

Abstract
We describe a 27-month-old girl with COG6 deficiency. She is the first child of healthy consanguineous Moroccan parents. She presented at birth with dysmorphic features including microcephaly, post-axial polydactyly, broad palpebral fissures, retrognathia, and anal anteposition. The clinical phenotype was further characterised by multiorgan involvement including mild psychomotor retardation, and microcephaly, chronic inflammatory bowel disease, micronodular liver cirrhosis, associated with life-threatening and recurrent infections due to combined T- and B-cell dysfunction and neutrophil dysfunction.
Mutation analysis showed the patient to be homozygous for the c.G1646T mutation in the COG6 gene. She is the second reported patient with a deficiency of subunit 6 of the COG complex. Although both patients are homozygous for the same mutation, they present a markedly different clinical picture. Indeed immunodeficiency as well as inflammatory bowel disease has not been described previously in patients with any COG-CDG.
Introduction
Congenital disorders of glycosylation (CDG) are a group of rare metabolic diseases caused by defects in the synthesis of glycans and in the attachment of glycans to proteins and lipids. The glycans on proteins are mostly N-linked or O-linked. Two types of protein N-glycosylation disorders can be distinguished: CDG-I (defects in the assembly of N-glycans in the cytosol and the endoplasmic reticulum (ER)) and CDG-II (defects in the processing and maturation of N-glycans in the ER and subsequently in the Golgi apparatus).
Patients with CDG form a rapidly growing group with a broad spectrum of clinical manifestations (Jaeken and Matthijs 2007; Grünewald et al. 2002; Jaeken 2010).
In 2004, Wu et al. described a new type of CDG due to a defect in subunit 7 of the conserved oligomeric Golgi (COG) complex (Wu et al. 2004). Subsequently, defects in other subunits (COG1, COG4, COG5, COG6, and COG8) have been reported (Foulquier et al. 2006; Reynders et al. 2009; Paesold-Burda et al. 2009; Lübbenhusen et al. 2010; Foulquier et al. 2007; Kranz et al. 2007). The COG is a hetero-octameric peripheral Golgi protein complex (COG1–COG8) and presents as a bi-lobed structure, bridged through the COG1–COG8 interaction. This complex appears to be required for the preservation of the integrity of the Golgi, to facilitate transport of proteins within the Golgi and for retrograde transport from the Golgi to the ER. The exact mechanism by which the COG complex influences the Golgi structure and function is still unclear. However, it seems that COG defects affect glycosylation through altered trafficking of glycosyltransferases (Smith and Lupashin 2008; Zeevaert et al. 2008).
We describe a second patient with COG6-CDG. She presented with multi-system disease including dysmorphy and neurological, hepato-intestinal, immunological and renal involvement. The novel nomenclature will be used in this report (Jaeken et al. 2008, 2009).
Case Report
We report a 27-month-old girl, born at 37 weeks of gestation after an uncomplicated pregnancy. She is the first child of healthy consanguineous Moroccan parents without remarkable family history. Her birth weight and height were respectively 2,860 g (−1.1 SD) and 50 cm (−0.1 SD). She presented with dysmorphic features including microcephaly (head circumference was 31.5 cm (−2.5 SD)), a post-axial polydactyly without syndactyly on the right hand, broad palpebral fissures, retrognathia, and anal anteposition. She showed a normal female karyotype.
