

Abstract
The predominant tumor cell of Kaposi’s Sarcoma (KS) is the spindle cell, a cell of endothelial origin that expresses markers of lymphatic endothelium. In culture, Kaposi’s Sarcoma-associated herpesvirus (KSHV) infection of blood endothelial cells drives expression of lymphatic endothelial cell specific markers, in a process that requires activation of the gp130 receptor and the JAK2/STAT3 and PI3K/AKT signaling pathways. While expression of each of the KSHV major latent genes in endothelial cells failed to increase expression of lymphatic markers, the viral homolog of human IL-6 (vIL-6) was sufficient for induction and requires the JAK2/STAT3 and PI3K/AKT pathways. Therefore, activation of gp130 and downstream signaling by vIL-6 is sufficient to drive blood to lymphatic endothelial cell differentiation. While sufficient, vIL-6 is not necessary for lymphatic reprogramming in the context of viral infection. This indicates that multiple viral genes are involved and suggests a central importance of this pathway to KSHV pathogenesis.
Keywords: KSHV, HHV-8, Kaposi’s Sarcoma, vIL-6, Lymphatic endothelial cells, Lymphatic endothelial cell differentiation
Introduction
Kaposi’s Sarcoma (KS) is the leading cancer in parts of sub-Saharan Africa and in AIDS patients world-wide (Antman and Chang, 2000). KS is histopathologically characterized by having a high degree of angiogenesis, infiltrating immune and red blood cells, and is predominantly comprised of spindle cells, which are of endothelial origins and specifically express markers of lymphatic endothelium (Boshoff et al., 1995; Jussila et al., 1998; Skobe et al., 1999). In addition, KS tumors have elevated levels of many cytokines and growth factors, including interleukin-1 (IL-1), IL-6, basic fibroblast growth factor (bFGF), platelet derived growth factor (PDGF), tumor necrosis factor (TNF), interferon gamma (IFN-γ), and vascular endothelial growth factor (VEGF) (Ensoli et al., 2001, 2000; Wang et al., 2004).
Kaposi’s Sarcoma-associated herpesvirus (KSHV or human herpesvirus-8) is the etiologic agent of KS, and is classified as a gamma-2-herpesvirus (Aluigi et al., 1996; Chang et al., 1994). The KS spindle cell is infected by KSHV, with greater than 95% of cells latently infected and 1–5% of cells undergoing lytic replication (Staskus et al., 1997). KSHV encodes over 90 genes, but only a few genes are expressed during latency in KS tumors and KSHV-infected endothelial cells in culture: latency associated nuclear antigen (LANA), viral Cyclin, viral FLICE inhibitory protein (vFLIP) (Dittmer et al., 1998; Staskus et al., 1997), the kaposin family (Sadler et al., 1999), and 17 or more microRNAs (Cai et al., 2005; Grundhoff et al., 2006; Pfeffer et al., 2005; Samols et al., 2005). Additionally, several KSHV lytic genes have been proposed to contribute to KS pathogenesis in an autocrine or paracrine fashion from the small percentage of lytically infected cells found in KS tumors. Some of these genes include: a constitutively active viral G-protein coupled receptor (vGPCR); K1, which signals through a constitutive ITAM (immunoreceptor tyrosine-based activation motif), K15, a membrane protein with SH2 and SH3 protein binding domains and vIL-6, a viral homolog of human IL-6 (Brinkmann et al., 2003; Couty and Gershengorn, 2004; Lagunoff et al., 1999; Molden et al., 1997). Interestingly, transcripts for K1 and vIL-6 were recently shown to be detectable in dilutions of primary effusion lymphoma cells where other lytic genes could no longer be detected, indicating there may be some previously unappreciated low-level of expression of these genes during latency (Chandriani and Ganem, 2010). Additionally, there are specific situations where vIL-6 expression can be induced during latency. For example, it was shown that interferon could activate expression of vIL-6 in primary effusion lymphoma (PEL) cells (Chatterjee et al., 2002).
KSHV infection of blood endothelial cells (BECs) induces their differentiation to lymphatic endothelial cells (LECs) (Carroll et al., 2004; Hong et al., 2004; Wang et al., 2004). BECs and LECs line the blood and lymphatic vasculatures respectively, and differentially express around 300 genes (Hirakawa et al., 2003). During embryonic development, the blood vessel system develops first, then BECs differentiate into LECs in the cardinal vein when an unknown signal induces the expression of the homeobox transcription factor Prox1 (Hong and Detmar, 2003; Hong et al., 2002; Petrova et al., 2002). Prox1 is a transcriptional repressor and activator, and is the master regulator of lymphatic differentiation (Hong and Detmar, 2003; Hong et al., 2002; Petrova et al., 2002). Following Prox1 induction, vascular endothelial growth factor receptor 3 (VEGFR-3) is expressed on LECs and is responsible for budding and formation of early lymph vessels through binding its ligand VEGF-C (Veikkola et al., 2001). Podoplanin, a cell surface glycoprotein induced by Prox1 expression, is involved in later stages of lymphatic vasculature development (Schacht et al., 2003). KSHV infection of primary BECs or telomerase-immortalized microvascular endothelial (TIME) cells, which resemble BECs in gene expression profile, induces the expression of Prox1, VEGFR-3 and podoplanin (Carroll et al., 2004; Hong et al., 2004; Wang et al., 2004).
The viral mechanism of KSHV-induced endothelial cell differentiation is poorly understood. Previously, we found that KSHV induction of lymphatic differentiation is dependent on the activation of the cytokine receptor gp130, and the resulting initiation of JAK2/STAT3 and PI3K/AKT intracellular signaling pathways (Morris et al., 2008; Punjabi et al., 2007). In addition, the latently expressed kaposin B was recently shown to stabilize Prox1 mRNA, leading to an increase in expression of Prox1 and its downstream-activated genes, such as VEGFR-3 (Yoo et al., 2010). However, kaposin B expression was unable to induce the expression of Prox1 in cells that do not already express Prox1, namely blood endothelial cells. This suggests that there must be a viral gene that initially induces the expression of Prox1 and lymphatic endothelial cell differentiation, and kaposin B subsequently increases or maintains expression of genes involved in the differentiation phenotype.
KSHV-encoded vIL-6 shares ~25% homology with human IL-6, but can bind the gp130 receptor directly and signal independent of the cognate IL-6 receptor (gp80) to initiate similar intracellular signaling pathways (Li et al., 2001; Molden et al., 1997; Moore et al., 1996; Nicholas et al., 1997; Wan et al., 1999). In PEL cells, which are KSHV-infected, vIL-6 promotes proliferation of cells, protects cells from apoptosis, and also promotes immune evasion of interferon activity (Chatterjee et al., 2002; Foussat et al., 1999; Jones et al., 1999). vIL-6 can also promote angiogenesis through induction of VEGF A (Aoki et al., 1999). In this report, we show that the genes from the major latency locus do not have the capacity to induce lymphatic endothelial cell markers when expressed individually. Interestingly, expression of KSHV vIL-6 is sufficient to induce the LEC-specific markers Prox1, VEGFR-3, and podoplanin. We also extend previous findings that vIL-6 is not necessary in the context of KSHV infection, and demonstrate that vIL-6 is not the soluble factor released by KSHV-infected cells that activates gp130 receptor signaling (Morris et al., 2008; Punjabi et al., 2007). Thus, while vIL-6 is sufficient for endothelial cell differentiation, another unidentified viral protein directly activates or induces a novel cytokine that activates the same pathway during latency. The presence of multiple viral factors that induce the same pathway suggests that this cytokine-signaling pathway plays a central role in KSHV pathogenesis and KS tumor formation.
