

Abstract
Most patients with Wilson’s disease (WD) are compound heterozygote, which complicates establishing genotype–phenotype correlations. We identified five patients who presented with early and/or severe hepatic disease who are homozygous for W939C missense mutation on exon 12 of ATP7B. We therefore conducted a meta-analysis to determine the phenotype of patients homozygous for missense or nonsense mutations in all ATP7B exons.
The meta-analysis showed that 69% and 31% of patients are homozygous for H1069Q and non-H1069Q mutations, respectively. Compared to patients with H1069Q, those with non-H1069Q mutations were significantly more likely to have a hepatic phenotype, severe liver disease, a mixed phenotype, and less likely to have a neurologic phenotype. Compared to patients with nonsense mutations, those with non-H1069Q ones were equally likely to present with a hepatic phenotype and to have severe liver disease. Mean age at symptom onset in the non-H1069Q versus the H1069Q group was 15.5 versus 20.5years (p<0.001).
Our data suggest that mutation W939C and other non-H1069Q missense mutations are associated with early disease onset, a hepatic phenotype, and a high risk of hepatic failure in homozygous patients. Early identification of such patients by genetic screening is important for timely initiation of treatment and prevention of complications.
Introduction
Wilson’s disease (WD) (MIM #277900) is a disorder of copper metabolism caused by mutations in the ATP7B gene, resulting in impaired function of the copper transporter Cu-ATPase and deposition of copper in various organs, particularly the liver and brain (Ala et al. 2007; Ferenci 2004). Clinical presentation of WD is variable. Patients present usually with hepatic, neurologic, or mixed hepatic and neurologic manifestations. Some are asymptomatic at diagnosis (Taly et al. 2007). The onset age of WD can range widely (2–71 years), but liver disease is more often the initial presentation in children and young adults compared to older patients (Ferenci et al. 2007; Manolaki et al. 2009).
WD is characterized by mutational diversity with more than 400 mutations identified thus far, and many of which are population specific. Establishing genotype–phenotype correlations is important for proper patient management, for initiation of early treatment in asymptomatic individuals to prevent certain complications, and for predicting the efficacy of treatment. Furthermore, it enhances understanding the molecular pathogenesis of the disease.
Reported associations between a given mutation and a specific WD phenotype remain preliminary and inconclusive (Liu et al. 2004; Margarit et al. 2005; Panagiotakaki et al. 2004). Attempts to establish genotype–phenotype correlations in WD have been hampered by several factors including: the small number of patients, the large number of mutations, and the fact that most patients are compound heterozygote making it difficult to ascribe a clinical phenotype to one mutated allele versus the other (Loudianos et al. 1999; Riordan and Williams 2001). Phenotypic diversity occurs within the same family, among the same genotype siblings (Riordan and Williams 2001; Takeshita et al. 2002), and even among monozygotic twins (Fraga et al. 2005; Machin 1996; Kegley et al. 2010). One meta-analysis (Stapelbroek et al. 2004) associated H1069Q with late-onset disease and neurological symptoms in contrast to other studies (Duc et al. 1998; Ivanova-Smolenskaya et al. 1999; Shah et al. 1997) that found no correlation between this mutation and clinical presentation. In addition, findings that nonsense and frameshift mutations are associated with early and severe liver disease (Gromadzka et al. 2005; Merle et al. 2010) were not replicated by others (Angius et al. 1998; Deguti et al. 2004; Okada et al. 2000; Palsson et al. 2001). Missense mutations were presumed to result in milder phenotypes compared to frameshift and nonsense mutations, but this too was not substantiated (Deguti et al. 2004; Firneisz et al. 2002; Loudianos et al. 1999; Shah et al., 1997; Wu et al. 2001). Finally, late-onset neurologic WD can occur without any evidence of liver involvement (Ferenci et al. 2007).
Establishing genotype–phenotype correlations is facilitated by studying patients who are homozygous for specific mutations (Barada et al. 2007, 2010; Thomas et al. 1995).We report here the genotypic and phenotypic profile of five patients from two unrelated Lebanese families who are homozygous for W939C, a missense mutation in exon-12 of ATP7B, and who had early and/or severe liver disease. To elucidate this finding, we conducted a comprehensive meta-analysis that included 448 and 29 patients homozygous for missense and nonsense mutations in the ATP7B exons, respectively. We propose the association of homozygous missense non-H1069Q mutations with early and severe hepatic involvement.
Materials and Methods
Twelve subjects from two Lebanese families were studied (Fig. 1): Family-T (n = 5, T1–T5) and Family-B (n = 7, B1–B6, B8). Three members of Family-T [2 in USA, 1 at American University of Beirut Medical Center (AUBMC)] and two members of Family-B [1 at AUBMC, 1 elsewhere] were diagnosed with WD. Evaluation of patients included history, physical exam, slit-lamp examination, abdominal ultrasound, liver function tests, serum copper, ceruloplasmin and 24 h urinary-Cu levels. Subsequently, DNA screening for mutations or single-nucleotide polymorphisms (SNPs) was performed. DNA extraction, amplification of the various exons by PCR, and sequencing methods were carried on as described before (Barada et al. 2010) using blood samples from members of Family-B and -T. Subjects or their guardian signed a consent form (protocol # BioCh.JU.01) approved by the Institutional Review Board (IRB) at AUB-MC.
Fig. 1.