During the first 3.5 months of life, she was hospitalised four times because of recurrent infections, diarrhoea, and poor weight gain. At the age of 8 months, the girl was admitted to the intensive care unit of our hospital with respiratory failure due to an interstitial pneumonia. A positive PCR for Pneumocystis carinii and Rhinovirus was found in the bronchoalveolar lavage. Laboratory tests revealed elevated transaminases (up to a 20-fold increase, ASAT more than ALAT) and slightly increased γ-GT. PTT and aPTT were normal and the child had no bleeding tendency. Serum lactate and ammonia, plasma and urinary amino acids, organic acids, and urinary oligosaccharides were normal. Infectious serology (EBV, hepatitis, and toxoplasmosis), a sweat test, and abdominal ultrasound were normal. Significant hypogammaglobulinaemia (IgG 0.89 g/l, IgA < 0.06 g/l, and IgM 0.06 g/l, normal values for age, respectively, 5.4–13.3 g/l, 0.3–1.85 g/l, and 0.52–1.93 g/l) were found. Monthly treatment with immunoglobulin was started. Further immunologic studies confirmed the presence of a combined immunodeficiency. In addition to antibody deficiency, cellular immunity was impaired with a poor response to mitogen (Phytohemagglutinin and CD3) and absence of lymphocyte proliferation to antigen (Candidine and tetanus toxin) after immunisation and despite recurrent skin and gut colonisation with Candida albicans. Nevertheless, there was no B or T lymphopenia and all the lymphocyte populations were within the normal range for age. In addition to antibody deficiency with T-cell dysfunction, granulocyte function assays showed poor dihydrorhodamine reduction and a reduced chemiluminescence (50% of controls). Chemotaxis, adhesion molecules and phagocytosis assays were normal.
At the age of 10 months, nutritional support by total parenteral nutrition followed by hyper-caloric feeding was started because of poor weight gain, chronic diarrhoea, and recurrent perianal fissures, but without significant weight gain. Normal albuminemia made protein loosing enteropathy unlikely. There was no proteinuria. The gastro- and colonoscopy performed at that time were normal.
Because of the hepatomegaly, the hepatic cytolysis, and failure to thrive, a liver biopsy was performed. This showed signs of micronodular cirrhosis and minimal macrovesicular steatosis with negative viral staining and no iron overload. Additional metabolic investigations, including a screening for CDG, were performed. The type 2 isoelectric focusing (IEF) pattern of serum transferrin and the abnormal IEF of serum apolipoprotein C-III in our patient pointed to a CDG-II, affecting the biosynthesis of both N- and O-linked glycans. Matrix-assisted laser desorption/ionisation-time of flight (MALDI-TOF) analysis of serum transferrin glycans showed mainly hyposialylation and a little hypogalactosylation (Fig. 1). Sequencing of the COG subunit genes revealed a homozygous mutation (c.G1646T) in COG6 leading to amino acid exchange p.G549V in the COG6 protein.
Fig. 1.
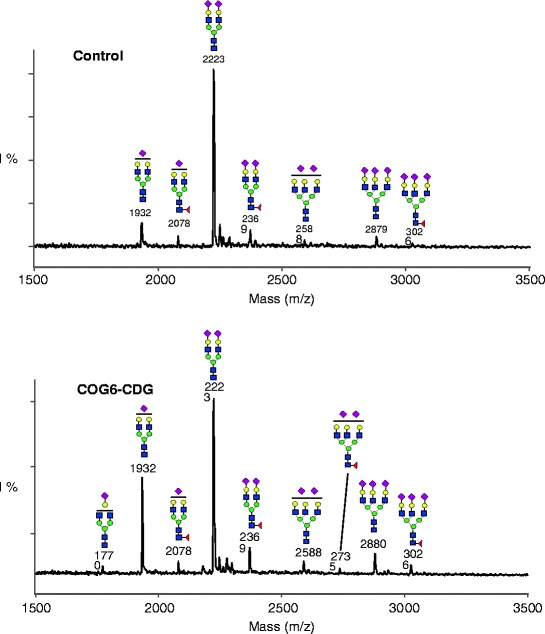
MALDI-TOF analysis of serum transferrin acidic glycans showing a clear increase of the biantennary glycan with only 1 sialic acid (mass 1932) and a small increase of the biantennary glycan with only 1 sialic acid and 1 galactose (mass 1770) compared to a control. Red diamonds sialic acid; yellow circles galactose; blue squares N-acetylglucosamine; green circles mannose
At the age of 2 years, severe failure to thrive with microcephaly was still present (weight 7,700 g, −4.6 SD; length 75 cm, −3.1 SD; head circumference 45 cm, −3.1 SD). Neurological examination showed axial hypotonia associated with mild neurodevelopmental delay. She was able to stand alone at the age of 16 months and said her first words at 2 years. One episode of febrile tonic-clonic seizures has been observed. Ophthalmologic examination, EEG, and cerebral MRI were normal.
