

Abstract
Case: A 34-year-old woman was referred to our hospital with progressive movement disorders and neurodegeneration with brain iron accumulation and enlargement of the frontal diploe on the MRI. Metabolic testing revealed that she had α-mannosidosis (AMD), a lysosomal storage disorder.
Background: AMD is a rare genetic disorder that causes α-mannosidase deficiency resulting in lysosomal accumulation of undigested oligosaccharides. The symptoms of AMD consist of facial and skeletal deformities combined with progressive psychiatric and neurological complaints, especially ataxia and mental retardation. Bilateral patellar dislocation and hearing impairment are frequent.
Discussion: The movement disorders we found in our patient have not been reported previously, but they are likely late symptoms of this progressive disorder. The iron deposits in the basal ganglia have also not been reported in AMD and are yet of unknown significance. Lysosomal storage disorders, such as AMD, should be considered in patients with progressive neurologic conditions and neurodegeneration with brain iron accumulation on MRI.
Electronic supplementary material The online version of this article (doi:10.1007/8904_2011) contains supplementary material, which is available to authorized users.
Introduction
α-Mannosidosis (AMD) is a rare autosomal recessive genetic disorder of the LAMAN gene (MAN2B1) that causes α-mannosidase deficiency. This results in lysosomal accumulation of undigested oligosaccharides. The incidence is 1 in 500,000 live births. Diagnosis is made by measuring acid α-mannosidase activity in leucocytes. The symptoms of AMD consist of facial and skeletal deformities combined with progressive psychiatric and neurological complaints, especially ataxia and mental retardation. Bilateral patellar dislocation and hearing impairment are frequent (Malm and Nilssen2008).
Three clinical types of AMD are recognized: Type 1, mild form with slow progression, clinically recognized after 10 years of age, without skeletal abnormalities; Type 2, the most frequent form with slow progression, recognized before 10 years of age, with skeletal abnormalities and ataxia from age 20 to 30; Type 3, severe form, immediately recognized and leading to an early death (Malm and Nilssen 2008). We report a case of an adult diagnosed patient with juvenile onset of AMD (Type 2) and striking neurodegeneration with brain iron accumulation (NBIA) on MRI.
Case Report
A 34-year-old woman was referred for a progressive movement disorder. She was born to healthy nonconsanguineous Dutch parents; pregnancy and birth were unremarkable. Developmental milestones were delayed, walking started at age 2.5 years. Since puberty, she suffered from progressive coordination problems of all limbs, leading to repetitive falls. In the same period she also lost the ability to ambulate and her cognition and memory progressively declined. Depressive symptoms developed over the years, for which she used sertraline and pipamperon, and hearing diminished. Medical history revealed repetitive knee surgery for dislocations and a laminectomy for lumbar stenosis. Family history was negative.
Her face showed the characteristic coarse facial features of AMD (see Video 1). On neurological examination, a mentally retarded woman was seen with impersistence of ocular movements and dysarthria. Mild atrophy of the hand interossei muscles and hammer toes was noted. Arm movements were bilaterally ataxic. During action, mild dystonia of the hands and positive and negative myoclonus on both arms were observed, distally more severe than proximally. On walking, a broad-based, ataxic gait was seen. Reflexes were symmetrical with bilaterally suspect plantar responses (Video 1).
A 1.5 T MRI of the cerebrum showed cerebral and cerebellar atrophy (Fig. 1), enlargement of the frontal diploe and bilateral hypointensities in the pulvinar of the thalamus, globus pallidus and putamen on T2-(turbo spin echo; TSE)-weighted images, suggestive for iron accumulation (Fig. 1; compared to healthy age- and sex-matched control in Fig. 2). The substantia nigra and red nucleus were also hypointense on T2-(TSE)-weighted images (Fig. 3). The basal ganglia were isointense on T1-weighted images. Other iron-rich nuclei (e.g., dentate nucleus) were less hypointense compared to the thalamus, globus pallidus, and putamen. A CT-scan ruled out calcifications. The combination of brain atrophy and basal ganglia hypointensities, suggestive for iron accumulation, is known as NBIA (Gregory et al. 2009). Of the usual causes of NBIA (also see Discussion), pantothenate kinase-associated neurodegeneration, neuroferritinopathy, and aceruloplasminaemia were excluded with genetic and laboratory tests.
Fig. 1.