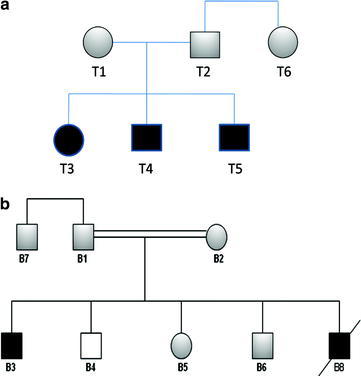
(a) Pedigree of family-T, which consists of six members with T3, T4, and T5 diagnosed with WD. (b) Pedigree of family-B, which consists of seven members with B3 and B8 diagnosed with WD. Patients were homozygous for W939C, whereas parents were heterozygous. B8 passed away of fulminant hepatic failure. B4 had a normal sequence
Meta-analysis: Phenotypes of Patients Homozygous for Missense and Nonsense Mutations in ATP7B Gene
We did a comprehensive literature review of all the articles mentioned in the University of Alberta database and retrieved all articles published in PubMed and Medline between 1993 and 2010 using the following index terms: Wilson disease, mutation, phenotype, and genotype. We included all articles in which the phenotypes of patients homozygous for missense or nonsense mutations were clearly stated. We considered patients to have severe liver disease if they had cirrhosis or fulminant hepatic failure.
Inclusion and Exclusion Criteria
To establish associations between genotype and phenotype, the analysis included only patients in whom the phenotype was clearly stated in the published articles and who are homozygous for missense or nonsense mutations—including the five patients presented in this study. Patients whose phenotype was not clearly stated as hepatic, neurologic, mixed or asymptomatic were excluded. Patients were considered to have severe liver disease if it was clearly indicated that they have cirrhosis or fulminant hepatic failure. All other patients (asymptomatic, neurologic, mixed, and other hepatic presentations) were considered as nonsevere. For comparing age at presentation among the various groups, we used the individual patient age when provided or repeated the mean or median age provided by the authors as appropriate. Patients were excluded from the age analysis if neither individual nor averaged age data were available.
Statistical Analysis
Data on our patients were combined with data abstracted from the literature. The Chi square test or Fisher Exact Test was used to compare the prevalence rates among the cohorts of (a) hepatic versus nonhepatic phenotype, and (b) severe versus nonsevere hepatic phenotype as defined above. Patient cohort comparisons included: (1) homozygous for non-H1069Q missense mutations anywhere along ATP7B versus homozygous for H1069Q, (2) homozygous missense (Any) versus homozygous nonsense mutations, and (3) homozygous for non-H1069Q missense versus homozygous nonsense mutations. Age comparisons among the same patient subcohorts were done using the two-tailed unpaired t-test or Mann–Whitney rank sum test on median age as appropriate. Determination of significance was set at the 5% level. All analyses were done using SigmaPlot version 11 (2008; Systat Software Inc., San Jose, CA).
Results
The clinical profile of Family-T and Family-B patients is shown in Table 1. Family-T consists of five-members (Fig. 1a). The parents are not consanguineous. Three members (T3, T4 and T5) have WD, all were asymptomatic at diagnosis. Family-B consists of seven-members (Fig. 1b). The parents are first-degree relatives. B3 has WD and B8 died at the age of 5 years of fulminant hepatic failure due to WD. Both B3 and B8 had severe liver involvement based on clinical, laboratory, and radiological criteria at the ages of 5 and 8 years, respectively. A presumptive diagnosis of WD was made for B8 at the age of 5 years based on a very low serum ceruloplasmin level, a borderline low serum Cu, and a positive family history. He developed fulminant liver failure and died of severe gastrointestinal bleeding. Unaffected members of both families had a full evaluation and were asymptomatic and their physical exam, serum ceruloplasmin, serum copper, and 24 h urine Cu were all normal.
Table 1.
Clinical and biochemical profile of five newly diagnosed Lebanese WD Patients, homozygous for W939C mutation
| T3 | T4 | T5 | B3 | B8 (deceased) | |
|---|---|---|---|---|---|
| Year of birth | 1998 | 2001 | 2006 | 1992 | 2002 |
| Age of symptoms onset | Asymptomatic | Asymptomatic | Asymptomatic | 8 | 5 |
| Age at diagnosis | 7 (incidental) | 8 (by screening) | 3 (by screening) | 13 | 5 |
| GI manifestations | Asymptomatic transaminitis | Asymptomatic transaminitis | Asymptomatic transaminitis | At age 8: hepatosplenomegaly cirrhosis, ascites |
Jaundice, abdominal distention hematemesis, shifting dullness, hepatomegaly |
| Neurological manifestations | None | None | None | Drooping of the jaw hypersalivation, slurred speech narrow-based gait, intention tremors |
Hepatic encephalopathy |
| Kayser Fleischer ring | Absent | Absent | Absent | Present | Absent |
| Serum copper | 5 | 6 | 9 | 2 | 72 |
| Serum ceruloplasmin | <0.02 | <0.02 | <0.02 | <0.0211 | 0.04 |
| 24-h-urine copper | 77.6 | 20 | 41.5 | 744 | Not done |
| Alkaline phosphatase | 288 | 286 | 257 | 191 | 131 |
| GGT | 89 | 23 | 26 | 230 | 21 |
| SGPT | 248 | 152 | 59 | 25 | 74.1 |
| SGOT | 112 | 93 | 54 | 33 | 150 |
| Bilirubin T/D | 0.6/0.2 | 0.8/0.2 | 0.7/<0.1 | 0.8/0.3 | 38.06/21.44 |
| PT/INR | 12.9/1.10 | 13.2/1.10 | 11.4/1.00 | 14.3/1.2 | >8.31/>120 |
| Albumin | 47 | 48 | 44 | 40 | 34.08 |
| Globulin | 29 | 31 | 21 | 26 | 20 |
| Ultrasound of abdomen | Hepatomegaly echogenic liver |
Increase in the liver echotexture, irregular contour, cirrhosis | Increase in the liver echotexture, irregular contour, cirrhosis | Hepatosplenomegaly (chronic liver disease or cirrhosis) Small amount of ascites |
Mild-to-moderate ascites |
| MRI of brain | Normal | Normal | Not done | Bilateral symmetrical areas of abnormal signals in the putamen, thalami, caudate, midbrain, superior cerebellar hemispheres and dentate nuclei | Not done |
| Liver histopathology | Chronic liver disease, bridging fibrosis, macro-vesicular fatty changes |
Fatty change portal fibrosis |
Not done | Not done | Not done |
Normal ranges of: Serum ceruloplasmin: 0.20–0.60 g/L; Serum Cu is 70–150 μg/dL; Urine Cu (μg/24 h): 15–50 μg/24 h
Mutation Analysis
Patients, T3, T4, T5, and B3 were homozygous for the disease-causing mutation in exon-12 (W939C) Unaffected members T1, T2, B1, B2, B5, and B6 were heterozygous for this mutation. Patient B8 passed away before any genotypic analysis was done. Furthermore, all patients were homozygous for the following SNPs: K834R (E10), R952K (E12), A1003A (E13), and V1140A (E16). Members of Family-T, but not Family-B, were also homozygous for S1166S.