At 24 months of age, a significant proximal tubulopathy and bloody diarrhoea were noted for the first time. An upper gastro-intestinal and lower endoscopy documented the presence of deep serpiginous ulcerations in the sigmoid and aphthous ulcers in the left part of the colon (Figs. 2 and 3). Biopsies showed an aspecific inflammatory infiltrate of the mucosa. No granuloma or viral inclusions were found on immunohisto-chemical staining. The findings were compatible with inflammatory bowel disease. On parenteral nutrition followed by progressive enteral feeding (Modulen IBD, Nestlé) and treatment with oral budesonide (0.75 mg per day), the gastro-intestinal symptoms improved and she started to gain some weight.
Fig. 2.
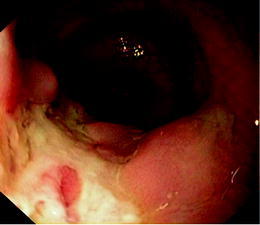
Presence of deep serpiginous ulcerations in the sigmoid before treatment
Fig. 3.
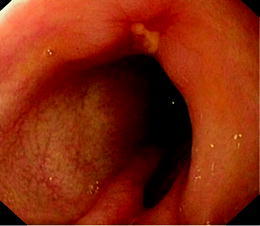
Aphthous ulceration present in the left part of the colon before treatment
Discussion
Our patient has a clinical phenotype characterised by multi-system involvement including chronic inflammatory bowel disease, liver cirrhosis, mild psychomotor retardation and microcephaly, associated with life-threatening and recurrent infections due to combined T and B immunodeficiency and neutrophil dysfunction. She presents with a homozygous mutation (c.G1646T) in the COG6 gene and is the second reported patient with a deficiency of COG6. The first patient described in 2010 by Lübbenhusen et al., suffered from intractable focal seizures, vomiting, and loss of consciousness due to intracranial bleedings. Cholestasis and vitamin K deficiency were found on metabolic investigation and considered, at least partly, as a cause for the intracranial haemorrhage. The patient died at 5 weeks of age due to brain oedema (Lübbenhusen et al. 2010). Despite marked differences in clinical presentation, both patients presented with the same mutation c.G1646T. This mutation produces instability of mRNA rather than a degradation of mutated protein (Lübbenhusen et al. 2010), indicating that individual factors could perhaps interfere with the degradation speed of mRNA and contribute to the interindividual phenotypical variation. Furthermore, MALDI-TOF analysis of transferrin revealed in both children hyposialylation associated with hypogalactosylation. Compared to our patient, there was more hyposialylation than hypogalactosylation in the first reported patient (Fig. 1). Another possible explanation for the different clinical picture might be the early death of the first patient before additional symptoms could have developed. Also, another genetic disease in our patient cannot be excluded taking into account the consanguinity of the parents, possibly explaining some unusual clinical features in particular the immunodeficiency.
Since the first description of COG7 deficient patients by Wu et al. in 2004, mutations in the genes encoding subunits COG 1, 4–6, and 8 of the COG complex have been identified. All these patients, including the present one, show a combination of a type 2 serum transferrin IEF pattern together with an abnormal serum apoC-III IEF profile. The clinical phenotypes of these patients vary from mild to severe with the COG7-CDG patients being the most severely affected (Zeevaert et al. 2008). Our patient showed some features that overlap with those of the previously described COG deficient patients (Table 1). The phenotypic differences correlate probably with differences in the severity of the trafficking and glycosylation abnormalities in patients’ fibroblasts (Reynders et al. 2009). A defect of sialylation, galactosylation and demannosylation has been found in COG7 deficient cells (the most severe form), while in COG5-CDG (mild form) only the terminal sialylation is affected (Reynders et al. 2009; Paesold-Burda et al. 2009).
