

Abstract
A library of approximately 40 N1-acylated (aza)indole alkanoic esters and acids was prepared employing a microwave-assisted approach. The optimized synthetic route allows for parallel synthesis, variation of the indole substitution pattern and high overall yield. Additionally, the procedure has been scaled up to yield multi-gram amounts of preferred indole compounds, e.g.: 2′-des-methyl indomethacin 2. The reported compounds were designed as biomedical tools for primary and secondary in vitro and in vivo studies at relevant molecular targets.
Keywords: Heterocyles, Indoles, (2′-Des-methyl) indomethacin, Microwave synthesis, Synthesis scale-up
1. Introduction
N-Acylated indole acetic acids are drawing continued attention due to their wide-ranging pharmacological activities.1–5 For example, indomethacin (INDO, 1, Fig. 1), a traditional non-steroidal anti-inflammatory drug (NSAID), is a potent, time-dependent inhibitor of cyclooxygenases (COX-1 and COX-2) with a long history of clinical use in human subjects.6 It promotes anti-inflammatory, anti-pyretic and analgesic activity, but also contributes to severe stomach irritation and ulcers due to secondary pharmacodynamic effects.7,8 Recently, 1 and some of its derivatives have been shown to hold interesting and potentially useful ‘off-target’ activities, e.g. regulation of Th2-mediated asthma and other allergic diseases or chemoprevention of tumors by addressing targets different from COX.9–14 Of these INDO derivatives, those that do not (strongly) interfere with arachidonic acid (AA) metabolism, have greater potential to be valuable drug candidates because of their presumably lower gastrointestinal toxicity. Very recently we have probed 2′-des-methyl indomethacin (2′-DM-INDO, 2, Fig. 1),15 a structurally edited version of 1, which exhibits drastically reduced inhibitory potency against both COX enzymes and reduced gastric toxicity relative to 1 in C57BL6 mice.16,10 To further probe the effects of indoles related to 1 and 2, we required rapid production of iterative series of N-acyl indole alkanoic acids and esters in good overall yield.
Fig. 1.
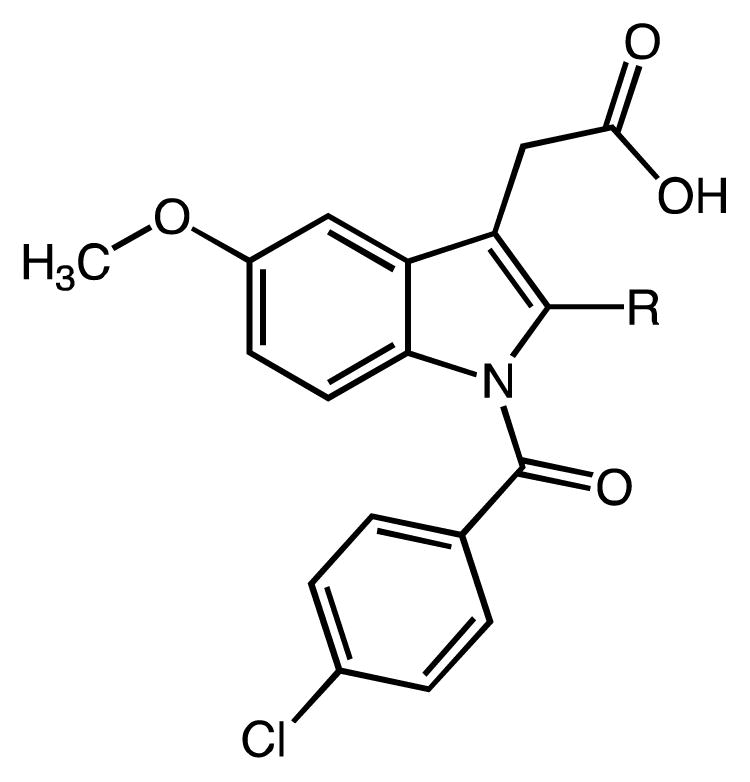
In the present manuscript we report a very efficient indolization/acylation approach toward N-substituted (aza)indole compounds and we describe the successful scale-up of the synthesis of two prototype indole analogs.