Results
KSHV latent genes do not activate gp130 receptor signaling or lymphatic differentiation of endothelial cells
In previous reports, we determined that viral gene expression is necessary to maintain KSHV-induced STAT3 activation and lymphatic reprogramming (Morris et al., 2008; Punjabi et al., 2007). These results suggest that viral genes are required for the constant activation of the gp130 receptor signaling pathway. Since latently infected TIME cells express LEC-specific markers and display persistent STAT3 activation (Carroll et al., 2004; Punjabi et al., 2007), we hypothesized that one or more of the latent genes are responsible for KSHV-induced activation of gp130 receptor signaling and thus lymphatic differentiation of endothelial cells. The latent genes viral Cyclin (vCyc), kaposin A (KapA), kaposin B (KapB), and kaposin C (KapC) were cloned into a mammalian expression vector with an N-terminal 3XFLAG epitope-tag and expressed in TIME cells. Additionally, we obtained a mammalian expression vector encoding a highly expressed codon-optimized FLAG-tagged vFLIP, which was also expressed in TIME cells (P. Bellare and D. Ganem, unpublished data). Previous studies have verified the functionality of N-terminally tagged vCyc, vFLIP, and KapA (Montaner et al., 2003). We verified the expression of all constructs by transient transfection into TIME cells followed by immunoblot analysis with an anti-FLAG antibody (Fig. 1A, bottom panel). Anti-vCyc and anti-kaposin antibodies were used to verify the expression of the FLAG-tagged vCyc, KapB, and KapC constructs (data not shown). The transfection efficiency was 50–70%, as determined by transfection of TIME cells with a plasmid encoding green fluorescent protein (GFP) and monitoring expression by fluorescence microscopy (data not shown). At 48 h post infection, cells were harvested for Western blot analysis. None of the latent viral proteins tested activated STAT3, as determined by STAT3 phosphorylation at Tyr705 (pSTAT3), VEGFR-3 or podoplanin expression (Fig. 1A and not shown). We also created a stable pooled TIME cell line expressing LANA or an empty vector control cell line (pZIP) using retroviral transduction, followed by puromycin selection. Although the expression of LANA in these cells was confirmed by immunoblot analysis with an anti-LANA antibody (Fig. 1A, lower right panel), LANA expression was not sufficient to induce persistent STAT3 phosphorylation or VEGFR-3 expression in TIME cells when compared to the vector control cell line. These results indicate that transient expression of the individual latent genes, LANA, vCyc, KapA, KapB, KapC, or vFLIP, is not sufficient to induce gp130 receptor activation or lymphatic differentiation. Furthermore, we did not see STAT3 phosphorylation when all of the latent gene constructs were co-transfected into TIME cells, although expression levels were lower than when they were transfected individually (data not shown).
Fig. 1.

Transient expression of latent genes do not induce lymphatic differentiation. (A) Constructs expressing individual latent viral genes were transiently transfected into TIME cells. 48 h post transfection, cell lysates were harvested and subjected to immunoblot analysis with the indicated antibodies. In addition, stable pooled TIME cells expressing LANA or a retroviral vector control were similarly analyzed. While expression of individual latent viral genes was confirmed by FLAG or LANA immunoblot, no viral gene activated STAT3 or induced VEGFR-3 expression. (B) TIME cells (2 × 105) were transduced with lentiviral preps of the indicated KSHV latent genes or pCGSW, a control lentivirus that expresses GFP. 72 hpi, cell lysates were harvested and subjected to immunoblot analysis with the indicated antibodies. TIME cells were mock or KSHV-infected and harvested 48 hpi for positive and negative controls respectively.
To examine longer-term expression of the latent genes in a higher percentage of cells, we obtained lentiviral vectors of the latent genes, including K12 (KapA), ORF71 (vFLIP), ORF72 (vCyc), and ORF73 (LANA). Infectious lentivirus containing each major latent gene was used to infect TIME cells. TIME cells were transduced with the KSHV lentiviral library and expression of the viral genes was confirmed for each by qRT-PCR with latent gene specific primers (see Supplemental Fig. 1). TIME cells were infected with the different lentiviral preparations in parallel, and protein lysates were harvested at 72 hpi. Mock- and KSHV-infected TIME cell lysates harvested 48 hpi were used as negative and positive controls respectively. As with the transient transfections, none of the genes from the latent locus were able to induce STAT3 activation, AKT activation as determined by phoshphorylation at Ser473 or lymphatic endothelial cell markers (Fig. 1B). KapB was previously reported to stabilize the mRNA of Prox-1 and subsequently increase VEGFR3 expression in LECs, though it did not induce the expression of Prox-1 in BECs (Yoo et al., 2010). To confirm this in TIME cells, we used a lentivirus vector to express a FLAG-tagged KapB protein. As expected, KapB expression did not induce any Prox-1 transcription in TIME cells as determined by quantitative RT-PCR (data not shown).
KSHV vIL-6 induces LEC-specific markers in TIME cells
Since expression of KSHV latent genes did not activate STAT3 or induce LEC-specific markers, we postulated that the small percentage of lytically infected TIME cells could be producing a paracrine factor to activate the gp130 receptor. Additionally, a recent publication found that vIL-6 mRNA could be detected by PCR in dilutions of PEL cells where many other lytic mRNAs could not be detected (Chandriani and Ganem, 2010). Therefore, we tested vIL-6 and a select number of KSHV lytic genes for activation of the gp130 receptor by transient transfection. These include K14 (vOX2), hemagglutinin (HA)-tagged K3 (MIR1), FLAG-tagged K5 (MIR2), or the vector controls pBMN-ZIN and pLNCX2. Of the genes tested, only vIL-6 was able to activate STAT3 and turn on expression of VEGFR3 (Fig. 2A). To determine if KSHV ORF50 (Rta), the lytic switch gene, activates STAT3 and lymphatic differentiation, we infected TIME cells with an adenovirus vector expressing Rta. While the levels of Rta used were sufficient to induce lytic reactivation in latently infected cells, in uninfected cells Rta expression did not lead to activation of STAT3 and did not increase Prox-1 expression (data not shown).
Fig. 2.

Expression of vIL-6 but not other lytic genes that activate AKT induce lymphatic differentiation. (A) TIME cells expressing the lytic viral proteins vIL-6, vOX2, HA-K3, or FLAG-K5 were created by retroviral transduction and antibiotic selection. Cell lysates were assayed by immunoblot analysis with the indicated antibodies. Stable vIL-6 expression induced VEGFR-3 expression and activated STAT3. (B) TIME cells (2 × 105) were transiently transduced with lentiviral preps of the indicated KSHV lytic genes or a control lentivirus (pCGSW). 72 hpi, cell lysates were harvested and subjected to immunoblot analysis with the indicated antibodies. TIME cells were mock or KSHV-infected and harvested 48 hpi for positive and negative controls respectively.
