestinalis A genes in Ensembl.
We found that ∼30% of the reads aligned to the reference. A similar percentage in matching reads was also obtained when sequences were trimmed for low-quality bases before mapping or when the whole genomic sequence of C. intestinalis A was used as a reference (data not shown). This was expected knowing the high level of molecular divergence between C. intestinalis A and B (up to 12.5%), previously reported (Nydam and Harrison 2010, 2011a, 2011b). Another explanation for the relatively low percentage of matching reads could be attributed to the potential occurrence of foreign genetic material in the RNA matrix used for sequencing. Ciona is a filter-feeding species, so contaminations from the marine environment during the dissection of animals are difficult to avoid. However, a de novo assembly of the reads into predicted cDNAs did not reveal a substantial contribution of foreign RNA to this data set (Cahais et al. 2012). It should be noted that 30% of the successfully mapped reads collectively targeted 19,934 cDNAs of the reference, i.e., 99% of the C. intestinalis A transcriptome content.
We next focused on a subset of 14,547 cDNAs representing the longest cDNA sequence of each C. intestinalis A gene. The coverage was highly variable across genes, as expected from non-normalized cDNA libraries. An average coverage of 5X or more (per individual) was achieved in ∼2,500 genes and an average coverage of 10X or more in ∼1,500 genes. These numbers varied across individuals, as presented in table 1.
For the aims of this study, we sought to select genes showing a high coverage level in many individuals. Among the 14,547 genes of C. intestinalis A, 8,080 had at least one read mapped in each of the eight individuals of our data set. This number dropped dramatically when constraints on coverage were introduced (table 2). Only 612 genes had a coverage of 5X or higher in all eight individuals and 317 a coverage of 10X. When high coverage was required in fewer individuals, the numbers of acceptable genes were higher (table 2). Facing this trade-off between coverage, number of individuals, and number of genes, we decided to focus on a subset of 1,669 genes for which the longest cDNA sequence was present in at least four of the eight C. intestinalis B individuals with a minimum coverage of 10X per individual. Finally, after the UTR sequence removal and unambiguously aligned site cleaning, we retained 1,602 data sets with a length above 100 bp. The average length of these sequences was 1,089 bp.
Table 2.
Target gene sharing across individuals
| No. of individuals | Transcripts (all) | Genes (all) | Genes ≥ 5X | Genes ≥ 10X |
|---|---|---|---|---|
| 8 | 11,466 | 8,080 | 612 | 317 |
| 7 | 13,939 | 9,800 | 1,170 | 689 |
| 6 | 15,639 | 11,015 | 1,590 | 955 |
| 5 | 16,939 | 11,998 | 2,063 | 1,280 |
| 4 | 17,970 | 12,754 | 2,509 | 1,669 |
| 3 | 18,789 | 13,361 | 3,054 | 2,067 |
| 2 | 19,425 | 13,854 | 3,649 | 2,532 |
| 1 | 19,934 | 14,282 | 4,744 | 3,261 |
Genotype Calling and Error Model Assessment
An ML method similar in spirit to those of Lynch (2009), Hohenlohe et al. (2010), and Keightley and Halligan (2011) was developed here to call SNPs and genotypes from short sequence reads. Described in detail earlier, the method includes two error models, M1 and M2. Both models take into consideration base reading errors (ε) in the data, with M2 additionally accounting for expression bias between alleles (γ). The two copies of a gene need not be expressed at equal rate within an individual (Wagner et al. 2010). This is taken into account under model M2, in which the expected read frequency of a given allele in a heterozygote individual may be different from 50%.
When the M1 model was used, the estimated error rate ranged from 0.0017 to 0.085 across genes and averaged 0.0217. This 2% error rate reflects a combination of Illumina sequencing errors and base misspecifications from incorrectly read mappings. A very similar estimate of the error rate was obtained when the M2 model was used (mean: 0.0206). The allelic expression bias, assessed through parameter γ, varied greatly among genes. In 375 genes, representing 16% of the data set, the ML estimate for γ reached its maximal value of 1, which implies zero expression of one of the two alleles. The average γ across genes was found to be 0.562. With respect to the model fit, likelihood ratio tests provided strong statistical support for the M2 model, the M1 model being rejected in >96% of the analyzed genes.
Focusing on the predicted genotypes, we found that the results of M1 and M2 differed in 58,852 cases, representing 0.2% of the ∼29 million individual genotypes predicted in total. Most of these differences (86%) were cases in which M1 inferred a genotype, whereas M2 did not, because the posterior probability for the inferred genotypes under M2 was below the defined confidence threshold. Only in 2,337 cases (0.008% of the data) did M1 and M2 predict distinct genotypes. In 2,330 of these 2,337 cases (99.7%), M1 predicted a homozygous genotype and M2 a heterozygous one. Therefore, despite statistically significant differences in model fit, the inferred genotypes under models M1 and M2 were very similar, M2 differing slightly from M1 by a higher level of uncertainty and a greater number of predicted heterozygotes.