2. Results and Discussion
2.1. Small-scale approach
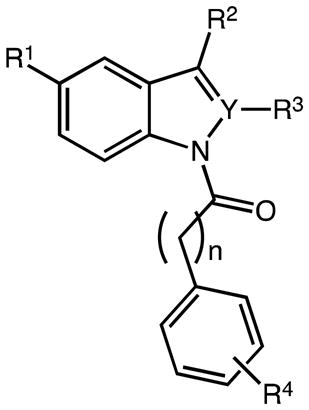
We accomplished a robust synthetic routine and generated a series of novel INDO (1) analogues 18–31 (Tables 2 and 3) with individual compounds emerging in milligram or multi-gram quantities and good overall yield (>55%). The general reaction sequence leading to indoles 18 through 31, followed three simple steps: (a) Fischer indolization of substituted phenylhydrazine hydrochlorides condensed with selected ketoacids/esters, (b) base-catalyzed indole N1-acylation reactions (SN2 substitutions) with different acid chlorides, and (c) ester hydrolysis of the intermediate methyl or cleavage of allyl N-acyl indole alkanoates in the presence of a metal organic catalyst. Structural modifications with respect to 1 (and 2) were made at the C2, C3, C5, COOH and N1-position of the indole ring. Previous procedures using similar synthetic strategies have not been reported on large scale and oftentimes proceed in low overall yield.
Table 2.
Synthesis of N-acyl methyl (aza)indole(cyclo)alkanoates
| ||||||||
|---|---|---|---|---|---|---|---|---|
| # | Y | R1 | R2 | R3 | n | R4 | method | yield (%)a |
| 18a | C | OMe | CH2C(O)OMe | Me | 0 | p-F | C2 | 25 |
| 18b | C | OMe | CH2C(O)OMe | Me | 0 | m-CF3 | C2 | 32 |
| 18c | C | OMe | CH2C(O)OMe | Me | 0 | p-CF3 | C2 | 33 |
| 18d | C | OMe | CH2C(O)OMe | Me | 0 | p-Me | C2 | 57 |
| 18e | C | OMe | CH2C(O)OMe | Me | 0 | p-CH2Cl | C2 | 42 |
| 18f | C | OMe | CH2C(O)OMe | Me | 0 | p-OMe | C2 | 32 |
| 18g | C | OMe | CH2C(O)OMe | H | 0 | p-Cl | C1 | 18 |
| 18h | C | OMe | CH2C(O)OMe | H | 0 | p-F | C2 | 22 |
| 18i | C | OMe | CH2C(O)OMe | H | 0 | m-CF3 | C2 | 13 |
| 18j | C | OMe | CH2C(O)OMe | H | 0 | p-CF3 | C2 | 29 |
| 18k | C | OMe | CH2C(O)OMe | H | 1 | p-OMe | C1 | (13)* |
| 18l | C | OMe | CH2C(O)OMe | H | 0 |
|
C1 | 21 |
| 18m | C | F | CH2C(O)OMe | Me | 0 | p-Cl | C2 | 42 |
| 18n | C | F | CH2C(O)OMe | Me | 0 | m-CF3 | C2 | 33 |
| 18o | C | F | CH2CH2C(O)OMe | Me | 0 | p-Cl | C2 | 49 |
| 18p | C | F | CH2C(O)OMe | Me | 1 | p-Cl | C2 | 4 |
| 18q | C | F | CH2C(O)OMe | H | 0 | p-Cl | C2 | 42 |
| 19a | C | OMe |
|
0 | m-CF3 | C2 | 24 | |
| 19b | C | F |
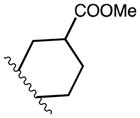
|
0 | p-Cl | C2 | 14 | |
| 20a | C | OMe | H | Me | 0 | p-Cl | C2 | 25 |
| 20b | C | OMe |
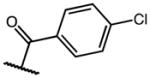
|
Me | 0 | p-Cl | C2 | 8b |
| 21 | N | OMe | CH2C(O)OMe | --- | 0 | p-Cl | C2 | 37 |
Reagents and Conditions: [method C1]: NaH, DMF, acyl chloride, 0 °C to rt, overnight; [method C2]: tBuONa, THF, acyl chloride, 0 °C to rt, overnight.