Table 1.
Clinical phenotypes of patients with COG deficiency
| COG-CDG (number of patients) | COG1–CDG (1) | COG4-CDG (1) | COG5-CDG (1) | COG6-CDG (2) | COG7-CDG (6) | COG8-CDG (2) | |
|---|---|---|---|---|---|---|---|
| Early death | Early death | ||||||
| Dysmorphic features | Present | Mild | Absent | Present | Absent | Present | Mild |
| Microcephaly | Absent | Present | Absent | Present | Absent | Severe | Present |
| Failure to thrive | Present | No | No | Present | – | Present | Present |
| Growth retardation | Present | Not Reported | Absent | Present | – | Present | Present |
| Liver involvement | Hepatosplenomegaly | Elevated TA; decreased clotting factors | Absent | Hepatomegaly; elevated TA; normal clotting factors; cirrhosis | Bleeding; mildly elevated TA | Cholestasis; fibrotis; normal coagulation | Bleeding and thrombosis; TA increased inconsistently |
| Neurological signs | Hypotonia | Hypotonia; epilepsia; ataxia | Hypotonia; ataxia | Hypotonia; febrile seizures | Seizures | Hypotonia; seizures; neuropathy | Regression; hypotonia; seizures (once); neuropathy; ataxia |
| Psychomotor retardation | Mild | Mild | Moderate | Mild | – | Severe | Moderate to severe |
| Other | Recurrent infections | Combined immunodeficiencies; inflammatory bowel disease | Hyperthermia; intestinal pseudo-obstruction | Recurrent infections; PFAPA | |||
| Origin | Portuguese | Portuguese | Iraqui | Moroccan | Turkish | Moroccan and Tunisian | Spanish |
| Publication | Foulquier et al. (2006) | Reynders et al. (2009) | Paesold-Burda et al. (2009) | This publication | Lübbenhusen et al. (2010) | Wu et al. (2004), Spaapen et al. (2005), Morava et al. (2007), Ng et al. (2007) | Foulquier et al. (2007), Kranz et al. (2007) |
PFAPA: Periodic Fever Aphtous Stomatitis and Adenitis; TA: serum transaminases
Despite recurrent infections reported in patients with COG 4 and COG8 defects, no immunodeficiencies have previously been described in COG-CDG. Nevertheless, immunologic abnormalities, especially humoral deficiencies (hypogammaglobulinaemia associated with increased or decreased B-lymphocytes), have already been described in different types of CDG-1 like PMM2-CDG (CDG-Ia) (Blank et al. 2006), ALG12-CDG (CDG-Ig); (Chantret et al. 2002; Eklund et al. 2005; Zdebska et al. 2003) and ALG1-CDG (CDG-Ik) (Kranz et al. 2004). In addition to antibody deficiency, a diminished neutrophil chemotaxis associated with normal expression of adhesion molecules, has been found in PMM2-CDG. Our patient presents with a combined B- and T-cell dysfunction despite normal B- and T-cell counts for age. Moreover, we found a diminished neutrophil chemiluminescence with normal chemotaxis and expression of adhesion molecules. This could reflect a defect in glycosylation of the gp91phox subunit of the phagocyte NADPH-oxidase flavocytochrome b 558, involved in the production of free radicals in neutrophils (Parkos et al. 1987). Further studies are needed.
Protein loosing enteropathy coupled with intractable diarrhoea is a leading sign in MPI-CDG, ALG6-CDG and, less, in PMM2-CDG and may be due to altered mucosal integrity because of defects in membranous glycoproteins. The diagnosis was based on an increased α-1-antitrypsin in the stools or low serum albumin without proteinuria (Damen et al. 2004; Kelly et al. 2001; Mention et al. 2001). In our patient, we found mucosal lesions in the left part of the colon identical to those found in inflammatory bowel disease and responding to a treatment with oral steroids and enteral nutrition. There were no signs of protein loosing enteropathy. This has never been reported before in patients with COG defects and could be related to a functional defect in the immune system. Further studies are needed to characterise the immunological processes involved in the pathogenesis of the digestive lesions.