Polymorphism analyses of the coding data sets were conducted following various genotype calling approaches. The main results of these are listed in table 3. Columns 1 and 2 describe the SNP calling method (model M1 or M2) and the missing data cleaning stringency (site_p), respectively. Six combinations of model and site_p are presented. The reliability of the inferred SNPs and genotypes was assessed using two indices: 1) number of predicted premature stop codons, stop% (column 3) and 2) heterozygote excess, FIS (column 4).
Table 3.
Error model assessment in SNP and genotype calls
| Model | site_p | Stop% | FIS | No. of codon sites |
|---|---|---|---|---|
| M2 | 0.75 | 6.9 | −0.054 | 133 |
| M1 | 0.75 | 4.8 | −0.017 | 138 |
| M1 | 0.5 | 4.8 | −0.017 | 137 |
| M1 | 0.25 | 4.8 | −0.009 | 122 |
| M1 | 0.125 | 4.8 | −0.006 | 104 |
| M2 | 0.125 | 6.9 | −0.036 | 101 |
We observed an increased frequency of premature stop codon predictions near the start and end positions of the C. intestinalis A reference sequence in Ensembl, which probably reflects annotation errors or true biological variation. It has been reported that gene annotations for Ciona intestinalis A genome were often inconsistent with the experimental cDNA-based sequence data, because of the unusual operon gene structures found in the Ciona genome and the limited accuracy of gene prediction programs (Satou et al. 2008). To minimize such biases, only stop codons at positions >4 and codons away from the start and end positions were considered here. Depending on the model, the percentage of genes with a stop codon in at least one individual was very low, ranging from 4.8 to 6.9% (table 3; column 3). Again, we note that not all inferred premature codons need to be erroneous: some must still reflect imperfections in the coding sequence annotation.
The FIS (table 3; column 4) measures departure from the Hardy–Weinberg equilibrium. A negative FIS indicates a genome-wide excess in heterozygote genotypes, which we interpret here as reflecting errors in genotype calls. Such heterozygote excess is expected in case of undetected sequencing errors and of erroneous mapping of paralog sequences or splicing variants. The average absolute value of FIS was low (less than 1%) when the M1 model and stringent site_p were used, with only 13% of the genes showing a FIS value below −0.2. This could suggest that our data set is negligibly affected by spurious heterozygote prediction due to undetected sequencing or mapping errors—even though one cannot rule out the possibility that the FIS in C. intestinalis B is truly positive, and our estimate biased downward because of genotype calling errors.
The first two rows of table 3 compare the M1 and M2 model predictions under a low stringency threshold for missing data (site_p is set to 0.75). Both the number of predicted premature stop codons and the excess level of heterozygotes (−FIS) were found to be higher under M2 than under M1, suggesting that the M1 predictions were the most reliable. A similar conclusion was drawn by comparing the last two rows, in which site_p was set to 0.125 (high stringency regarding missing data). The effect of an increased stringency regarding missing data (rows 2–5) was a decrease in heterozygote excess, at the cost of reduced sequence length, as shown by the average number of complete codon sites (with no missing data) per gene (table 3, column 5).
Ciano intestinalis Population Genomics
Table 4 summarizes the population genomics of C. intestinalis, calculated from the ∼30,000 coding SNPs identified in this study. Importantly, the estimates of πN, πS, dN, dS, and α were essentially unaffected by the error model and the missing data cleaning stringency applied. The single noticeable discrepancy was observed in the estimate of α, which was 25% higher under M1 than under M2. We note that this difference disappeared when α0.2 was computed, i.e., when low-frequency variants were excluded from the calculation. This again suggests that the M2 model tends to predict a proportion of erroneous heterozygotes at truly invariable sites because of undetected sequencing/mapping errors. These incorrect predictions are expected to affect nonsynonymous sites as frequently as synonymous sites, slightly increasing the πN/πS ratio and decreasing α. The M1 model appeared more robust to missing data.
Table 4.
Main population genomic statistics calculated under various genotype calling methods
| Model | site_p | 103 πN | 102 πS | πN/πS | dN/dS | α | α0.2 | αKEW |
|---|---|---|---|---|---|---|---|---|
| M2 | 0.75 | 2.60 | 5.48 | 0.048 | 0.074 | 0.208 | 0.546 | 0.792 |
| M1 | 0.75 | 2.62 | 5.70 | 0.046 | 0.074 | 0.256 | 0.542 | 0.782 |
| M1 | 0.5 | 2.62 | 5.70 | 0.046 | 0.074 | 0.257 | 0.542 | 0.782 |
| M1 | 0.25 | 2.64 | 5.73 | 0.046 | 0.074 | 0.251 | 0.541 | 0.790 |
| M1 | 0.125 | 2.69 | 5.64 | 0.048 | 0.078 | 0.260 | 0.532 | 0.812 |
| M2 | 0.125 | 2.67 | 5.42 | 0.049 | 0.077 | 0.215 | 0.535 | 0.820 |
| CIa | 0.5 | ±0.14 | ±0.21 | ±0.003 | ±0.005 | ±0.05 | ±0.04 | ±0.15 |
aThe 95% confidence intervals (CIs, 90% for αKEW) around the estimates obtained under model M1, site_p = 0.5 (bolded).