Meta-analysis Findings
The meta-analysis included a total of 448 patients with clearly stated phenotype who were homozygous for missense mutations along ATP7B, 311 with H1069Q and 137 with non-H1069Q mutations (Table 2). It also included 29 patients who are homozygous for nonsense mutations anywhere along ATP7B. Hepatic phenotype was reported in 85 patients (62%) of the non-H1069Q group compared to 115 (37%) in the H1069Q group (p < 0.001). Furthermore, severe liver disease occurred in 24.8% of the non-H1069Q versus 8.4% in the H1069Q group (p < 0.001). The mixed phenotype was also more common in the non-H1069Q group (12.4 versus 6.1%, p = 0.038). Conversely, neurologic phenotype was much more likely to occur in the H1069Q group (54.0 versus 14.6%, p < 0.001). There were 29 patients homozygous for nonsense mutations in whom the phenotype could be clearly identified. Of these, 13 (44.8%) had a hepatic phenotype and 3 (10.3%) had severe liver disease (Table 3). There was no significant difference in the prevalence of hepatic phenotype, severe liver disease, or neurologic phenotype between patients with missense and those with nonsense mutations. Age at onset of symptoms was available for 356 of the 448 (79.5%) patients that are homozygous for missense mutations, and it was significantly greater for H1069Q (n = 293; 20.5 years) versus non-H1069Q (n = 63; 15.1 years) mutations (p < 0.001). Mean onset age for non-H1069Q mutations was also greater than those with nonsense mutations (n = 8; 10.8 years), yet this difference only approached significance (p = 0.068) possibly because of the limited onset age data among nonsense mutation patients.
Table 2.
Phenotypes of patients homozygous for missense mutations in the WD gene
| Exon | Mutation | # Hepatic (%) | # Neurologic (%) | # Mixed (%) | # Asymptomatic (%) | References |
|---|---|---|---|---|---|---|
| E5 | R616Q | 1 (100%) | – | – | – | Santhosh et al. (2006) |
| E7 | P690L | 1 (100%) | – | – | – | Margarit et al. (2005) |
| G691R | 2 (40%) | – | – | 3 (60%) | Barada et al. (2007) | |
| E8 | R778L | 20 (71.4%) | 5 (17.9%) | – | 3 (10.7%) | Butler et al. (2001), Okada et al. (2000), Thomas et al. (1995), Wu et al. (2001), Yoo (2002) |
| L708P | 3 (25%) | 6 (50%) | – | 3 (25%) | Garcia-Villarreal et al. (2000) | |
| G710A | 1 (100%) | – | – | – | Duc et al. (1998) | |
| G710S | 1 (50%) | 1 (50%) | – | – | Waldenstrom et al. (1996) | |
| L708P | 2 (50%) | – | 2 (50%) | – | Deguti et al. (2004) | |
| D765N | – | – | 1 (100%) | – | Deguti et al. (2004) | |
| P768L | 1 (100%) | – | – | – | Santhosh et al. (2006) | |
| E10 | I857T | – | – | 1 (100%) | – | Folhoffer et al. (2007) |
| E11 | G875R | 3 (100%) | – | – | – | Santhosh et al. (2008) |
| A874V | – | – | 1 (100%) | – | Kusuda et al. (2000) | |
| E12 | G943S | – | 1 (100%) | – | – | Thomas et al. (1995) |
| W939C | 4 (80%) | – | 1 (20%) | – | Our study | |
| E13 | R969Q | 1 (100%) | – | – | – | Santhosh et al. (2006) |
| R969W | 4 (57%) | – | – | 3 (43%) | Panagiotakaki et al. (2004) | |
| P992L | 1 (33%) | 2 (67%) | – | – | Gu et al. (2003), Wu et al. (2006) | |
| A1003V | 1 (100%) | – | – | – | Santhosh et al. (2006) | |
| K1010R | 3 (75%) | – | – | 1 (25%) | Santhosh et al. (2006), Santhosh et al. (2008) | |
| E14 | G1061E | 2 (100%) | – | – | – | Margarit et al. (2005), Santhosh, et al. (2006) |
| A1065P | 1 (100%) | – | – | – | Brage et al. (2007) | |
| H1069Q | 115 (37%) | 168 (54%) | 19 (6%) | 9 (3%) | Brage et al. (2007), Butler et al. (2001), Caca et al. (2001), Caprai et al. (2006), Curtis et al. (1999), Duc et al. (1998), Ferenci et al. (2007), Firneisz et al. (2002), Houwen et al. (1995), Kucinskas et al. (2008), Loudianos et al. (2003), Maier-Dobersberger et al. (1997), Panagiotakaki et al. (2004), Shah et al. (1997), Stapelbroek et al. (2004), Tarnacka et al. (2000), Thomas et al. (1995), Vrabelova et al. (2005) | |
| E15 | G1101R | 1 (50%) | – | – | 1 (50%) | Thomas et al. (1995) |