isolated yield;
20b emerged as one side product in the synthesis of 20a;
compound after repeated chromatography only about 50% pure
Table 3.
Synthesis of N-acyl indole(cyclo)alkanoic acids
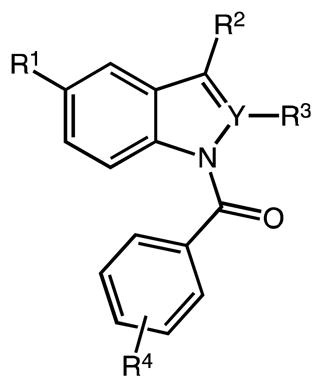
| |||||||
|---|---|---|---|---|---|---|---|
| # | Y | R1 | R2 | R3 | R4 | method | yield (%)a |
| 2 | C | OMe | CH2C(O)OH | H | p-Cl | D | 81 |
| 22a | C | OMe | CH2C(O)OH | Me | p-F | D | 90 |
| 22b | C | OMe | CH2C(O)OH | Me | m-CF3 | D | 97 |
| 22c | C | OMe | CH2C(O)OH | Me | p-CF3 | D | 90 |
| 22d | C | OMe | CH2C(O)OH | Me | p-Me | D | 82 |
| 22e | C | OMe | CH2C(O)OH | Me | p-CH2Cl | D | 98 |
| 22f | C | OMe | CH2C(O)OH | Me | p-OMe | D | 93* |
| 22g | C | OMe | CH2C(O)OH | H | p-F | D | 76 |
| 22h | C | OMe | CH2C(O)OH | H | m-CF3 | D | 96 |
| 22i | C | OMe | CH2C(O)OH | H | p-CF3 | D | 99 |
| 22j | C | OMe | CH2C(O)OH | H |
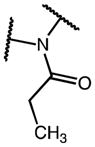
|
D | 94 |
| 22k | C | F | CH2C(O)OH | Me | p-Cl | D | 87 |
| 22l | C | F | CH2C(O)OH | Me | m-CF3 | D | 95 |
| 22m | C | F | CH2CH2C(O)OH | Me | p-Cl | D | 95 |
| 22n | C | F | CH2C(O)OH | H | p-Cl | D | 78 |
| 23 | C | OMe |
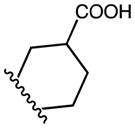
|
m-CF3 | D | 98* | |
| 24 | N | OMe | CH2C(O)OH | --- | Cl | D | No product (decomposition) |
Reagents and Conditions: [method D]: trimethyltin hydroxide, 1,2-DCE, microwave, 130 °C, 30 min.
isolated yield;
crude educts used; impurity of the starting material taken into account for the calculation of the percent yield (see Supporting Information)!
Several creative methods for assembly of N1-substituted indole compounds have been reported. These include one-pot Stille couplings of N-acyl-2-iodoanilines,17 catalytic dehydrogenative N1-couplings of indoles with primary alcohols,18 distinct indolization reactions with cyclic enol ethers and enol lactones,19 and variations of the Fischer Indole synthesis.20
In the refined standard procedure of Yamamoto for compound 1 (Scheme 1),21,22 an arylhydrazone precursor 4 is benzoylated at the N1 position to give the N1,N1-disubstituted hydrazine 5. Typically, commercial hydrazine hydrochloride starting materials 3 need to be deprotonated prior to reaction. The intermediate 5 is then hydrolyzed with hydrogen chloride to give the hydrazine hydrochloride salt 6, which is cyclized in the final reaction step with levulinic acid to afford 1.23 The synthesis of 2′-des-methyl INDO 2 has been conducted accordingly with the exception that succinic semialdehyde was used for the ring-closure.