The genomic average synonymous diversity in C. intestinalis B was estimated to be 0.057 per site. This is a very large number, which makes Ciona one of the most genetically diverse animal species known until now. The πN/πS ratio (ratio of average πN to average πS) was below 0.05, which indicates a strong influence of purifying selection on genomic variations, in concordance with conclusions drawn by the Ciona intestinalis A genome project (Dehal et al. 2002). In table 5, we calculated the average population genomic statistics in H. sapiens and D. simulans using either all available genes or only orthologs to the C. intestinalis genes analyzed in this study. Figure 1 shows that the distributions of πs and πN/πS across genes in C. intestinalis B are essentially similar to those of D. simulans (data from Begun et al. 2007) but very different from those of H. sapiens (data from Bustamante et al. 2005, see averages in table 5). A high average πS and a low average πN/πS suggest a large population-sized species, in which genetic drift is reduced when compared with, e.g., humans. On the basis of the population haplotypic structure and a direct estimate of the recombination rate, Small et al. (2007) inferred that the effective population size in the congeneric Ciona savignyi was 1.5 × 106, i.e., the same order of magnitude as in D. simulans. Our data suggest that the population size in C. intestinalis is even larger than in D. simulans (table 5). This indicates that the accelerated evolution of amino acid sequences in tunicates is clearly not the consequence of relaxed purifying selection.
Table 5.
Comparison of major population genomic statistics across three animal species
| C. intestinalis | D. simulans (all genes) | D. simulans (orthologs) | H. sapiens (all genes) | H. sapiens (orthologs) | |
|---|---|---|---|---|---|
| No. of genes | 1,602 | 10,996 | 1,431 | 6,530 | 980 |
| 102 πS | 5.70 | 3.27 | 2.88 | 0.164 | 0.089 |
| πN/πS | 0.046 | 0.085 | 0.058 | 0.241 | 0.303 |
| dN/dS | 0.074 | 0.115 | 0.100 | 0.229 | 0.181 |
Fig. 1.—
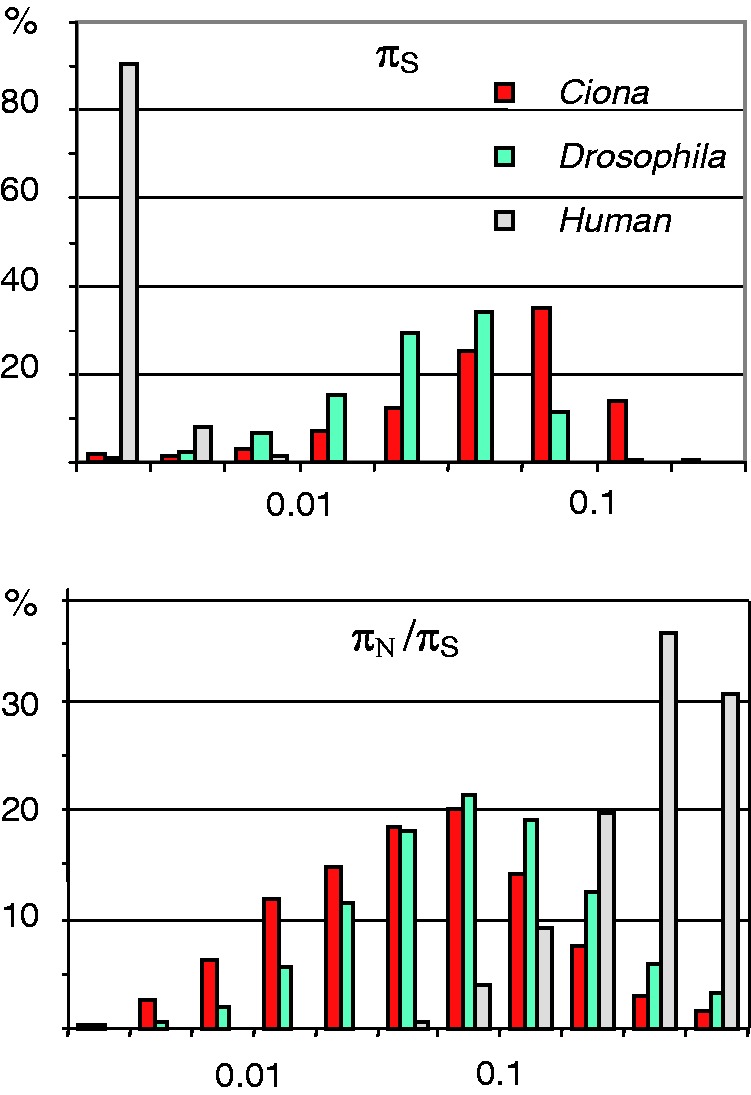
Distribution of πS and πN/πS across genes in three animal species. In D. simulans and H. sapiens, the subsets of orthologs to the C. intestinalis genes analyzed in this study were used here.