| I1102T | 2 (100%) | – | – | – | Butler et al. (2001), Thomas et al. (1995) | |
| C1104F | 1 (100%) | – | – | – | Loudianos et al. (1999) | |
| F1094L | – | – | 1 (100%) | – | Deguti et al. (2004) | |
| E16 | I1148T | 2 (100%) | – | – | – | Panagiotakaki et al. (2004) |
| E17 | T1232P | 1 (100%) | – | – | – | Margarit et al. (2005) |
| E18 | V1262F | 1 (100%) | – | – | – | Loudianos et al. (1999) |
| G1266K | 2 (40%) | 2 (40%) | – | 1 (20%) | Tarnacka et al. (2000), Thomas et al. (1995) | |
| N1270T | 1 (50%) | – | 1 (50%) | – | Santhosh et al. (2006), Yoo (2002) | |
| N1270S | 2 (50%) | – | 2 (50%) | – | Barada et al. (2010), Santhosh et al. (2006) | |
| P1273L | 2 (100%) | – | – | – | Barada et al. (2010) | |
| E19 | G1341S | – | 2 (66%) | 1 (34%) | – | Santhosh et al. (2006), Santhosh et al. (2008) |
| E20 | S1363F | – | 1 (100%) | – | – | Loudianos et al. (1999) |
| –a | – | 18 (75%) | – | 6 (25%) | – | Abdelghaffar et al. (2008) |
Note: No patients with homozygous missense mutations in Exons: 1–4, 6, and 9 with a clearly defined phenotype were reported
aNineteen different missense mutations were reported in 24 patients (Abdelghaffar et al. 2008)
Table 3.
Prevalence of various phenotypes in patients homozygous for missense or nonsense mutations in the ATP7B gene
| Disease | Homozygous missense mutations (ATP7B) | Nonsense | |||||
|---|---|---|---|---|---|---|---|
| H1069Q | Non-H1069Q | Overall | Mutationsa | ||||
| Presentation | N (%) | N (%) | P value | N (%) | N (%) | P value | |
| No. of patients | 311 | 137 | 448 | 29 | |||
| Hepatic onlyb | Yes | 115 (37.0%) | 85 (62.0%) | <0.001 | 200 (44.6%) | 13 (44.8%) | 0.86 |
| No | 196 (63.0%) | 52 (38.0%) | 248 (55.4%) | 16 (55.2%) | |||
| Severe hepaticb,c | Yes | 24 (8.4%) | 34 (24.8%) | <0.001 | 58 (13.7%) | 3 (10.3%) | 0.849 |
| No | 261 (91.6%) | 103 (75.2%) | 364 (86.3%) | 26 (89.7%) | |||
| Neurologic onlyb | Yes | 168 (54.0%) | 20 (14.6%) | <0.001 | 188 (42.0%) | 7 (24.1%) | 0.09 |
| No | 143 (46%) | 117 (85.4%) | 260 (58.0%) | 22 (75.9%) | |||
| Mixed | Yes | 19 (6.1%) | 17 (12.4%) | 0.038 | 36 (8.0%) | 9 (31.0%) | <0.001 |
| No | 292 (93.9%) | 120 (87.6%) | 412 (92.0%) | 20 (69.0%) | |||
| Asymptomatic | Yes | 9 (2.9%) | 13 (9.5%) | 0.006 | 22 (4.9%) | N/A | |
| No | 302 (97.1%) | (90.5%) | 426 (95.1%) | N/A | |||
aReferences: Abdelghaffar et al. (2008), Folhoffer et al. (2007), Gupta et al. (2005), Prella et al. (2001), Santhosh et al. (2006), Waldenstrom et al. (1996), Deguti et al. (2004), Thomas et al. (1995), Panagiotakaki et al. (2004)
bHepatic presentation (p = 0.132) and severity (p = 0.145) did not differ significantly for non-H1069Q versus Non-sense mutations, but this may be due to small number of patients
cSeverity data was unavailable in 26 patients and were excluded from analysis
Discussion
We report in this paper a missense mutation (W939C) that, in the homozygous state, was associated with early and/or severe hepatic disease in five WD patients. Although the W939C mutation was reported before in a single patient in the heterozygous state (Folhoffer et al. 2007), this is the first study to describe the clinical phenotype of a group of patients with this mutation in the homozygous state. The primary findings of the accompanying meta-analysis of all reported homozygous mutations in ATP7B are: (1) patients who are homozygous for non-H1069Q missense mutations are more likely to develop symptoms at a younger age, to have a hepatic phenotype, and to have severe liver disease on presentation than their H1069Q counterparts, (2) the phenotype–genotype analysis does not lend support to the notion that patients with nonsense mutations have more severe disease compared to missense mutation patients, and (3) patients who are homozygous for H1069Q mutations are more likely to present with later and neurologic manifestations as previously reported (Stapelbroek et al. 2004; Gromadzka et al. 2006; Ferenci et al. 2007).