We next used a lentiviral expression library of select KSHV genes: K1, K2 (vIL-6), K3 (MIR1), ORF28, K6 (vMIP), K8 (K-bZIP), K9 (vIRF1), K11.1 (vIRF2), ORF74 (vGPCR), and K15-P (LAMP) to test which KSHV lytic genes could induce lymphatic endothelial cell differentiation. This lentiviral library was previously used to identify the viral genes that induce Ang2 expression in endothelial cells (Vart et al., 2007). Again, K2 (vIL-6) was the only viral gene tested that induced persistent STAT3 phosphorylation (Fig. 2B and data not shown). Conditioned media from cells transduced with each KSHV gene examined was also tested for activation of STAT3 and again, only conditioned media from vIL-6 expressing cells led to phosphorylation of STAT3 (Supplemental Fig. 2). Interestingly, vGPCR and K1 induced low levels of AKT activation in this assay while in cells with long-term vIL-6 expression the levels of phosphorylated AKT were at our limits of detection. However, as described below and seen in Fig. 5B, treatment of cells with conditioned media containing vIL-6 induces stronger activation of AKT indicating that vIL-6 can transiently induce AKT. Overall, this data indicates that activation of AKT is important for blood to lymphatic endothelial cell differentiation, but activation of AKT alone is not sufficient to induce blood to lymphatic endothelial cell differentiation as both K1 and vGPCR induce the activation of AKT but do not activate STAT3 or significantly increase the expression of VEGFR3.
Fig. 5.

vIL-6 induction of lymphatic differentiation requires JAK2/STAT3 and PI3K/AKT. (A) The PI3K inhibitor LY294002 (LY) and the JAK2 inhibitor AG490 (AG) block VEGFR-3 and podoplanin expression in stable vIL-6-expressing TIME cells. The numbers below the lanes indicated the relative abundance of VEGFR-3 and podoplanin expression normalized to β-actin expression in each lane compared to expression in the untreated vIL-6 expression cells. (B) Conditioned media harvested from stable vIL-6-expressing TIME cells transiently induces AKT and STAT3 phosphorylation in serum starved TIME cells treated for 30 min.
We then selected for cells that had been transduced with a retrovirus expressing vIL-6 or a vector control to make a stable vIL-6 cell line and a passage matched control vector cell line (pBMN-ZIN). The stable vIL-6 TIME cell line induced long-term activation of STAT3 and long-term VEGFR-3 and podoplanin protein expression similar to levels seen in KSHV infected cells as determined by Western blot analysis (Fig. 3A). There were also highly induced levels of Prox1 mRNA in vIL-6 stable cells but not in the vector control cells (Fig. 3B). Prox1 is the transcription factor necessary for the induction of lymphatic differentiation in vivo and in vitro. Previously, we determined that gp130 receptor expression is induced by KSHV infection of TIME cells, and this induction was controlled by STAT3 activation (Morris et al., 2008; Punjabi et al., 2007). Accordingly, we also observed an increase in gp130 mRNA induction in the vIL-6-expressing TIME cells (Fig. 3B). These results demonstrate that vIL-6 expression is sufficient to activate gp130 receptor signaling and induce lymphatic differentiation of TIME cells.
Fig. 3.

Stable vIL-6 expression in TIME cells induces LEC-specific markers. (A) Stable vIL-6-expressing TIME cells induce VEGFR-3 and podoplanin expression compared to stable TIME cells expressing retroviral vector alone (pBMN-ZIN) as determined by immunoblot analysis. Mock and KSHV-infected TIME cells harvested 48 hpi were used as negative and positive controls. Stable vIL-6-expressing TIME cells also display persistent activation of STAT3 (B) Stable vIL-6-expressing TIME cells induce Prox1 and gp130 mRNA expression as determined by quantitative real-time RT-PCR. Error bars represent standard error of the mean (n=3).
KSHV vIL-6 induces LEC-specific markers in primary blood endothelial cells
The previous analysis was performed in an immortalized blood endothelial cell line, TIME cells. We also confirmed that vIL-6 was sufficient to reprogram primary human dermal blood microvascular endothelial cells (BECs) to express lymphatic endothelial cell markers. Primary BECs (Lonza) were transduced with a lentivirus expressing vIL-6 or the control vector lentivirus pCGSW and lysates were harvested 72 h after transduction. Immunoblot analysis shows that vIL-6 induces the activation of STAT3 and the expression of VEGFR3 in primary BECs (Fig. 4).
Fig. 4.
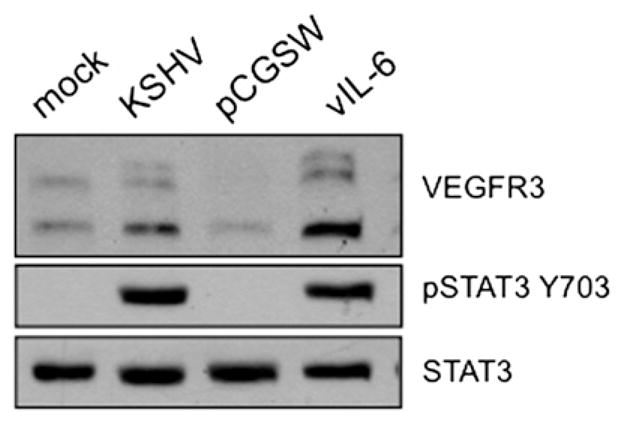
vIL-6 reprograms primary blood endothelial cells to express lymphatic markers. Primary human blood microvascular endothelial cells (BECs) (2 × 105) were transiently transduced with lentiviral preps of vIL-6 lytic or control lentivirus (pCGSW). 72 hpi, cell lysates were harvested and subjected to immunoblot analysis with the indicated antibodies. Primary BECs were mock or KSHV-infected and harvested 48 hpi for positive and negative controls respectively.
vIL-6 induction of lymphatic differentiation requires JAK2/STAT3 and PI3K/AKT signaling
We observed persistent phosphorylation of STAT3 in stable vIL-6 expressing TIME cells, but very weak phosphorylation of AKT (Fig. 2B). We therefore wanted to determine if JAK2/STAT3 and PI3K/AKT signaling were necessary for vIL-6 activation of lymphatic endothelial cell markers. TIME cells expressing vIL-6 were treated with pharmacological inhibitors of PI3K (LY294002), and of JAK2 (AG490). Both inhibitors blocked vIL-6 induced expression of VEGFR3 and podoplanin (Fig. 5A). Therefore, as with KSHV latent infection, both JAK2/STAT3 and PI3K/AKT signaling is necessary for induction of lymphatic endothelial cell reprogramming by vIL-6.