In D. simulans, we found that the subset of the orthologs to our C. intestinalis genes was biased toward a lower πS, and lower πN/πS and dN/dS ratio, when compared with the whole set of genes. This was expected given the universal relationship between the level of gene expression and the dN/dS ratio (Drummond et al. 2005; Koonin 2011) and the fact that the majority of genes studied here are evolutionary conserved, coding for binding proteins (GO:0005488), enzymes (GO:0003824), structural components of the ribosome, cytoskeleton and muscle (GO:0005200, GO:0008307, and GO:0003735), and transcription factors (GO:0030528). We assessed that this bias was of the order of 30% as far as πN/πS was concerned and of the order of 10% for πS and dN/dS. The human data set was more equivocal, with different biases depending on the statistics. At any rate, none of the biological conclusions drawn from this study are affected by these biases.
The dN/dS ratio (ratio of average dN to average dS) in C. intestinalis was relatively low but higher than the πN/πS ratio. This implies that a substantial fraction of amino acid substitutions has been driven to fixation by positive selection. This fraction α was estimated to be ∼0.54 when low-frequency SNPs were discarded and ∼0.78 when the whole site frequency spectrum was taken into account (table 4). This is similar to what was found in Drosophila (∼0.5, Smith and Eyre-Walker 2002; Bierne and Eyre-Walker 2004), wild mouse (0.57, Halligan et al. 2010), and rabbit (0.65, Carneiro et al. 2012) and much larger than the published estimates in plants (close to zero, with one exception, Gossmann et al. 2010), chicken (0.2, Axelsson and Ellegren 2009), and humans (0–0.15, Boyko et al. 2008 and references therein). Ciona intestinalis belongs to the set of species in which adaptive evolution strongly impacts amino acid sequence evolution. According to the method of Eyre-Walker and Keightley (2009), the estimated fraction of effectively neutral nonsynonymous mutations (population selection coefficient below 1) in C. intestinalis was 0.023 ± 0.001, i.e., quite low, in agreement with the hypothesis of a large population size in this species. Finally, the C. intestinalis B versus C. intestinalis A comparison revealed that, of 1,239 contigs longer than 100 codons, just six showed no fixed difference between the two species. This does not suggest that gene flow between the two species have impacted the genetic diversity of C. intestinalis B, even though more data from C. intestinalis A would be required to conclude.
In the above-described analysis of this study, reads from C. intestinalis B were directly mapped onto the reference C. intestinalis A genome. The reference being a bit distant, the proportion of mapped reads was only 30%. This procedure, furthermore, might have biased the sample toward genes showing a relatively low level of divergence between the two C. intestinalis species. To take this potential bias into account, we performed an additional analysis in which reads from C. intestinalis B were first assembled into contigs using the Abyss and Cap3 programs (Cahais et al. 2012). Reads were next mapped to the de novo assembly using BWA. The proportion of mapped reads was 67%. Then BLAST searches of assembled contigs against C. intestinalis A cDNA were performed, and contigs with exactly one hit were selected (Cahais et al. 2012). Of these, the 1,459 contigs that showed coverage >10X in at least four individuals were used for population genomic analysis (M1 model, site_p = 0.5)—note that this number is slightly lower than in the main analysis. The results of this control analysis were largely similar to the main ones (πS = 0.059, πN = 0.0029, πN/πS = 0.049, percentage of stop-codon containing genes = 2.2%, FIS = −0.027). The average dN/dS ratio was increased to 0.12 (when compared with 0.08 in the main analysis), which resulted in an even higher estimate of α (0.46) and α0.2 (0.62). In conclusion, the main results of this study were confirmed or reinforced when a distinct approach was followed for linking the reads from C. intestinalis B to the genome of C. intestinalis A.
The Causes of Accelerated Evolution in Tunicates
Based on these population genomic analyses, what can be concluded about the causes of the high amino acid substitution rate in tunicates? First, our results suggest that this increased rate is not due to relaxed functional constraints on proteins or less efficient purifying selection. The low values of both dN/dS and πN/πS indicate that selection against deleterious mutations is strong in C. intestinalis, as expected in a large population-sized species. So the elevated amino acid substitution rate in tunicates must be due to other causes, namely a higher rate of adaptive evolution and/or an increased mutation rate.