More than 400 mutations have been described in WD, and missense ones are the most common. Multiple difficulties in establishing clear and convincing genotype–phenotype correlations in WD have been described (Curtis et al. 1999; Kalinsky et al. 1998; Kegley et al. 2010; Margarit et al. 2005; Shah et al. 1997). To overcome them, others (Thomas et al. 1995) and we have proposed (Barada et al. 2010): (1) conducting family studies since their members are more likely to share the same genetic and environmental factors, (2) studying homogenous populations with high rates of consanguinity, which would increase the chance of identifying homozygous patients. Such approach facilitates establishing associations between a phenotype and a given mutation.
The parents in the two-described families are of the same ethnicity, and in one family they were consanguineous. All five WD patients had liver disease with evidence of hepatocellular injury, fibrosis or cirrhosis as demonstrated by laboratory tests, imaging or liver biopsy at a very young age. Three patients were asymptomatic on presentation, but had biochemical evidence of hepatitis and sonographic evidence of cirrhosis. However, on liver biopsy, patients T3 and T4 had fibrosis and fatty change but no frank cirrhosis. This discrepancy is explained by the fact that the liver biopsies were done 3 years before the abdominal ultrasounds revealing cirrhosis. In the case of T3, T4, T5, and B3, the diagnosis was unequivocal and based on clinical, biochemical, and radiologic findings, in addition to the genetic testing revealing that all four patients were homozygous for the missense mutation in exon-12. B8, however, had a presumptive diagnosis of WD presenting as fulminant hepatitis based on a very low serum ceruloplasmin level, a borderline low serum copper level, and a family history of WD.
Patients with WD may present with neurological manifestations without ever experiencing liver disease, as demonstrated by normal liver tests, imaging and histopathology (Ferenci et al. 2007; Horslen and Hahn 2010). Thus, the course of WD may either start with hepatic or with isolated neurological manifestations. The disease may be mild, severe, or life threatening depending on the ATP7B mutation and other genetic and environmental factors. Our meta-analysis suggests that about 62% of patients homozygous for non-H1069Q mutations present with a hepatic phenotype, and about 24.8% have severe disease, i.e., cirrhosis or fulminant hepatic failure. This diverges with other reports (Duc et al. 1998; Ivanova-Smolenskaya et al. 1999; Shah et al. 1997; Takeshita et al. 2002) because only patients homozygous for missense mutations were considered. However, some non-H1069Q mutations may be associated with neurologic manifestations in the homozygous state, and this may not be statistically apparent due to the small number of patients.
Assigning a specific phenotype to a specific mutation helps predict the pattern of disease progression, the approximate age of symptom onset, and facilitates monitoring the response to therapy. The missense mutation that is reported in this article is associated with early and/or severe liver disease that could lead to early liver failure and death if left untreated. If treated, however, those manifestations may be prevented like in Family-T patients. Hence, it is important to initiate treatment directly at genetic diagnosis in patients homozygous for this mutation and to offer genetic screening to all their family members.
Our findings, in part, confirm and expand those of Stapelbroek et al. (2004). However, we differ from them in several ways: (1) our analysis was restricted to patients homozygous for specific mutations; (2) we compared patients homozygous for H1069Q (common in Europe but rare in the Arab World) to patients homozygous for non-H1069Q mutations reported in the Arab World and elsewhere; (3) we addressed acute and chronic hepatic failure on presentation which is of obvious clinical importance; and (4) we compared patients with missense and nonsense mutations.
There are limitations to our study. The first is the small number of patients with the identified W939C mutation in the homozygous state. The second is that our analysis of the literature was restricted to patients whose phenotype was clearly identified. The third is that the number of patients who are homozygous for missense and nonsense mutations at specific exons is relatively small, rendering exon or mutation specific statistical analysis inapplicable. The relatively small number of WD patients who are homozygous for nonsense mutations limits the statistical power of comparisons with missense counterparts, and hence negative findings or lack of significance must be interpreted with caution. Finally, the non-H1069Q group in our study is a heterogeneous group with missense mutations in various exons of ATP7B. Studies involving larger numbers of patients who are homozygous for missense mutations at specific exons may clarify further genotype/phenotype correlations.
Conclusions
The mutation W939C and other non-H1069Q missense mutations in homozygous patients are associated with early disease onset, a hepatic phenotype, and a high risk of hepatic failure on presentation. Early identification of such affected individuals via genetic screening is important for timely initiation of treatment and prevention of sequelae.
Acknowledgements
This research was funded by grants from the University Research Board and the Medical Practice Plan of the American University of Beirut.
Competing Interest
The authors declare no competing interest
Take-Home Message (Synopsis)
Mutation W939C and other non-H1069Q missense mutations are associated with early disease onset, a hepatic phenotype, and a high risk of hepatic failure in homozygous patients.
Footnotes
Competing interests: None declared.