Scheme 1.

Yamamoto’s procedure for the synthesis of (2′-des-methyl) indomethacin; R = CH3 or H
Isolated product yields are often unsatisfactory using this literature procedure due to multiple reaction steps, the formation of by-products, and the requirement for extensive purification of intermediates.19 The early insertion of the N1-acyl moiety in the reaction sequence is another limitation of this entry, which involves the addition and subsequent displacement of an ethylidene protection group (N2). Therefore, in order to vary the N-acyl substituent for a series of indole analogues 18–21 (Table 2), each derivative needed to be constructed individually. To overcome this limitation and enhance the flexibility toward the introduction of multiple substituents in the indole scaffold, we pursued a strategy (Scheme 2) that has not been intensively investigated for the serial production of substituted indoles because it has proven to be rather uneconomical, especially for larger scales. This strategy employs indolization first followed by late-stage N1-substitution and release of the carboxylic acid under optimized reaction conditions. Cyclization reactions of arylhydrazines with different ketoacids/esters were conducted in AcOH or refluxing methanol/H2SO4. We used methyl or allyl esters for our small-scale and large-scale reactions, respectively, because of their stability to the reaction conditions and ease of removal. Utilization of t-BuONa in THF or DMAP/Et3N in DCM as reaction medium facilitated indole N-acylation with select aryl acid chlorides. Final ester cleavage was done in 1,2-DCE with trimethyltin hydroxide24 (methyl esters) or in THF using Pd(PPh3)4/morpholine as catalyst complex (allyl esters). Concurrent N-deacylation was not observed with these expedient procedures, which clearly privileges them over standard saponification methods with metal alkaline hydroxide/water. We also investigated the synthesis under microwave irradiation for numerous compounds with a view to speed up reaction rates and increase isolable yields.
Scheme 2.

Esterification of (2′-aza-/2′-des-methyl) indole-3′-acetic acids
2.1. 1. Preparation of different indole methyl-alkanoates
Initially, we generated a set of 10 different C2/C3/C5-substituted (aza)indole intermediates. The structures, specific conditions and yields are summarized in Table 1. Detailed experimental data can be also found in Supporting Information.
Table 1.
Preparation of different (aza)indole alkanoic acid ester precursors
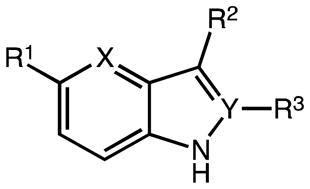
| |||||||
|---|---|---|---|---|---|---|---|
| # | X | Y | R1 | R2 | R3 | method | yield (%)a |
| 9a | C | C | OMe | CH2C(O)OMe | Me | A1 | >99 |
| 9b | C | C | OMe | CH2C(O)OMe | H | A1 | 85 |
| 9c | C | C | F | CH2C(O)OMe | Me | B2 | 84 |
| 9d | C | C | F | CH2CH2C(O)OMe | Me | B2 | 76 |
| 9e | C | C | F | CH2C(O)OMe | H | B2b | 86 |
| 14 | C | C | F | Me | CH2CH2C(O)OMe | B2 | 46 |
| 15a | C | C | OMe |
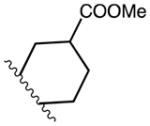
|
B1 | 90 | |
| 15b | C | C | F |
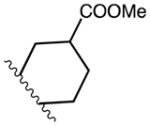
|
B2 | 99 | |
| 10 | C | N | H | CH2C(O)OMe | --- | A2 | 77 |
| 17 | N | C | OMe | CH2C(O)OMe | H | other | 60 |
Reagents and Conditions: [method A1]: BOP-Cl, triethylamine, CH2Cl2, MeOH, rt, overnight; [method A2]: MeOH, H2SO4, reflux, overnight; [method B1]: arylhydrazine HCl, ketoacid ester, AcOH, 80 °C, 3h; [method B2]: arylhydrazine HCl, ketoacid or ketoacid ester, MeOH, H2SO4, microwave, 120 °C, 10 min.