McDonald–Kreitman-based analyses show that adaptive processes substantially affect the evolutionary rate of protein sequences in C. intestinalis. No such effect was detected in humans, in which the estimated proportion of adaptive amino acid substitutions is below 0.15 (Boyko et al. 2008). This suggests that the accelerated amino acid sequence evolution in tunicates might be explained by a higher adaptive rate in this group. Published estimates of α in mice and rabbits, however, are similar to that we report in C. intestinalis. To tentatively quantify the relative influence of adaptive rate and mutation rate on tunicate proteic rate, let us model the per-year nonsynonymous substitution rate, dN, as:
| (1) |
where dN_a is the per-year rate of adaptive amino acid substitution, and µ P the rate of effectively neutral amino acid substitution, written as the product of the per-year mutation rate, µ, by the proportion of effectively neutral mutations, P. Equation (1) can be rewritten as:
| (2) |
with α = dN_a/dN. Adding subscript T for tunicates and V for vertebrates, and dividing the two equations, we obtain an expression for the tunicate versus vertebrate mutation rate ratio:
| (3) |
The [(dN)T/(dN)V] ratio was estimated to ∼2 by Tsagkogeorga et al. (2010) based on 35 high-expressed genes. P was approached here by the proportion of nonsynonymous mutations whose population selection coefficient is between 0 and −1. This proportion has been estimated, together with α, in humans (α = 0.1, P = 0.21, Boyko et al. 2008), mice (α = 0.57, P = 0.1, Halligan et al. 2010), and rabbit (α = 0.65, P = 0.03, Carneiro et al. 2012) using the method of Eyre-Walker and Keightley (2009), as we did for C. intestinalis in this study. It is important to note that, because our estimates of dN have been obtained in a per-year basis, the µT/µV ratio in equation (3) is meant per year too.
Table 6 gives estimates of the µT/µV ratio, using the C. intestinalis estimates of PT and αT, and the human, mouse, and rabbit estimates of PV and αV, respectively. Table 6 indicates that the long-term average per-year tunicate mutation rate could be 2–6 times as high as the vertebrate one, depending on which species best represents the long-term vertebrate average. These figures suggest that the higher amino acid substitution rate in tunicate is explained in the first place by an increased per-year mutation rate in this lineage. Please note that the whole rationale is dependent to some extent on a battery of assumptions, among which constant in time mutation rate and population size in the C. intestinalis lineage.
Table 6.
Estimates of the tunicate versus vertebrate mutation rate ratio obtained using population genomics parameters of three distinct vertebrate species
| Rabbit | Mouse | Human | |
|---|---|---|---|
| (dN)T/(dN)V | 2 | 2 | 2 |
| α V | 0.65 | 0.57 | 0.15 |
| (1 − α T)/(1 − α V) | 0.63 | 0.51 | 0.37 |
| PV | 0.03 | 0.1 | 0.21 |
| PV/PT | 1.34 | 4.34 | 9.13 |
| µT/µV | 1.69 | 4.43 | 6.76 |
If we assumed that the average generation time in vertebrates (approximately from 1 year to some tens of years) was ≥10 times as long as the average generation time in tunicates (approximately from 1 month to some years), then our results would predict a lower per-generation mutation rate in tunicates than in vertebrates, consistent with the suggestion that large populations achieve higher DNA-polymerase fidelity thanks to more efficient purifying selection (Lynch 2008).
Conclusions
This study demonstrates that NGS-based transcriptome analysis is a convenient way to obtain reliable population genomic data at a relatively low cost—our approach yielded ∼30,000 SNPs in ∼1,600 genes. Regarding SNP and genotype calling, of the two error models we tested in a novel ML framework, the more complex M2, which accounts for potential allelic expression biases, was found to be statistically better but empirically less robust than the simpler M1. This suggests that the additional γ parameter captures some signal in the data, which is unrelated to true allelic expression bias, thus making inferences less accurate. However, both models yielded very similar estimates of the population genomic parameters. The very low number of inferred stop codons, nearly zero FIS and low value of πN/πS suggest that our data set was not strongly affected by sequencing or read mapping errors. This work also confirms that transcriptome-based gene sampling tends to bias the subset of analyzed genes toward evolutionarily conserved proteins. Although this issue deserves to be taken into account, it certainly does not disqualify transcriptome-based data for population genomic studies.
Our analyses indicate that the increased amino acid substitution rate of tunicates, when compared with vertebrates, is not due to a relaxing of purifying selection but rather reflects a stronger effect of adaptive evolution and a 2–6 times higher per-year mutation rate. Quantifying more precisely the relative importance of these two factors would require obtaining similar data in a large number of tunicate and vertebrate species, to measure the average strength of positive and negative selection in both groups. In this direction, the planktonic tunicate Oikopleura dioica would be a good candidate, as the acceleration in evolutionary rate is particularly marked in this species (Tsagkogeorga et al. 2009, 2010) and its complete genome has already been sequenced (Denoeud et al. 2010).