References
- Abdelghaffar TY, Elsayed SM, Elsobky E, Bochow B, Buttner J, Schmidt H. Mutational analysis of ATP7B gene in Egyptian children with Wilson disease: 12 novel mutations. J Hum Genet. 2008;53(8):681–687. doi: 10.1007/s10038-008-0298-7. [DOI] [PubMed] [Google Scholar]
- Ala A, Walker AP, Ashkan K, Dooley JS, Schilsky ML. Wilson’s disease. Lancet. 2007;369(9559):397–408. doi: 10.1016/S0140-6736(07)60196-2. [DOI] [PubMed] [Google Scholar]
- Angius A, Dessi V, Lovicu M, De Virgiliis S, Pirastu M, Cao A. Early and severe neurological features in a Wilson disease patient compound heterozygous for two frameshift mutations. Eur J Pediatr. 1998;157(2):128–129. doi: 10.1007/s004310050783. [DOI] [PubMed] [Google Scholar]
- Barada K, Nemer G, ElHajj II, et al. Early and severe liver disease associated with homozygosity for an exon 7 mutation, G691R, in Wilson’s disease. Clin Genet. 2007;72(3):264–267. doi: 10.1111/j.1399-0004.2007.00853.x. [DOI] [PubMed] [Google Scholar]
- Barada K, El-Atrache M, El H, et al. Homozygous mutations in the conserved ATP hinge region of the Wilson disease gene: association with liver disease. J Clin Gastroenterol. 2010;44(6):432–439. doi: 10.1097/MCG.0b013e3181ce5138. [DOI] [PubMed] [Google Scholar]
- Brage A, Tome S, Garcia A, Carracedo A, Salas A. Clinical and molecular characterization of Wilson disease in Spanish patients. Hepatol Res. 2007;37(1):18–26. doi: 10.1111/j.1872-034X.2007.00010.x. [DOI] [PubMed] [Google Scholar]
- Butler P, McIntyre N, Mistry PK. Molecular diagnosis of Wilson disease. Mol Genet Metab. 2001;72(3):223–230. doi: 10.1006/mgme.2000.3143. [DOI] [PubMed] [Google Scholar]
- Caca K, Ferenci P, Kuhn HJ, et al. High prevalence of the H1069Q mutation in East German patients with Wilson disease: rapid detection of mutations by limited sequencing and phenotype-genotype analysis. J Hepatol. 2001;35(5):575–581. doi: 10.1016/S0168-8278(01)00219-7. [DOI] [PubMed] [Google Scholar]
- Caprai S, Loudianos G, Massei F, Gori L, Lovicu M, Maggiore G. Direct diagnosis of Wilson disease by molecular genetics. J Pediatr. 2006;148(1):138–140. doi: 10.1016/j.jpeds.2005.07.036. [DOI] [PubMed] [Google Scholar]
- Curtis D, Durkie M, Balac P, et al. A study of Wilson disease mutations in Britain. Hum Mutat. 1999;14(4):304–311. doi: 10.1002/(SICI)1098-1004(199910)14:4<304::AID-HUMU5>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Deguti MM, Genschel J, Cancado EL, et al. Wilson disease: novel mutations in the ATP7B gene and clinical correlation in Brazilian patients. Hum Mutat. 2004;23(4):398. doi: 10.1002/humu.9227. [DOI] [PubMed] [Google Scholar]
- Duc HH, Hefter H, Stremmel W, et al. His1069Gln and six novel Wilson disease mutations: analysis of relevance for early diagnosis and phenotype. Eur J Hum Genet. 1998;6(6):616–623. doi: 10.1038/sj.ejhg.5200237. [DOI] [PubMed] [Google Scholar]
- Ferenci P. Pathophysiology and clinical features of Wilson disease. Metab Brain Dis. 2004;19(3–4):229–239. doi: 10.1023/B:MEBR.0000043973.10494.85. [DOI] [PubMed] [Google Scholar]
- Ferenci P, Czlonkowska A, Merle U, et al. Late-onset Wilson’s disease. Gastroenterology. 2007;132(4):1294–1298. doi: 10.1053/j.gastro.2007.02.057. [DOI] [PubMed] [Google Scholar]
- Firneisz G, Lakatos PL, Szalay F, Polli C, Glant TT, Ferenci P. Common mutations of ATP7B in Wilson disease patients from Hungary. Am J Med Genet. 2002;108(1):23–28. doi: 10.1002/ajmg.10220. [DOI] [PubMed] [Google Scholar]
- Folhoffer A, Ferenci P, Csak T, et al. Novel mutations of the ATP7B gene among 109 Hungarian patients with Wilson’s disease. Eur J Gastroenterol Hepatol. 2007;19(2):105–111. doi: 10.1097/01.meg.0000223904.70492.0b. [DOI] [PubMed] [Google Scholar]
- Fraga MF, Ballestar E, Paz MF, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 2005;102(30):10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Villarreal L, Daniels S, Shaw SH, et al. High prevalence of the very rare Wilson disease gene mutation Leu708Pro in the Island of Gran Canaria (Canary Islands, Spain): a genetic and clinical study. Hepatology. 2000;32(6):1329–1336. doi: 10.1053/jhep.2000.20152. [DOI] [PubMed] [Google Scholar]
- Gromadzka G, Schmidt HH, Genschel J, et al. Frameshift and nonsense mutations in the gene for ATPase7B are associated with severe impairment of copper metabolism and with an early clinical manifestation of Wilson’s disease. Clin Genet. 2005;68(6):524–532. doi: 10.1111/j.1399-0004.2005.00528.x. [DOI] [PubMed] [Google Scholar]