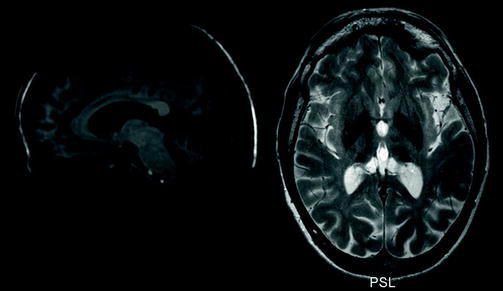
Combined MRI image. The left side is a sagittal T1-weighted image showing cranial dysmorphia and thickening of the calvaria with enlargement of the frontal diploe, mild cerebral atrophy, and severe cerebellar atrophy. The right side is a transversal T2-(TSE)-weighted image showing hypointensities consistent with bilateral iron accumulation in the pulvinar of the thalamus, globus pallidus, and putamen
Fig. 2.
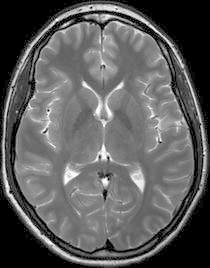
T2-weighted MRI-scan of a healthy age- and sex-matched control at the same level as the right side of Fig. 1
Fig. 3.
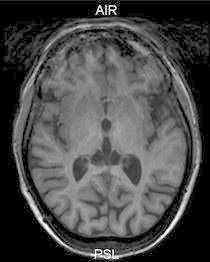
T1-weighted image of the patient at the level of the basal ganglia showing an isointense signal
Based on the patients’ clinical picture, extensive biochemical and genetic tests were performed including a muscle biopsy and genetic tests for mitochondrial disorders, spinocerebellar ataxias (SCA1, 2, 3, and 17) and Friedrich’s ataxia (FRDA) and all tests were negative. However, laboratory testing of the urine showed high concentrations of mannose-rich oligosaccharides, indicative of AMD. This diagnosis was confirmed by a deficient α-mannosidase activity of 1 nmol/mg h in leucocytes (n = 70–270).
Discussion
We diagnosed AMD in this patient at the age of 34 years, despite the juvenile onset and typical clinical phenotype. The movement disorders with dystonia and positive and negative myoclonus have not been reported previously, but they are likely late symptoms of this progressive disorder. The NBIA detected in our patient has also not been described previously. In 12 patients with AMD, the MRI has been described, showing several nonspecific abnormalities in bone structure and gray and white matter (Ara et al. 1999; Dietemann et al. 1990; Niemann et al. 1996; Gutschalk et al. 2004; Patlas et al. 2001). Interestingly, in a 27-year-old man with AMD bilateral hypointensities of unknown origin in the basal ganglia, especially the thalamus, on T2-weighted images were described (Ara et al. 1999). Furthermore, two AMD patients showed bilateral hypointensities in the thalamic region on T2-weighted images, but the authors state that the intensity of the basal ganglia was normal (Gutschalk et al. 2004). Iron accumulation has not been shown in postmortem pathology studies of young patients with AMD (Kjellman et al. 1969; Sung et al. 1977). As far as we know, there are no published pathology studies of older patients with AMD.The combination of brain atrophy and hypointensities in the basal ganglia on MRI is referred to as NBIA. Iron is hypointense on T2-weigthed images and isointense on T1-weighted images and CT-scan, differentiating it from calcifications that are hypointense on T2-weighted and hyperintense on T1-weighted images. Until now, in only 50–70% of the NBIA cases, the cause is known and associated with a genetic disorder leading to iron accumulation in the basal ganglia; e.g., pantothenate kinase-associated neurodegeneration (PKAN), infantile neuroaxonal dystrophy (INAD), neuroferritinopathy and aceruloplasminaemia (Gregory et al. 2009). The remaining 30–50% of NBIA cases is idiopathic. In our patient, three of these four causes of NBIA were excluded (PKAN, neuroferritinopathy, and aceruloplasminaemia) with genetic and laboratory tests. INAD was not excluded, because it is not possible to test this condition in the Netherlands and we already made a suitable diagnosis. This case illustrates that lysosomal storage disorders should be considered. Hypointensities in the basal ganglia and the thalamus have been described in other lysosomal storage diseases, such as fucosidosis and mucoliposidosis, but were not classified as NBIA (Autti et al. 2007). The pathogenetic mechanisms underlining the basal ganglia abnormalities in AMD remain to be determined. In mucolipidosis, the hypointensities in the basal ganglia were suggested to be caused by iron accumulation based on the inability of lysosomes to release iron because of lipofuscin accumulation (Johnstone and Milward 2010). Comparable mechanisms could play a role in AMD. Alternatively, the hypointensities could be caused by alterations in tissue viscosity by accumulation of lysosomal storage material and give similar resonances on MRI as that of iron (Autti et al. 2007). However, one would expect the abnormalities to be present in almost all lysosomal storage disorders, instead of in a selected number of disorders (Autti et al. 2007).
In conclusion, in patients with a progressive neurologic condition and features of NBIA on the MRI not only ferritinopathies but also AMD and other lysosomal storage diseases should be considered.