The C. intestinalis population genomics appears typical of a large population sized, short-generation time species, with a high level of genetic diversity, low πN/πS and dN/dS ratio, a substantial fraction of adaptive amino acid substitutions, and an elevated per-year mutation rate. This is reasonably consistent with the ecology and life history traits of this species: C. intestinalis is a widespread and invasive broadcast spawner, occurring in large numbers of abundant colonies in all the temperate coasts of the Atlantic and Pacific oceans, and its generation time is of 1 year (Lambert 2005). This possible link between amino acid substitution rate and species life history traits and ecology would deserve to be confirmed by similar analyses in various animal phyla. Next-generation sequencing of whole transcriptomes gives the opportunity for such a comparative approach across animals, at a reasonable cost.
Supplementary Material
Supplementary Materials are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors are grateful to D. Jiang and B. Vercaemer for providing samples, to P. Gayral, M. Ballenghien, F. Delsuc, and M. Tilak for their help, and to four reviewers for helpful comments. This work was supported by a European Research Council grant to N.G. (ERC PopPhyl 232 971). This is publication number ISEM 2012-078.
Literature Cited
- Axelsson E, Ellegren H. Quantification of adaptive evolution of genes expressed in avian brain and the population size effect on the efficacy of selection. Mol Biol Evol. 2009;26:1073–1079. doi: 10.1093/molbev/msp019. [DOI] [PubMed] [Google Scholar]
- Begun DJ, et al. Population genomics: whole-genome analysis of polymorphism and divergence in Drosophila simulans. PLoS Biol. 2007;5:e310. doi: 10.1371/journal.pbio.0050310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierne N, Eyre-Walker A. The genomic rate of adaptive amino acid substitution in Drosophila. Mol Biol Evol. 2004;21:1350–1360. doi: 10.1093/molbev/msh134. [DOI] [PubMed] [Google Scholar]
- Bourlat SJ, et al. Deuterostome phylogeny reveals monophyletic chordates and the new phylum Xenoturbellida. Nature. 2006;444:85–88. doi: 10.1038/nature05241. [DOI] [PubMed] [Google Scholar]
- Boyko AR, et al. Assessing the evolutionary impact of amino acid mutations in the human genome. PLoS Genet. 2008;4:e1000083. doi: 10.1371/journal.pgen.1000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromham L. Why do species vary in their rate of molecular evolution? Biol Lett. 2009;5:401–404. doi: 10.1098/rsbl.2009.0136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustamante CD, et al. Natural selection on protein-coding genes in the human genome. Nature. 2005;437:1153–1157. doi: 10.1038/nature04240. [DOI] [PubMed] [Google Scholar]
- Cahais V, et al. Reference-free transcriptome assembly in non-model animals from next-generation sequencing data. Mol Ecol Resour. 2012 doi: 10.1111/j.1755-0998.2012.03148.x. Advance Access published April 30, 2012, doi: 10.1111/j.1755-0998.2012.03148.x. [DOI] [PubMed] [Google Scholar]
- Caputi L, et al. Cryptic speciation in a model invertebrate chordate. Proc Natl Acad Sci U S A. 2007;104:9364–9369. doi: 10.1073/pnas.0610158104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carneiro M, et al. Evidence for widespread positive and purifying selection across the European rabbit (Oryctolagus cuniculus) genome. Mol Biol Evol. 2012;29:1837–1849. doi: 10.1093/molbev/mss025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- Dehal P, et al. The draft genome of Ciona intestinalis: insights into chordate and vertebrate origins. Science. 2002;298:2157–2167. doi: 10.1126/science.1080049. [DOI] [PubMed] [Google Scholar]
- Delsuc F, Brinkmann H, Chourrout D, Philippe H. Tunicates and not cephalochordates are the closest living relatives of vertebrates. Nature. 2006;439:965–968. doi: 10.1038/nature04336. [DOI] [PubMed] [Google Scholar]
- Delsuc F, Brinkmann H, Philippe H. Phylogenomics and the reconstruction of the tree of life. Nat Rev Genet. 2005;6:361–375. doi: 10.1038/nrg1603. [DOI] [PubMed] [Google Scholar]
- Delsuc F, Tsagkogeorga G, Lartillot N, Philippe H. Additional molecular support for the new chordate phylogeny. Genesis. 2008;46:592–604. doi: 10.1002/dvg.20450. [DOI] [PubMed] [Google Scholar]
- Denoeud F, et al. Plasticity of animal genome architecture unmasked by rapid evolution of a pelagic tunicate. Science. 2010;330:1381–1385. doi: 10.1126/science.1194167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond DA, Bloom JD, Adami C, Wilke CO, Arnold FH. Why highly expressed proteins evolve slowly. Proc Natl Acad Sci U S A. 2005;102:14338–14343. doi: 10.1073/pnas.0504070102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn CW, et al. Broad phylogenomic sampling improves resolution of the animal tree of life. Nature. 2008;452:745–749. doi: 10.1038/nature06614. [DOI] [PubMed] [Google Scholar]