- Gromadzka G, Schmidt HH, Genschel J, et al. p.H1069Q Mutation in ATP7B and biochemical parameters of copper metabolism and clinical Manifestation of Wilson’s disease. Movement Disord. 2006;21(2):245–248. doi: 10.1002/mds.20671. [DOI] [PubMed] [Google Scholar]
- Gu YH, Kodama H, Du SL, Gu QJ, Sun HJ, Ushijima H. Mutation spectrum and polymorphisms in ATP7B identified on direct sequencing of all exons in Chinese Han and Hui ethnic patients with Wilson’s disease. Clin Genet. 2003;64(6):479–484. doi: 10.1046/j.1399-0004.2003.00179.x. [DOI] [PubMed] [Google Scholar]
- Gupta A, Aikath D, Neogi R, et al. Molecular pathogenesis of Wilson disease: haplotype analysis, detection of prevalent mutations and genotype-phenotype correlation in Indian patients. Hum Genet. 2005;118(1):49–57. doi: 10.1007/s00439-005-0007-y. [DOI] [PubMed] [Google Scholar]
- Horslen S, Hahn SH. Genotype-phenotype correlation in Wilson disease. J Clin Gastroenterol. 2010;44(6):387–388. doi: 10.1097/MCG.0b013e3181d96ac4. [DOI] [PubMed] [Google Scholar]
- Houwen RH, Juyn J, Hoogenraad TU, Ploos van Amstel JK, Berger R. H714Q mutation in Wilson disease is associated with late, neurological presentation. J Med Genet. 1995;32(6):480–482. doi: 10.1136/jmg.32.6.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova-Smolenskaya IA, Ovchinnikov IV, Karabanov AV, et al. The His1069Gln mutation in the ATP7B gene in Russian patients with Wilson disease. J Med Genet. 1999;36(2):174. [PMC free article] [PubMed] [Google Scholar]
- Kalinsky H, Funes A, Zeldin A, et al. Novel ATP7B mutations causing Wilson disease in several Israeli ethnic groups. Hum Mutat. 1998;11(2):145–151. doi: 10.1002/(SICI)1098-1004(1998)11:2<145::AID-HUMU7>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Kegley KM, Sellers MA, Ferber MJ, Johnson MW, Joelson DW, Shrestha R. Fulminant Wilson’s disease requiring liver transplantation in one monozygotic twin despite identical genetic mutation. Am J Transplant. 2010;10(5):1325–1329. doi: 10.1111/j.1600-6143.2010.03071.x. [DOI] [PubMed] [Google Scholar]
- Kucinskas L, Jeroch J, Vitkauskiene A, et al. High frequency of the c.3207C>A (p.H1069Q) mutation in ATP7B gene of Lithuanian patients with hepatic presentation of Wilson’s disease. World J Gastroenterol. 2008;14(38):5876–5879. doi: 10.3748/wjg.14.5876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusuda Y, Hamaguchi K, Mori T, Shin R, Seike M, Sakata T. Novel mutations of the ATP7B gene in Japanese patients with Wilson disease. J Hum Genet. 2000;45(2):86–91. doi: 10.1007/s100380050017. [DOI] [PubMed] [Google Scholar]
- Liu XQ, Zhang YF, Liu TT, et al. Correlation of ATP7B genotype with phenotype in Chinese patients with Wilson disease. World J Gastroenterol. 2004;10(4):590–593. doi: 10.3748/wjg.v10.i4.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loudianos G, Dessi V, Lovicu M, et al. Mutation analysis in patients of Mediterranean descent with Wilson disease: identification of 19 novel mutations. J Med Genet. 1999;36(11):833–836. [PMC free article] [PubMed] [Google Scholar]
- Loudianos G, Kostic V, Solinas P, et al. Characterization of the molecular defect in the ATP7B gene in Wilson disease patients from Yugoslavia. Genet Test. 2003;7(2):107–112. doi: 10.1089/109065703322146786. [DOI] [PubMed] [Google Scholar]
- Machin GA. Some causes of genotypic and phenotypic discordance in monozygotic twin pairs. Am J Med Genet. 1996;61(3):216–228. doi: 10.1002/(SICI)1096-8628(19960122)61:3<216::AID-AJMG5>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Maier-Dobersberger T, Ferenci P, Polli C, et al. Detection of the His1069Gln mutation in Wilson disease by rapid polymerase chain reaction. Ann Intern Med. 1997;127(1):21–26. doi: 10.7326/0003-4819-127-1-199707010-00004. [DOI] [PubMed] [Google Scholar]
- Manolaki N, Nikolopoulou G, Daikos GL, et al. Wilson disease in children: analysis of 57 cases. J Pediatr Gastroenterol Nutr. 2009;48(1):72–77. doi: 10.1097/MPG.0b013e31817d80b8. [DOI] [PubMed] [Google Scholar]
- Margarit E, Bach V, Gomez D, et al. Mutation analysis of Wilson disease in the Spanish population – identification of a prevalent substitution and eight novel mutations in the ATP7B gene. Clin Genet. 2005;68(1):61–68. doi: 10.1111/j.1399-0004.2005.00439.x. [DOI] [PubMed] [Google Scholar]