Footnotes
Competing interests: None declared.
References
- Blank C, Smith L, Hammer D, et al. Recurrent infections and immunological dysfunction in congenital disorder of glycosylation Ia (CDG Ia) J Inherit Metab Dis. 2006;26:592. doi: 10.1007/s10545-006-0275-2. [DOI] [PubMed] [Google Scholar]
- Chantret I, Dupre T, Delenda C, et al. Congenital disorders of glycosylation type Ig is defined by a deficiency in dolichyl-P-mannose: Man7GlcNac2-PP-dolichyl mannosyltransferase. J Biol Chem. 2002;277:25815–25822. doi: 10.1074/jbc.M203285200. [DOI] [PubMed] [Google Scholar]
- Damen G, de Klerk H, Huijmans J, et al. Gastrointestinal and other clinical manifestations in 17 children with congenital disorders of glycosylation type Ia, Ib, Ic. J Pediatr Gastroenterol Nutr. 2004;38:282–287. doi: 10.1097/00005176-200403000-00010. [DOI] [PubMed] [Google Scholar]
- Eklund EA, Newell JW, Sun L. Molecular and clinical description of the first US patients with congenital disorder of glycosylation Ig. Mol Genet Metab. 2005;84:25–31. doi: 10.1016/j.ymgme.2004.09.014. [DOI] [PubMed] [Google Scholar]
- Foulquier F, Vasile E, Schollen, et al. Conserved oligomeric Golgi complex subunit 1 deficiency reveals a previously uncharacterized congenital disorder of glycosylation type II. Proc Natl Acad Sci USA. 2006;103:3764–3769. doi: 10.1073/pnas.0507685103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foulquier F, Ungar D, Reynders E, et al. A new error of glycosylation due to a Cog8 deficiency reveals a critical role for the Cog1-Cog8 interaction in COG complex formation. Hum Mol Genet. 2007;16:717–730. doi: 10.1093/hmg/ddl476. [DOI] [PubMed] [Google Scholar]
- Grünewald S, Matthijs G, Jaeken J. Congenital disorders of glycosylation. Pediatr Res. 2002;52:618–624. doi: 10.1203/00006450-200211000-00003. [DOI] [PubMed] [Google Scholar]
- Jaeken J. Congenital disorders of glycosylation. Ann N Y Acad Sci. 2010;1214:190–198. doi: 10.1111/j.1749-6632.2010.05840.x. [DOI] [PubMed] [Google Scholar]
- Jaeken J, Matthijs G. Congenital disorders of glycosylation: a rapid expanding disease family. Annu Rev Genomics Hum Genet. 2007;8:261–278. doi: 10.1146/annurev.genom.8.080706.092327. [DOI] [PubMed] [Google Scholar]
- Jaeken J, Hennet T, Freeze HH, Matthijs G. On the nomenclature of congenital disorders of glycosylation (CDG) J Inherit Metab Dis. 2008;31:669–672. doi: 10.1007/s10545-008-0983-x. [DOI] [PubMed] [Google Scholar]
- Jaeken J, Hennet T, Matthijs G, Freeze HH. CDG nomenclature: time for a change! Biochim Biophys Acta. 2009;1792:825–826. doi: 10.1016/j.bbadis.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly DF, Boneh A, Pitsch S, et al. Carbohydrate-deficient glycoprotein syndrome 1b: a new answer to an old diagnostic dilemma. J Paediatr Child Health. 2001;37:510–512. doi: 10.1046/j.1440-1754.2001.00671.x. [DOI] [PubMed] [Google Scholar]
- Kranz C, Denecke J, Lehle L, et al. Congenital disorders of glycosylation type Ik (CDG-Ik): A defect of mannosyltransferase I. Am J Hum Genet. 2004;74:545–551. doi: 10.1086/382493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranz C, Ng B, Sun L, et al. COG8 deficiency causes a new congenital disorder of glycosylation type IIh. Hum Mol Genet. 2007;16:731–741. doi: 10.1093/hmg/ddm028. [DOI] [PubMed] [Google Scholar]