- Dutheil J, et al. Bio++: a set of C++ libraries for sequence analysis, phylogenetics, molecular evolution and population genetics. BMC Bioinformatics. 2006;7:188. doi: 10.1186/1471-2105-7-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyre-Walker A, Keightley PD. Estimating the rate of adaptive molecular evolution in the presence of slightly deleterious mutations and population size change. Mol Biol Evol. 2009;26:2097–2108. doi: 10.1093/molbev/msp119. [DOI] [PubMed] [Google Scholar]
- Fay JC, Wyckoff GJ, Wu CI. Positive and negative selection on the human genome. Genetics. 2001;158:1227–1234. doi: 10.1093/genetics/158.3.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gayral P, et al. Next-generation sequencing of transcriptomes: a guide to RNA isolation in nonmodel animals. Mol Ecol Resources. 2011;11:650–661. doi: 10.1111/j.1755-0998.2011.03010.x. [DOI] [PubMed] [Google Scholar]
- Gossmann TI, et al. Genome wide analyses reveal little evidence for adaptive evolution in many plant species. Mol Biol Evol. 2010;27:1822–1832. doi: 10.1093/molbev/msq079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halligan DL, Oliver F, Eyre-Walker A, Harr B, Keightley PD. Evidence for pervasive adaptive protein evolution in wild mice. PLoS Genet. 2010;6:e1000825. doi: 10.1371/journal.pgen.1000825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohenlohe PA, et al. Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags. PLoS Genet. 2010;6:e1000862. doi: 10.1371/journal.pgen.1000862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland LZ, Gibson-Brown JJ. The Ciona intestinalis genome: when the constraints are off. Bioessays. 2003;25:529–532. doi: 10.1002/bies.10302. [DOI] [PubMed] [Google Scholar]
- Iannelli F, Pesole G, Sordino P, Gissi C. Mitogenomics reveals two cryptic species in Ciona intestinalis. Trends Genet. 2007;23:419–422. doi: 10.1016/j.tig.2007.07.001. [DOI] [PubMed] [Google Scholar]
- Keightley PD, Eyre-Walker A. What can we learn about the distribution of fitness effects of new mutations from DNA sequence data? Philos Trans R Soc Lond B Biol Sci. 2010;365:1187–1193. doi: 10.1098/rstb.2009.0266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley PD, Halligan DL. Inference of site frequency spectra from high-throughput sequence data: quantification of selection on nonsynonymous and synonymous sites in humans. Genetics. 2011;188:931–940. doi: 10.1534/genetics.111.128355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV. Are there laws of genome evolution? PLoS Comput Biol. 2011;7:e1002173. doi: 10.1371/journal.pcbi.1002173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert G. Ecology and natural history of the protochordates. Can J Zool. 2005;83:34–50. [Google Scholar]
- Lartillot N, Brinkmann H, Philippe H. Suppression of long-branch attraction artefacts in the animal phylogeny using a site-heterogeneous model. BMC Evol Biol. 2007;7(1 Suppl):S4. doi: 10.1186/1471-2148-7-S1-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lartillot N, Philippe H. A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol Biol Evol. 2004;21:1095–1109. doi: 10.1093/molbev/msh112. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. The cellular, developmental and population-genetic determinants of mutation-rate evolution. Genetics. 2008;180:933–943. doi: 10.1534/genetics.108.090456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. Estimation of allele frequencies from high-coverage genome-sequencing projects. Genetics. 2009;182:295–301. doi: 10.1534/genetics.109.100479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991;351:652–654. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- Nabholz B, Glémin S, Galtier N. Strong variations of mitochondrial mutation rate across mammals—the longevity hypothesis. Mol Biol Evol. 2008;25:120–130. doi: 10.1093/molbev/msm248. [DOI] [PubMed] [Google Scholar]
- Nikolaev SI, et al. Life-history traits drive the evolutionary rates of mammalian coding and noncoding genomic elements. Proc Natl Acad Sci U S A. 2007;104:20443–20448. doi: 10.1073/pnas.0705658104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nydam ML, Harrison RG. Polymorphism and divergence within the ascidian genus Ciona. Mol Phylogenet Evol. 2010;56:718–726. doi: 10.1016/j.ympev.2010.03.042. [DOI] [PubMed] [Google Scholar]