Since the levels of AKT activation in the vIL-6 cell line was at the limit of detection, we tested whether vIL-6 could transiently activate AKT in TIME cells. Conditioned media from vIL-6-expressing or control pBMN-ZIN TIME cells were harvested, and used to treat serum-starved TIME cells for 30 min. Both AKT and STAT3 were transiently phosphorylated in cells treated with conditioned media from vIL-6-expressing TIME cells (Fig. 5B). This result indicates that while vIL-6 expression persistently activates STAT3 in TIME cells, AKT activation is mostly transient. However, there appears to be some low-level activation of AKT in long term vIL-6 expressing endothelial cells as the PI3 kinase inhibitor, LY294002 inhibited vIL-6 activation of VEGFR3. However, because long-term activation is at our limit of detection, it was not seen consistently in every experiment. Nonetheless, this indicates that vIL-6 can, at least transiently, activate AKT signaling and this activation is necessary for the induction of lymphatic markers by vIL-6.
vIL-6 is not essential for activation of gp130 receptor signaling in KSHV-infected endothelial cells
While vIL-6 expression alone is sufficient to induce STAT3 activation in TIME cells, we previously showed that vIL-6 is not necessary for activation of STAT3 nor the induction of LEC-specific markers in the context of KSHV infection (Punjabi et al., 2007). For those experiments, we used a recombinant virus where more than half of the ORFK2 gene encoding vIL-6 is deleted, and infected TIME cells with either vIL-6-positive or negative-recombinant KSHV isolates. In both cases, we observed phosphorylation of STAT3 and induction of VEFGR-3 and podoplanin (Chen and Lagunoff, 2007; Morris et al., 2008; Punjabi et al., 2007). However, it is still possible that vIL-6 plays a significant role in the activation of STAT3 and VEGFR3 that is masked by comparing the infections of the recombinant virus versus the control virus. Additionally, the deletion of vIL-6 leaves a small portion of the gene intact, though we could not detect expression of the vIL-6 transcript in this recombinant (Chen and Lagunoff, 2007). To determine if vIL-6 is the dominant gp130-activating factor and that the infection with the recombinant is not equivalent to infection with the control, we used lentivirus-mediated delivery of shRNA specific to vIL-6. TIME cells were infected with KSHV, and subsequently infected with lentivirus expressing vIL-6 shRNA or a negative control nonspecific shRNA (NS) (Chen et al., 2009). By quantitative real-time RT-PCR, we observed a 60% decrease in vIL-6 mRNA in the vIL-6 versus the NS shRNA transduced KSHV-infected cells, normalizing to cellular GAPDH mRNA levels (data not shown and (Chen et al., 2009)). Importantly, there was no decrease in mRNA levels of the latent gene LANA, indicating the establishment of KSHV infection was not impaired (data not shown). However, vIL-6 shRNA did not significantly decrease KSHV induction of VEGFR-3 or STAT3 activation (Fig. 6A)
Fig. 6.

Knockdown of vIL-6 inhibits conditioned media from vIL-6 expression cells from inducing STAT3 but not from KSHV infected cells. (A) TIME cells were infected with KSHV, 24 hpi cells were transduced with lentiviruses encoding nonspecific shRNA (NS) or a vIL-6-specific shRNA. Cell lysates were harvest 48 h after transduction and subjected to immunoblot analysis with the indicated antibodies. Knockdown of vIL-6 had little or no effect on expression of lymphatic genes or STAT3 activation in infected cells. The numbers below the lanes indicated the relative abundance of phospho-STAT3 normalized to total STAT3 expression in each lane compared to expression in the NS control cells. (B) TIME cells were infected and transduced with vIL-6 specific or nonspecific shRNAs as in A. At 40 h post lentiviral transduction, the media was changed, conditioned media was harvested 8 h later and used to treat serum-starved TIME cells for 30 mins. As a control for vIL-6 knockdown, vIL-6 expressing TIME cells were concurrently infected with the shRNA lentiviruses and either pCGSW or vIL-6 expressing lentivirus. The vIL-6 shRNA decreased the phosphorylation of STAT3 by conditioned media harvested from control vIL-6 expressing cells (lanes 7 and 8), but not from KSHV-infected cell conditioned media (lanes 3 and 4). The numbers below the lanes indicated the relative abundance of phospho-STAT3 normalized to total STAT3 expression in each lane compared to expression in the NS control cells.
In the next set of experiments we examined the ability of the shRNA to block activation of STAT3 by conditioned media from KSHV infected TIME cells and from vIL-6 expressing TIME cells to determine if vIL-6 is the dominant factor in transient activation of STAT3 in KSHV infected cells. TIME cells were co-transduced with a K2 (vIL-6) expressing lentivirus and either vIL-6 specific or NS shRNA lenti-viruses and the conditioned media was harvested 48 h later. The vIL-6 specific shRNA strongly inhibited the activation of STAT3 by conditioned media from vIL-6 expressing cells (Fig. 6). However, there was little or no decrease in the activation of STAT3 by conditioned media from KSHV-infected cells in the presence of vIL-6 specific shRNA, indicating that vIL-6 is not necessary for a significant portion of the activation of gp130 by conditioned media during de novo KSHV infection. This set of experiments shows that the vIL-6-independent activation of STAT3 and lymphatic differentiation is likely the predominant pathway in latent KSHV-infected cells.
Discussion
KSHV infection of endothelial cells, the main cell type of KS tumors, induces the aberrant activation of the cytokine receptor gp130, resulting in the persistent activation of the JAK2/STAT3 and PI3K/AKT signaling pathways. These pathways result in KSHV-mediated transcriptional reprogramming of endothelial cells to a LEC differentiation state (Morris et al., 2008). In this study we sought to determine whether specific viral proteins could activate gp130 receptor signaling or LEC-specific genes. Using transient transfection or lentiviral transduction of individual viral genes, we could not identify a latent protein that activated STAT3 or induced LEC-specific markers. We went on to test a number of lytic genes that have been previously shown to play a role in KSHV pathogenesis during latency through cytokine signaling or other paracrine effects. Interestingly, although the KSHV lytic proteins vGPCR and K1 can activate PI3K/AKT signaling in other systems (Sodhi et al., 2004; Wang et al., 2006), individual expression of these viral genes did not induce lymphatic reprogramming in TIME cells indicating that activation of PI3K/AKT signaling alone is not sufficient for BEC to LEC differentiation. A number of other lytic genes that can act as cytokines like the vMIPs were also unable to activate STAT3 signaling or induce LEC-specific markers. However, we found that KSHV-encoded vIL-6, which can bind and activate the gp130 receptor, induces LEC-specific markers through activation of both JAK2/STAT3 and PI3K/AKT signaling pathways. Interestingly, both STAT3 and AKT are phosphorylated early after KSHV infection of endothelial cells, dephosphorylated at 4–8 hpi and then constitutively activated after 12 h (Punjabi et al., 2007; Sadagopan et al., 2007). The early activation of STAT3 is due to viral binding and entry while the late activation of STAT3 is due to viral gene expression as UV-irradiated virus only activates the early wave of STAT phosphorylation (Punjabi et al., 2007). We previously published that in latently infected cells gp130 signaling was necessary for KSHV induced differentiation of blood endothelial cells to lymphatic. Here we found that activation of these pathways by vIL-6 expression alone was sufficient to induce lymphatic reprogramming of both TIME and primary microvascular blood endothelial cells. Additionally, while it was known that vIL-6 could activate STAT3 through the gp130 receptor, we found here that at least transiently, vIL-6 could activate AKT. While vIL-6 has been shown to predominantly signal through intracellular pathways, we found that conditioned media from cells expressing vIL-6 could also activate STAT3 and AKT indicating it can act both cell autonomously and as a paracrine factor.