Acknowledgments
The authors would like to thank Marieke Sprengers for her helpful comments on the MRI-scan.
Take Home Message
In patients with a progressive neurologic condition and features of iron depositions on the MRI also think of AMD.
Supplemental Data
Video of the neurological exam (Zoons et al video1.mpg).
Video legend
Zoons et al video 1.mpg: The video consists of 5 segments, divided by transitions.
Segment 1 shows the patient at rest. The typical coarse facial features are visible and there is mild dysarthria.
Segment 2 shows myoclonus of the hands (left>right) in rest.
Segment 3 shows the patient during extension of the arms and wrists, first both and then one arm at a time. During this segment there is mild dystonia and both positive and negative myoclonus on both arms.
Segment 4 shows ataxia when the patient moves her finger from her knee to her nose and back, on both arms. During the second part of this segment the patient attempts to hold up both arms in front of her chest with flexion in the elbows. Mild dystonia and positive and negative myoclonus can be noted.
Segment 5 shows the patient walking with a broad-based ataxic gait. Mild dystonia of the arms can be noted.
Author Roles
E Zoons: literature search, writing of the first draft, and revise the manuscript
TJ de Koning: review and critique
NGGM Abeling: review and critique, metabolic testing
MAJ Tijssen: review and critique, corresponding author
Financial Disclosures
This study was not sponsored.
Dr. Zoons reports no disclosures.
Dr. de Koning was sponsored by Milupa Metabolics to visit the “40th European Metabolic Group Meeting” in Heidelberg, Germany, in May 2008 and by Swedish Orphan to attend a symposium titled “Treatment of Urea Cycle Defects” in 2009.
Dr. Abeling reports no disclosures.
Dr. Tijssen reports no personal disclosures; however, the dystonia nurse working both in patient care and in the research group of which Dr. Tijssen is the principal investigator is supported by a grant from Ipsen Pharmaceuticals.
Footnotes
Competing interests: None declared.
References
- Ara JR, Mayayo E, Marzo ME, Guelbenzu S, Chabas A, Pina MA, Calderon C. Neurological impairment in alpha-mannosidosis: a longitudinal clinical and MRI study of a brother and sister. Childs Nerv Syst. 1999;15:369–371. doi: 10.1007/s003810050416. [DOI] [PubMed] [Google Scholar]
- Autti T, Joensuu R, Aberg L. Decreased T2 signal in the thalami may be a sign of lysosomal storage disease. Neuroradiology. 2007;49:571–578. doi: 10.1007/s00234-007-0220-6. [DOI] [PubMed] [Google Scholar]
- Dietemann JL, Filippi de la Palavesa MM, Tranchant C, Kastler B. MR findings in mannosidosis. Neuroradiology. 1990;32:485–487. doi: 10.1007/BF02426460. [DOI] [PubMed] [Google Scholar]
- Gregory A, Polster BJ, Hayflick SJ. Clinical and genetic delineation of neurodegeneration with brain iron accumulation. J Med Genet. 2009;46:73–80. doi: 10.1136/jmg.2008.061929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutschalk A, Harting I, Cantz M, Springer C, Rohrschneider K, Meinck HM. Adult alpha-mannosidosis: clinical progression in the absence of demyelination. Neurology. 2004;63:1744–1746. doi: 10.1212/01.WNL.0000143057.25471.4F. [DOI] [PubMed] [Google Scholar]
- Johnstone D, Milward EA. Molecular genetic approaches to understanding the roles and regulation of iron in brain health and disease. J Neurochem. 2010;113(6):1387–1402. doi: 10.1111/j.1471-4159.2010.06697.x. [DOI] [PubMed] [Google Scholar]
- Kjellman B, Gamstorp I, Brun A, Ockerman PA, Palmgren B. Mannosidosis: a clinical and histopathologic study. J Pediatr. 1969;75:366–373. doi: 10.1016/S0022-3476(69)80260-X. [DOI] [PubMed] [Google Scholar]
- Malm D, Nilssen O. Alpha-mannosidosis. Orphanet J Rare Dis. 2008;3:21. doi: 10.1186/1750-1172-3-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemann S, Beck M, Seidel G, Spranger J, Vieregge P. Neurology of adult alpha-mannosidosis. J Neurol Neurosurg Psychiatry. 1996;61:116–117. doi: 10.1136/jnnp.61.1.116-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patlas M, Shapira MY, Nagler A, Sheffer R, Gomori JM. MRI of mannosidosis. Neuroradiology. 2001;43:941–943. doi: 10.1007/s002340100607. [DOI] [PubMed] [Google Scholar]
- Sung JH, Hayano M, Desnick RJ. Mannosidosis: pathology of the nervous system. J Neuropathol Exp Neurol. 1977;36:807–820. doi: 10.1097/00005072-197709000-00004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.