- Nydam ML, Harrison RG. Reproductive protein evolution in two cryptic species of marine chordate. BMC Evol Biol. 2011a;11:18. doi: 10.1186/1471-2148-11-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nydam ML, Harrison RG. Introgression despite substantial divergence in a broadcast spawning marine invertebrate. Evolution. 2011b;65:429–442. doi: 10.1111/j.1558-5646.2010.01153.x. [DOI] [PubMed] [Google Scholar]
- Ohta K. Mechanisms of molecular evolution. Philos Trans R Soc Lond B Biol Sci. 2000;355:1623–6162. doi: 10.1098/rstb.2000.0724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paps J, Baguna J, Riutort M. Bilaterian phylogeny: a broad sampling of 13 nuclear genes provides a new Lophotrochozoa phylogeny and supports a paraphyletic basal Acoelomorpha. Mol Biol Evol. 2009;26:2397–2406. doi: 10.1093/molbev/msp150. [DOI] [PubMed] [Google Scholar]
- Philippe H, Lartillot N, Brinkmann H. Multigene analyses of bilaterian animals corroborate the monophyly of Ecdysozoa, Lophotrochozoa, and Protostomia. Mol Biol Evol. 2005;22:1246–1253. doi: 10.1093/molbev/msi111. [DOI] [PubMed] [Google Scholar]
- Philippe H, et al. Acoelomorph flatworms are deuterostomes related to Xenoturbella. Nature. 2011;470:255–258. doi: 10.1038/nature09676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popadin K, Polishchuk LV, Mamirova L, Knorre D, Gunbin K. Accumulation of slightly deleterious mutations in mitochondrial protein-coding genes of large versus small mammals. Proc Natl Acad Sci U S A. 2007;104:13390–13395. doi: 10.1073/pnas.0701256104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam NH, et al. The amphioxus genome and the evolution of the chordate karyotype. Nature. 2008;453:1064–1071. doi: 10.1038/nature06967. [DOI] [PubMed] [Google Scholar]
- Rand DM, Kann LM. Excess amino acid polymorphism in mitochondrial DNA: contrasts among genes from Drosophila, mice, and humans. Mol Biol Evol. 1996;13:735–748. doi: 10.1093/oxfordjournals.molbev.a025634. [DOI] [PubMed] [Google Scholar]
- Satoh N. The ascidian tadpole larva: comparative molecular development and genomics. Nat Rev Genet. 2003;4:285–295. doi: 10.1038/nrg1042. [DOI] [PubMed] [Google Scholar]
- Satou Y, et al. Improved genome assembly and evidence-based global gene model set for the chordate Ciona intestinalis: new insight into intron and operon populations. Genome Biol. 2008;9:R152. doi: 10.1186/gb-2008-9-10-r152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh TR, et al. Tunicate mitogenomics and phylogenetics: peculiarities of the Herdmania momus mitochondrial genome and support for the new chordate phylogeny. BMC Genomics. 2009;10:534. doi: 10.1186/1471-2164-10-534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small KS, Brudno M, Hill MM, Sidow A. A haplome alignment and reference sequence of the highly polymorphic Ciona savignyi genome. Genome Biol. 2007;8:R41. doi: 10.1186/gb-2007-8-3-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith NG, Eyre-Walker A. Adaptive protein evolution in Drosophila. Nature. 2002;415:1022–1024. doi: 10.1038/4151022a. [DOI] [PubMed] [Google Scholar]
- Thomas JA, Welch JJ, Lanfear R, Bromham L. A generation time effect on the rate of molecular evolution in invertebrates. Mol Biol Evol. 2010;27:1173–1180. doi: 10.1093/molbev/msq009. [DOI] [PubMed] [Google Scholar]
- Tsagkogeorga G, Turon X, Galtier N, Douzery EJ, Delsuc F. Accelerated evolutionary rate of housekeeping genes in tunicates. J Mol Evol. 2010;71:153–167. doi: 10.1007/s00239-010-9372-9. [DOI] [PubMed] [Google Scholar]
- Tsagkogeorga G, et al. An updated 18S rRNA phylogeny of tunicates based on mixture and secondary structure models. BMC Evol Biol. 2009;9:187. doi: 10.1186/1471-2148-9-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner JR, et al. Computational analysis of whole-genome differential allelic expression data in human. PLoS Comput Biol. 2010;6:e1000849. doi: 10.1371/journal.pcbi.1000849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HC, Susko E, Spencer M, Roger AJ. Topological estimation biases with covarion evolution. J Mol Evol. 2008;66:50–60. doi: 10.1007/s00239-007-9062-4. [DOI] [PubMed] [Google Scholar]
- Wiens JJ. Can incomplete taxa rescue phylogenetic analyses from long-branch attraction? Syst Biol. 2005;54:731–742. doi: 10.1080/10635150500234583. [DOI] [PubMed] [Google Scholar]
- Zhan A, Macisaac HJ, Cristescu ME. Invasion genetics of the Ciona intestinalis species complex: from regional endemism to global homogeneity. Mol Ecol. 2010;19:4678–4694. doi: 10.1111/j.1365-294X.2010.04837.x. [DOI] [PubMed] [Google Scholar]