Our previous studies found that blood to lymphatic differentiation only occurs in latently infected cells as determined by co-immunofluorescence with LANA and LEC-specific markers in KSHV infected TIME cells (Morris et al., 2008). Additionally, another study showed similar results with immunofluorescence to Prox1 (Yoo et al., 2010). Therefore, it is likely that there is a factor expressed only in latently infected cells that is critical for KSHV induced BEC to LEC differentiation. A recent study found that kaposin B, a KSHV gene that is expressed during latency, was able to stabilize Prox-1 mRNA through sequences in its 3′UTR (Yoo et al., 2010) However, kaposin B alone was not sufficient to induce expression of Prox1 in BECs. Therefore, two events must occur: (1) KSHV induces the expression of Prox-1 and (2) once expressed, kaposin B stabilizes and effectively increases expression of Prox-1 to drive differentiation. Therefore, a lytic gene could theoretically induce the expression of Prox-1 to low levels in a paracrine fashion while kaposin B stabilizes and effectively increases the expression of Prox-1 to levels sufficient to fully drive BEC to LEC differentiation in the latently infected cells but not the surrounding uninfected cells. While we demonstrate here that vIL-6 is certainly sufficient to turn on the expression of Prox-1 and could act as this paracrine factor, it is not essential for KSHV induced BEC to LEC differentiation. This indicates that KSHV expresses another gene that is capable of activating STAT3, AKT and lymphatic reprogramming. We found that vIL-6 is unlikely to play the predominant role in activation of gp130 signaling during latent infection as knockdown of vIL-6 did not decrease the activation of gp130 signaling in KSHV infected cells or from KSHV conditioned media but was able to significantly decrease para-crine signaling from vIL-6 expressing cells. This suggests that another viral gene(s) is expressed during latency that is responsible for gp130 receptor signaling, or there is another untested lytic gene capable of activating gp130 in a paracrine fashion. It is also possible that the 17 or more KSHV miRNAs play a significant role in the regulation of this pathway. Further work is underway to determine if expression of the KSHV miRNAs alter endothelial cell differentiation. Nonetheless, our findings imply that there are redundant methods used by KSHV to activate the gp130 receptor and lymphatic reprogramming, which would predict an important role for this signaling pathway in KSHV pathogenesis.
Materials and methods
Reagents and antibodies
Phospho-AKT (Ser473), pan AKT, phospho-STAT3 (Y705) and pan STAT3 antibodies (Cell Signaling Technologies) were used as specified by the manufacturer. Antibodies to β-actin (Sigma), ORF59 (Advanced Biotechnologies, Inc.), LANA (Advanced Biotechnologies, Inc. and kind gift of D. Ganem), gp130, VEGFR-3 (Santa Cruz Biotechnologies), and podoplanin (Abcam) were used in immunoblot or immunofluorescence analysis as outlined below. Kinase inhibitors AG490 and LY294002 (Calbiochem) were reconstituted in DMSO and used at the concentrations specified.
Cells
TIME cells and primary dermal blood microvascular endothelial cells (Lonza) were maintained as monolayer cultures in EGM-2 media [Lonza (Lagunoff et al., 2002; Venetsanakos et al., 2002)]. BCBL-1 cells (Renne et al., 1996) were maintained in RPMI 1640 media (Cellgro, Mediatech, Inc.) supplemented with 10% fetal bovine serum, penicillin, streptomycin, L-glutamine and β-mercaptoethanol. HEK293T cells and Phoenix-ampho packaging cell lines were obtained from American Type Culture Collection (ATCC) and were maintained in DMEM (Cellgro, Mediatech, Inc.) supplemented with 10% fetal bovine serum, penicillin, streptomycin, and L-glutamine.
KSHV viruses and infections
KSHV inoculum, from BCBL-1 cells, was used to infect TIME cells as previously described (Lagunoff et al., 2002; Punjabi et al., 2007). Briefly, KSHV infections of TIME cells were performed in serum free EBM-2 media for 3 h, after which the media was replaced with complete EGM-2 containing serum and supplements. Infection rates were assessed by immunofluorescence using antibodies against the latent protein LANA and the lytic protein ORF59 and only experiments with greater than 90% LANA positive cells and less than 1% ORF 59 positive cells were used.
Plasmids
The KSHV latent genes vCyclin and kaposin A were cloned by PCR into the 3XFLAG-CMV-10 expression vector (Sigma) using KpnI and XbaI restriction sites, and kaposin B and C were subcloned from pCR3.1 into the HindIII and EcoRI restriction sites (pCR3.1-kaposin B and pCR3.1-kaposin C plasmids obtained from Craig McCormick, Dalhousie University). FLAG-vFLIP with codons optimized for expression was a kind gift from Don Ganem (University of California San Francisco, P. Bellare and D. Ganem, unpublished data). A retroviral plasmid encoding LANA, pBABE-LANA, was a kind gift from Adam Grundhoff (Heinrich-Pette-Institute for Experimental Virology and Immunology, University of Hamburg). The following retroviral expression vectors were used: pLNCX2 (Clontech); hemaglutinin-tagged K3, pLNCX-HA-K3; FLAG-tagged K5, pBMN-FLAG-K5; pLNCX-K14 [kind gifts from Laurant Coscoy, University of California Berkley (Coscoy and Ganem, 2000)]; pBMN-ZIN; and pBMN-vIL-6 (vIL-6 cloned by PCR into the BamH1 and EcoR1 sites of pBMN-ZIN). Lentiviral packaging plasmids psPAX2 and pMD2.G were obtained from Addgene (supplied by Dr. Didier Trono). Lentiviral expression plasmids pSIN-MCS containing KSHV genes (Vart et al., 2007) or pCGSW (Godfrey et al., 2005) encoding GFP were described elsewhere. Lentiviral shRNA vectors to vIL-6 and nonspecific, nonsilencing (NS) control are described elsewhere [gifts from John Nicholas, Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University School of Medicine (Chen et al., 2009)].
Transient transfection of plasmids by Amaxa nucleofection
TIME cells (1 × 106 cells) were transfected with 4 μg of the plasmids encoding the latent genes using the Amaxa Nucleofector (Koeln, Germany) according to the manufacturer’s protocol. 72 h post transfection, cells were harvested and analyzed by immunoblot analysis.
Immunoblot analysis
Cells were harvested and resuspended in RIPA lysis buffer (50 mM Tris–HCl, pH 7.6, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.5% deoxycholate, 0.1% sodium dodecyl sulfate, 1 mM sodium orthovanadate, 1 mM sodium fluoride, 40 mM β-glycerophosphate, and Complete Mini Protease Inhibitor Tablet [Roche]), cell debris was removed after a 20 min incubation by centrifugation. Protein concentrations were determined by BCA assay (Pierce) and 10 μg protein was fractionated on a sodium dodecyl sulfate-polyacrylamide gel, transferred to polyvinyl difluoride membrane, blotted with the appropriate antibody (dilutions were 1:1000 for anti-pAKT, anti-AKT anti-pSTAT3, anti-STAT3, anti-gp130, anti-VEGFR-3; 1:2500 for anti-podoplanin; and 1:10,000 anti-β-actin), and subsequently probed with horse radish peroxidase-conjugated goat anti-mouse or -rabbit immunoglobulin G (Jackson ImmunoResearch Laboratories). Immunoreactive proteins were visualized by chemiluminescence using Amersham ECL Plus. Differences in band intensity were quantified by densitometric methods (ImageJ program).