- Merle U, Weiss KH, Eisenbach C, Tuma S, Ferenci P, Stremmel W. Truncating mutations in the Wilson disease gene ATP7B are associated with very low serum ceruloplasmin oxidase activity and an early onset of Wilson disease. BMC Gastroenterol. 2010;10:8. doi: 10.1186/1471-230X-10-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada T, Shiono Y, Hayashi H, et al. Mutational analysis of ATP7B and genotype-phenotype correlation in Japanese with Wilson’s disease. Hum Mutat. 2000;15(5):454–462. doi: 10.1002/(SICI)1098-1004(200005)15:5<454::AID-HUMU7>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Palsson R, Jonasson JG, Kristjansson M, et al. Genotype-phenotype interactions in Wilson’s disease: insight from an Icelandic mutation. Eur J Gastroenterol Hepatol. 2001;13(4):433–436. doi: 10.1097/00042737-200104000-00023. [DOI] [PubMed] [Google Scholar]
- Panagiotakaki E, Tzetis M, Manolaki N, et al. Genotype-phenotype correlations for a wide spectrum of mutations in the Wilson disease gene (ATP7B) Am J Med Genet A. 2004;131(2):168–173. doi: 10.1002/ajmg.a.30345. [DOI] [PubMed] [Google Scholar]
- Prella M, Baccala R, Horisberger JD, et al. Haemolytic onset of Wilson disease in a patient with homozygous truncation of ATP7B at Arg1319. Br J Haematol. 2001;114(1):230–232. doi: 10.1046/j.1365-2141.2001.02899.x. [DOI] [PubMed] [Google Scholar]
- Riordan SM, Williams R. The Wilson’s disease gene and phenotypic diversity. J Hepatol. 2001;34(1):165–171. doi: 10.1016/S0168-8278(00)00028-3. [DOI] [PubMed] [Google Scholar]
- Santhosh S, Shaji RV, Eapen CE, et al. ATP7B mutations in families in a predominantly Southern Indian cohort of Wilson’s disease patients. Indian J Gastroenterol. 2006;25(6):277–282. [PubMed] [Google Scholar]
- Santhosh S, Shaji RV, Eapen CE, et al. Genotype phenotype correlation in Wilson’s disease within families–a report on four south Indian families. World J Gastroenterol. 2008;14(29):4672–4676. doi: 10.3748/wjg.14.4672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah AB, Chernov I, Zhang HT, et al. Identification and analysis of mutations in the Wilson disease gene (ATP7B): population frequencies, genotype-phenotype correlation, and functional analyses. Am J Hum Genet. 1997;61(2):317–328. doi: 10.1086/514864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapelbroek JM, Bollen CW, van Amstel JK, et al. The H1069Q mutation in ATP7B is associated with late and neurologic presentation in Wilson disease: results of a meta-analysis. J Hepatol. 2004;41(5):758–763. doi: 10.1016/j.jhep.2004.07.017. [DOI] [PubMed] [Google Scholar]
- Takeshita Y, Shimizu N, Yamaguchi Y, et al. Two families with Wilson disease in which siblings showed different phenotypes. J Hum Genet. 2002;47(10):543–547. doi: 10.1007/s100380200082. [DOI] [PubMed] [Google Scholar]
- Taly AB, Meenakshi-Sundaram S, Sinha S, Swamy HS, Arunodaya GR. Wilson disease: description of 282 patients evaluated over 3 decades. Medicine (Baltimore) 2007;86(2):112–121. doi: 10.1097/MD.0b013e318045a00e. [DOI] [PubMed] [Google Scholar]
- Tarnacka B, Gromadzka G, Rodo M, Mierzejewski P, Czloonkowska A. Frequency of His1069Gln and Gly1267Lys mutations in Polish Wilson’s disease population. Eur J Neurol. 2000;7(5):495–498. doi: 10.1046/j.1468-1331.2000.t01-1-00112.x. [DOI] [PubMed] [Google Scholar]
- Thomas GR, Forbes JR, Roberts EA, Walshe JM, Cox DW. The Wilson disease gene: spectrum of mutations and their consequences. Nat Genet. 1995;9(2):210–217. doi: 10.1038/ng0295-210. [DOI] [PubMed] [Google Scholar]
- Vrabelova S, Letocha O, Borsky M, Kozak L. Mutation analysis of the ATP7B gene and genotype/phenotype correlation in 227 patients with Wilson disease. Mol Genet Metab. 2005;86(1–2):277–285. doi: 10.1016/j.ymgme.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Waldenstrom E, Lagerkvist A, Dahlman T, Westermark K, Landegren U. Efficient detection of mutations in Wilson disease by manifold sequencing. Genomics. 1996;37(3):303–309. doi: 10.1006/geno.1996.0564. [DOI] [PubMed] [Google Scholar]
- Wu ZY, Wang N, Lin MT, Fang L, Murong SX, Yu L. Mutation analysis and the correlation between genotype and phenotype of Arg778Leu mutation in Chinese patients with Wilson disease. Arch Neurol. 2001;58(6):971–976. doi: 10.1001/archneur.58.6.971. [DOI] [PubMed] [Google Scholar]
- Wu ZY, Zhao GX, Chen WJ, et al. Mutation analysis of 218 Chinese patients with Wilson disease revealed no correlation between the canine copper toxicosis gene MURR1 and Wilson disease. J Mol Med. 2006;84(5):438–442. doi: 10.1007/s00109-005-0036-y. [DOI] [PubMed] [Google Scholar]
- Yoo HW. Identification of novel mutations and the three most common mutations in the human ATP7B gene of Korean patients with Wilson disease. Genet Med. 2002;4(6 Suppl):43S–48S. doi: 10.1097/00125817-200211001-00009. [DOI] [PubMed] [Google Scholar]