- Lübbenhusen J, Thiel C, Rind N, et al. Fatal outcome due to deficiency of subunit 6 of the conserved oligomeric Golgi complex leading to a new type of congenital disorders of glycosylation. Hum Mol Genet. 2010;19:3623–3633. doi: 10.1093/hmg/ddq278. [DOI] [PubMed] [Google Scholar]
- Mention K, Michaud D, Dobbbelaere D, et al. Neonatal severe intractable diarrhoea as the presenting manifestation of an unclassified congenital disorder of glycosylation (CDG-x) Arch Dis Child Fetal Neonatal Ed. 2001;85:F217–F219. doi: 10.1136/fn.85.3.F217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morava E, Zeevaert R, Korsch E, et al. A common mutation in the COG7 gene with a consistent phenotype including microcephaly, adducted thumbs, growth retardation, VSD and episodes of hyperthermia. Eur J Hum Genet. 2007;15:638–645. doi: 10.1038/sj.ejhg.5201813. [DOI] [PubMed] [Google Scholar]
- Ng B, Kranz C, Hagebeuk E, et al. Molecular and clinical characterization of a Moroccan Cog7 deficient patient. Mol Genet Metab. 2007;91:201–204. doi: 10.1016/j.ymgme.2007.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paesold-Burda P, Maag C, Troxler H, et al. Deficiency in COG5 causes a moderate form of congenital disorders of glycosylation. Hum Mol Genet. 2009;18:4350–4356. doi: 10.1093/hmg/ddp389. [DOI] [PubMed] [Google Scholar]
- Parkos CA, Allen RA, Cochrane CG, et al. Purified cytochrome b from human granulocyte plasma membrane is comprised of two polypeptides with relevant molecular weights of 91,000 an 22,000. J Clin Invest. 1987;80:732–742. doi: 10.1172/JCI113128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynders E, Foulquier F, Leão Teles E, et al. Golgi function and dysfunction in the first COG4-deficient CDG type II patient. Hum Mol Genet. 2009;18:3244–3256. doi: 10.1093/hmg/ddp262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R, Lupashin V. Role of the conserved oligomeric Golgi (COG) complex in protein glycosylation. Carbohydr Res. 2008;343:2024–2031. doi: 10.1016/j.carres.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaapen L, Bakker J, van der Meer S, et al. Clinical and biochemical presentation of siblings with COG-7 deficiency, a lethal multiple O- and N-glycosylation disorder. J Inherit Metab Dis. 2005;28:707–714. doi: 10.1007/s10545-005-0015-z. [DOI] [PubMed] [Google Scholar]
- Wu X, Rush J, Karaoglu D, et al. Mutation of the COG complex subunit gene COG7 causes a lethal congenital disorder. Nat Med. 2004;10:518–523. doi: 10.1038/nm1041. [DOI] [PubMed] [Google Scholar]
- Zdebska E, Bader-Meunier B, Schischmanoff PO, et al. Abnormal glycosylation of red cell membrane band 3 in the congenital disorder of glycosylation Ig. Pediatr Res. 2003;54:224–229. doi: 10.1203/01.PDR.0000072327.55955.F7. [DOI] [PubMed] [Google Scholar]
- Zeevaert R, Foulquier F, Jaeken J, Matthijs G. Deficiencies in subunits of the conserved oligomeric golgi (COG) complex define a novel group of congenital disorders of glycosylation. Mol Genet Metab. 2008;93:15–21. doi: 10.1016/j.ymgme.2007.08.118. [DOI] [PubMed] [Google Scholar]