RNA isolation and quantitative RT-PCR
Total RNA was isolated from TIME cells using the RNeasy Plus kit (Qiagen). 100 or 500 ng of total RNA was used in SuperScript III, Platinum SYBR green, one-step quantitative reverse transcription PCR (RT-PCR) (Invitrogen) according to manufacturer’s protocols with the primers for either GAPDH (glyceraldehydes-3-phosphate dehydrogenase) (forward: 5′-GGA CTC ATG ACC ACA GTC CA-3′, reverse: 5′-CCA GTA GAG GCA GGG ATG AT-3′), Prox1 (forward: 5′-AGT GCA ATG CAG GAA GGA TT-3′, reverse: 5′-CCA CTT GAT GAG CTG AGA GG-3′), gp130 (forward: 5′-GAG CCA GAT TCC TCC TGA AG-3′, reverse: 5′-CCA CTT GCT TCT TCA CTC CA-3′). Relative abundances of the indicated mRNAs were normalized by the delta threshold cycle method to the abundance of GAPDH, with mock-infected TIME cells set to 1. Error bars reflect standard errors of the mean (three experiments).
Retrovirus production and infection of TIME cells
For production of replication-defective amphotropic murine leukemia virions, Phoenix-ampho packaging cells were seeded in a 6-cm dish and transfected with 5 μg of retroviral vector using TransIT 293 protocol (Mirus). 48 h after transfection, the medium containing virions was cleared of cell debris by spinning at 2000 rpm for 10 min, aliquotted directly, and stored at −80 °C. Retroviral infections were done by incubating 1 mL of virus preparation with 1 × 105 TIME cells and 8 μg/mL polybrene in a 6-well plate, first, by centrifugation at 2000 rpm for 2 h at 37 °C, and then for an additional 3 h in an incubator. 48 h after infection, stable pooled cells were selected with the appropriate antibiotic, 2.5 μg/mL puromycin, 500 μg/mL G418, or 100 μg/mL hygromycin (Cellgro, Mediatech, Inc.).
Lentivirus production and infection of TIME cells
For production of vesicular stomatitis virus-G envelope-pseu-dotyped lentiviral virions, HEK293T were seeded in a 10-cm dish (5.5 × 106 cells) and were cotransfected the next day with 8 μg psPAX2, 4 μg pMD2.G, and 8 μg of lentiviral (pSIN-MCS or pCGSW) construct using the TransIt 293 protocol (Mirus). 24 h after transfection, the medium was changed, and at 48 and 72 h post transfection, the medium containing the lentiviral virions was harvested, filtered through a 0.45-μM filter, aliquotted directly, and stored at −80 °C. Lentiviral infections were done by incubating 2 mL of virus preparation with 2 × 105 TIME cells overnight with 8 μg/mL polybrene. Cells were harvested 72 h post infection and GFP expression in the pCGSW-transduced cells was monitored by fluorescent microscopy (Supplemental Fig. 1).
RT-PCR to verify expression of KSHV genes from lentiviral constructs
Total RNA was isolated from TIME cells using the RNeasy Plus kit (Qiagen). Approximately 2 μg of total RNA was used to make cDNA with the SuperScript VILO cDNA Synthesis kit (Invitrogen). Reactions without the addition of reverse transcriptase were used as negative controls. Subsequently, 1 μL of cDNA was used in a PCR reaction with primers to specific KSHV genes or cellular GAPDH (Vart et al., 2007).
Conditioned medium
Conditioned medium from lentivirus-infected or KSHV-infected TIME cells was harvested 72 h or 24 h post infection respectively. Cell debris was removed by centrifugation in a tabletop centrifuge at 2000 rpm for 10 min, then free virions were removed via centrifugation at 15,000 rpm for 2 h and the clarified medium was stored at −80 °C. Uninfected TIME cells were serum starved for 2 h before treatment with conditioned medium for 30 min.
Supplementary Material
Acknowledgments
We would like to thank John Nicholas for the vIL-6-specific shRNA vectors, Didier Trono for the lentiviral packaging plasmids, Adam Grundhoff for the pZIP-LANA vector, Priya Bellare and Don Ganem for the FLAG-vFLIP plasmid, and Craig McCormick for the kaposin vectors. VAM was supported by a National Science Foundation Graduate Research Fellowship. ASP was supported by a postdoctoral training grant from the National Cancer Institute (T32CA09229). ML was supported by a Research Scholar grant from the American Cancer Society, a grant from the National Cancer Institute (R01CA097934) and a grant from the National institute for dental and craniofacial research (PO1DE021954).
Appendix A. Supplementary materials
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.virol.2012.03.013.
References
- Aluigi MG, Albini A, Carlone S, Repetto L, De Marchi R, Icardi A, Moro M, Noonan D, Benelli R. KSHV sequences in biopsies and cultured spindle cells of epidemic, iatrogenic and Mediterranean forms of Kaposi’s sarcoma. Res Virol. 1996;147:267–275. doi: 10.1016/0923-2516(96)82285-0. [DOI] [PubMed] [Google Scholar]
- Antman K, Chang Y. Kaposi’s sarcoma. N Engl J Med. 2000;342:1027–1038. doi: 10.1056/NEJM200004063421407. [DOI] [PubMed] [Google Scholar]
- Aoki Y, Jaffe ES, Chang Y, Jones K, Teruya-Feldstein J, Moore PS, Tosato G. Angiogenesis and hematopoiesis induced by Kaposi’s sarcoma-associated herpesvirus-encoded interleukin-6. Blood. 1999;93:4034–4043. [PubMed] [Google Scholar]
- Boshoff C, Schulz TF, Kennedy MM, Graham AK, Fisher C, Thomas A, McGee JO, Weiss RA, O’Leary JJ. Kaposi’s sarcoma-associated herpesvirus infects endothelial and spindle cells. Nat Med. 1995;1:1274–1278. doi: 10.1038/nm1295-1274. [DOI] [PubMed] [Google Scholar]
- Brinkmann MM, Glenn M, Rainbow L, Kieser A, Henke-Gendo C, Schulz TF. Activation of mitogen-activated protein kinase and NF-kappaB pathways by a Kaposi’s sarcoma-associated herpesvirus K15 membrane protein. J Virol. 2003;77:9346–9358. doi: 10.1128/JVI.77.17.9346-9358.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Lu S, Zhang Z, Gonzalez CM, Damania B, Cullen BR. Kaposi’s sarcoma-associated herpesvirus expresses an array of viral microRNAs in latently infected cells. Proc Natl Acad Sci USA. 2005;102:5570–5575. doi: 10.1073/pnas.0408192102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll PA, Brazeau E, Lagunoff M. Kaposi’s sarcoma-associated herpesvirus infection of blood endothelial cells induces lymphatic differentiation. Virology. 2004;328:7–18. doi: 10.1016/j.virol.2004.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandriani S, Ganem D. Array-based transcript profiling and limiting-dilution reverse transcription-PCR analysis identify additional latent genes in Kaposi’s sarcoma-associated herpesvirus. J Virol. 2010;84:5565–5573. doi: 10.1128/JVI.02723-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science (New York, NY) 1994;266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- Chatterjee M, Osborne J, Bestetti G, Chang Y, Moore PS. Viral IL-6-induced cell proliferation and immune evasion of interferon activity. Science (New York, NY) 2002;298:1432–1435. doi: 10.1126/science.1074883. [DOI] [PubMed] [Google Scholar]
- Chen D, Sandford G, Nicholas J. Intracellular signaling mechanisms and activities of human herpesvirus 8 interleukin-6. J Virol. 2009;83:722–733. doi: 10.1128/JVI.01517-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Lagunoff M. The KSHV viral interleukin-6 is not essential for latency or lytic replication in BJAB cells. Virology. 2007;359:425–435. doi: 10.1016/j.virol.2006.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coscoy L, Ganem D. Kaposi’s sarcoma-associated herpesvirus encodes two proteins that block cell surface display of MHC class I chains by enhancing their endocytosis. Proc Natl Acad Sci USA. 2000;97:8051–8056. doi: 10.1073/pnas.140129797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couty JP, Gershengorn MC. Insights into the viral G protein-coupled receptor encoded by human herpesvirus type 8 (HHV-8) Biol Cell. 2004;96:349–354. doi: 10.1016/j.biolcel.2004.03.011. [DOI] [PubMed] [Google Scholar]
- Dittmer D, Lagunoff M, Renne R, Staskus K, Haase A, Ganem D. A cluster of latently expressed genes in Kaposi’s sarcoma-associated herpesvirus. J Virol. 1998;72:8309–8315. doi: 10.1128/jvi.72.10.8309-8315.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ensoli B, Sgadari C, Barillari G, Sirianni MC, Sturzl M, Monini P. Biology of Kaposi’s sarcoma. Eur J Cancer. 2001;37:1251–1269. doi: 10.1016/s0959-8049(01)00121-6. [DOI] [PubMed] [Google Scholar]
- Ensoli B, Sturzl M, Monini P. Cytokine-mediated growth promotion of Kaposi’s sarcoma and primary effusion lymphoma. Semin Cancer Biol. 2000;10:367–381. doi: 10.1006/scbi.2000.0329. [DOI] [PubMed] [Google Scholar]
- Foussat A, Wijdenes J, Bouchet L, Gaidano G, Neipel F, Balabanian K, Galanaud P, Couderc J, Emilie D. Human interleukin-6 is in vivo an autocrine growth factor for human herpesvirus-8-infected malignant B lymphocytes. Eur Cytokine Network. 1999;10:501–508. [PubMed] [Google Scholar]
- Godfrey A, Anderson J, Papanastasiou A, Takeuchi Y, Boshoff C. Inhibiting primary effusion lymphoma by lentiviral vectors encoding short hairpin RNA. Blood. 2005;105:2510–2518. doi: 10.1182/blood-2004-08-3052. [DOI] [PubMed] [Google Scholar]
- Grundhoff A, Sullivan CS, Ganem D. A combined computational and microarray-based approach identifies novel microRNAs encoded by human gamma-herpesviruses. Rna. 2006;12:733–750. doi: 10.1261/rna.2326106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirakawa S, Hong YK, Harvey N, Schacht V, Matsuda K, Libermann T, Detmar M. Identification of vascular lineage-specific genes by transcriptional profiling of isolated blood vascular and lymphatic endothelial cells. Am J Pathol. 2003;162:575–586. doi: 10.1016/S0002-9440(10)63851-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong YK, Detmar M. Prox1, master regulator of the lymphatic vasculature phenotype. Cell Tissue Res. 2003;314:85–92. doi: 10.1007/s00441-003-0747-8. [DOI] [PubMed] [Google Scholar]
- Hong YK, Foreman K, Shin JW, Hirakawa S, Curry CL, Sage DR, Libermann T, Dezube BJ, Fingeroth JD, Detmar M. Lymphatic reprogramming of blood vascular endothelium by Kaposi sarcoma-associated herpesvirus. Nat Genet. 2004;36:683–685. doi: 10.1038/ng1383. [DOI] [PubMed] [Google Scholar]
- Hong YK, Harvey N, Noh YH, Schacht V, Hirakawa S, Detmar M, Oliver G. Prox1 is a master control gene in the program specifying lymphatic endothelial cell fate. Dev Dyn. 2002;225:351–357. doi: 10.1002/dvdy.10163. [DOI] [PubMed] [Google Scholar]
- Jones KD, Aoki Y, Chang Y, Moore PS, Yarchoan R, Tosato G. Involvement of interleukin-10 (IL-10) and viral IL-6 in the spontaneous growth of Kaposi’s sarcoma herpesvirus-associated infected primary effusion lymphoma cells. Blood. 1999;94:2871–2879. [PubMed] [Google Scholar]
- Jussila L, Valtola R, Partanen TA, Salven P, Heikkila P, Matikainen MT, Renkonen R, Kaipainen A, Detmar M, Tschachler E, Alitalo R, Alitalo K. Lymphatic endothelium and Kaposi’s sarcoma spindle cells detected by antibodies against the vascular endothelial growth factor receptor-3. Cancer Res. 1998;58:1599–1604. [PubMed] [Google Scholar]
- Lagunoff M, Bechtel J, Venetsanakos E, Roy AM, Abbey N, Herndier B, McMahon M, Ganem D. De novo infection and serial transmission of Kaposi’s sarcoma-associated herpesvirus in cultured endothelial cells. J Virol. 2002;76:2440–2448. doi: 10.1128/jvi.76.5.2440-2448.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagunoff M, Majeti R, Weiss A, Ganem D. Deregulated signal transduction by the K1 gene product of Kaposi’s sarcoma-associated herpesvirus. Proc Natl Acad Sci USA. 1999;96:5704–5709. doi: 10.1073/pnas.96.10.5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Wang H, Nicholas J. Detection of direct binding of human herpesvirus 8-encoded interleukin-6 (vIL-6) to both gp130 and IL-6 receptor (IL-6R) and identification of amino acid residues of vIL-6 important for IL-6R-dependent and -independent signaling. J Virol. 2001;75:3325–3334. doi: 10.1128/JVI.75.7.3325-3334.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molden J, Chang Y, You Y, Moore PS, Goldsmith MA. A Kaposi’s sarcoma-associated herpesvirus-encoded cytokine homolog (vIL-6) activates signaling through the shared gp130 receptor subunit. J Biol Chem. 1997;272:19625–19631. doi: 10.1074/jbc.272.31.19625. [DOI] [PubMed] [Google Scholar]
- Montaner S, Sodhi A, Molinolo A, Bugge TH, Sawai ET, He Y, Li Y, Ray PE, Gutkind JS. Endothelial infection with KSHV genes in vivo reveals that vGPCR initiates Kaposi’s sarcomagenesis and can promote the tumorigenic potential of viral latent genes. Cancer Cell. 2003;3:23–36. doi: 10.1016/s1535-6108(02)00237-4. [DOI] [PubMed] [Google Scholar]
- Moore PS, Boshoff C, Weiss RA, Chang Y. Molecular mimicry of human cytokine and cytokine response pathway genes by KSHV. Science (New York, NY) 1996;274:1739–1744. doi: 10.1126/science.274.5293.1739. [DOI] [PubMed] [Google Scholar]
- Morris VA, Punjabi AS, Lagunoff M. Activation of Akt through gp130 receptor signaling is required for Kaposi’s sarcoma-associated herpesvirus-induced lymphatic reprogramming of endothelial cells. J Virol. 2008;82:8771–8779. doi: 10.1128/JVI.00766-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas J, Ruvolo VR, Burns WH, Sandford G, Wan X, Ciufo D, Hendrickson SB, Guo HG, Hayward GS, Reitz MS. Kaposi’s sarcoma-associated human herpesvirus-8 encodes homologues of macrophage inflammatory protein-1 and interleukin-6. Nat Med. 1997;3:287–292. doi: 10.1038/nm0397-287. [DOI] [PubMed] [Google Scholar]
- Petrova TV, Makinen T, Makela TP, Saarela J, Virtanen I, Ferrell RE, Finegold DN, Kerjaschki D, Yla-Herttuala S, Alitalo K. Lymphatic endothelial reprogramming of vascular endothelial cells by the Prox-1 homeobox transcription factor. EMBO J. 2002;21:4593–4599. doi: 10.1093/emboj/cdf470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer S, Sewer A, Lagos-Quintana M, Sheridan R, Sander C, Grasser FA, van Dyk LF, Ho CK, Shuman S, Chien M, Russo JJ, Ju J, Randall G, Lindenbach BD, Rice CM, Simon V, Ho DD, Zavolan M, Tuschl T. Identification of microRNAs of the herpesvirus family. Nat Methods. 2005;2:269–276. doi: 10.1038/nmeth746. [DOI] [PubMed] [Google Scholar]
- Punjabi AS, Carroll PA, Chen L, Lagunoff M. Persistent activation of STAT3 by latent Kaposi’s sarcoma-associated herpesvirus infection of endothe-lial cells. J Virol. 2007;81:2449–2458. doi: 10.1128/JVI.01769-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renne R, Lagunoff M, Zhong W, Ganem D. The size and conformation of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) DNA in infected cells and virions. J Virol. 1996;70:8151–8154. doi: 10.1128/jvi.70.11.8151-8154.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadagopan S, Sharma-Walia N, Veettil MV, Raghu H, Sivakumar R, Bottero V, Chandran B. Kaposi’s sarcoma-associated herpesvirus induces sustained NF-kappaB activation during de novo infection of primary human dermal microvascular endothelial cells that is essential for viral gene expression. J Virol. 2007;81:3949–3968. doi: 10.1128/JVI.02333-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadler R, Wu L, Forghani B, Renne R, Zhong W, Herndier B, Ganem D. A complex translational program generates multiple novel proteins from the latently expressed kaposin (K12) locus of Kaposi’s sarcoma-associated her-pesvirus. J Virol. 1999;73:5722–5730. doi: 10.1128/jvi.73.7.5722-5730.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samols MA, Hu J, Skalsky RL, Renne R. Cloning and identification of a microRNA cluster within the latency-associated region of Kaposi’s sarcoma-associated herpesvirus. J Virol. 2005;79:9301–9305. doi: 10.1128/JVI.79.14.9301-9305.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schacht V, Ramirez MI, Hong YK, Hirakawa S, Feng D, Harvey N, Williams M, Dvorak AM, Dvorak HF, Oliver G, Detmar M. T1alpha/podoplanin deficiency disrupts normal lymphatic vasculature formation and causes lymphedema. EMBO J. 2003;22:3546–3556. doi: 10.1093/emboj/cdg342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skobe M, Brown LF, Tognazzi K, Ganju RK, Dezube BJ, Alitalo K, Detmar M. Vascular endothelial growth factor-C (VEGF-C) and its receptors KDR and flt-4 are expressed in AIDS-associated Kaposi’s sarcoma. J Invest Dermatol. 1999;113:1047–1053. doi: 10.1046/j.1523-1747.1999.00798.x. [DOI] [PubMed] [Google Scholar]
- Sodhi A, Montaner S, Patel V, Gomez-Roman JJ, Li Y, Sausville EA, Sawai ET, Gutkind JS. Akt plays a central role in sarcomagenesis induced by Kaposi’s sarcoma herpesvirus-encoded G protein-coupled receptor. Proc Natl Acad Sci USA. 2004;101:4821–4826. doi: 10.1073/pnas.0400835101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staskus KA, Zhong W, Gebhard K, Herndier B, Wang H, Renne R, Beneke J, Pudney J, Anderson DJ, Ganem D, Haase AT. Kaposi’s sarcoma-associated herpesvirus gene expression in endothelial (spindle) tumor cells. J Virol. 1997;71:715–719. doi: 10.1128/jvi.71.1.715-719.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vart RJ, Nikitenko LL, Lagos D, Trotter MW, Cannon M, Bourboulia D, Gratrix F, Takeuchi Y, Boshoff C. Kaposi’s sarcoma-associated herpesvirus-encoded interleukin-6 and G-protein-coupled receptor regulate angiopoietin-2 expression in lymphatic endothelial cells. Cancer Res. 2007;67:4042–4051. doi: 10.1158/0008-5472.CAN-06-3321. [DOI] [PubMed] [Google Scholar]
- Veikkola T, Jussila L, Makinen T, Karpanen T, Jeltsch M, Petrova TV, Kubo H, Thurston G, McDonald DM, Achen MG, Stacker SA, Alitalo K. Signalling via vascular endothelial growth factor receptor-3 is sufficient for lymphangiogenesis in transgenic mice. EMBO J. 2001;20:1223–1231. doi: 10.1093/emboj/20.6.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venetsanakos E, Mirza A, Fanton C, Romanov SR, Tlsty T, McMahon M. Induction of tubulogenesis in telomerase-immortalized human microvascular endothelial cells by glioblastoma cells. Exp Cell Res. 2002;273:21–33. doi: 10.1006/excr.2001.5424. [DOI] [PubMed] [Google Scholar]
- Wan X, Wang H, Nicholas J. Human herpesvirus 8 interleukin-6 (vIL-6) signals through gp130 but has structural and receptor-binding properties distinct from those of human IL-6. J Virol. 1999;73:8268–8278. doi: 10.1128/jvi.73.10.8268-8278.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HW, Trotter MW, Lagos D, Bourboulia D, Henderson S, Makinen T, Elliman S, Flanagan AM, Alitalo K, Boshoff C. Kaposi sarcoma herpesvirus-induced cellular reprogramming contributes to the lymphatic endothelial gene expression in Kaposi sarcoma. Nat Genet. 2004;36:687–693. doi: 10.1038/ng1384. [DOI] [PubMed] [Google Scholar]
- Wang L, Dittmer DP, Tomlinson CC, Fakhari FD, Damania B. Immortalization of primary endothelial cells by the K1 protein of Kaposi’s sarcoma-associated herpesvirus. Cancer Res. 2006;66:3658–3666. doi: 10.1158/0008-5472.CAN-05-3680. [DOI] [PubMed] [Google Scholar]
- Yoo J, Kang J, Lee HN, Aguilar B, Kafka D, Lee S, Choi I, Lee J, Ramu S, Haas J, Koh CJ, Hong YK. Kaposin-B enhances the PROX1 mRNA stability during lymphatic reprogramming of vascular endothelial cells by Kaposi’s sarcoma herpes virus. PLoS Pathog. 2010;6:e1001046. doi: 10.1371/journal.ppat.1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.