isolated yield;
conventional heating, 3h instead of microwave
Methyl esters 9a, and 9b of commercial 5-methoxy-2-methylindole-3-acetic acid 7a, and 5-methoxyindole-3-acetic acid 7b were generated overnight at room temperature in yields >85% using BOP-Cl/triethylamine and methanol (Scheme 2).25
The preparation of different C2/C3-substituted indole compounds 9c-e, 14 and 15 is summarized in Scheme 3. Substituted aryl hydrazine hydrochlorides 3 were directly subjected to reaction with selected ketoacids or esters 11 through 13. Particularly, indole compound 15a was generated in 90% yield under standard reaction conditions (AcOH, 80 °C, 3h) using 4-methoxyphenyl hydrazine hydrochloride 3a and methyl 4-ketocyclohexane-carboxylate. Next, in order to allow investigation of the significance of a fluoro substitution at the indole 5-position, we pursued Fischer indole synthesis of C2/C3-substituted indole analogues 9c-e, 14 and 15b. Whereas para-methoxylated phenylhydrazine hydrochlorides 3a (e.g. for compound 15a) readily dissolved in AcOH upon heating, this was not the case for fluorinated starting materials 3b (leaving unreacted starting materials). Therefore, AcOH was replaced by methanol/H2SO4 as solvent mixture for better solubility of the fluorinated precursors. In this case, selected ‘ketoacids’ rather than their low alkyl esters were employed in the reaction, as clean COOH-esterification usually occurred alongside the ‘indole’ ring-closure (compare experimental section for 9c vs. 9e). Reactions were accelerated by microwave irradiation and ran approx. 50 times faster (5 to 10 min instead 3h to 6h) than with conventional heating affording consistently high and reproducible yields (~80%). In many cases products precipitated upon pouring the reaction mixture on crushed ice or could be extracted from the resulting aqueous mixture, washed and obtained in acceptable quality after removal of solvent in vacuo and trituration with hexanes.
Scheme 3.

Fischer indolization reactions toward different 2′-/3′-alkylcarboxylic acid esters. Reagents and conditions: (a) AcOH, 80 °C, 3h; (b) MeOH/H2SO4, 5 min (μwave) to 3h (conventional heating)
The same results were achieved when using keto acids whose carboxylic acid group had been esterified beforehand (see experimental details for 9c). (Partial) re-esterification to the corresponding methyl esters 9 even occurred when starting for example with ethyl esters of 11 (data not shown). For compound 9d bearing an extended exocyclic alkyl chain on position C3 of the indole, 4-F-phenylhydrazine hydrochloride was converted with 5-oxohexanoic acid under the conditions described. Finally a 2-propionic acid derivative was realized with indole 14, using 4-oxohexanoic acid as cyclization reagent. In this case, however, the desired product could be isolated only in 46% yield.
Not only did this reaction eliminate the need for an N-protection, it also allowed the variation of all relevant substituents R1 thru R3 as well as the length and position of the carboxyalkyl chain at the indole framework within only one reaction step. Generally, linear ketones 11 or 12 were of importance for the later configuration of the carboxy alkyl substituted indoles. Certain ‘keto’ alkanoic acids/esters provided high regioselectivity in the 3,3-sigmatropic rearrangement in the course of the Fisher indole synthesis. In the case of 11 (acetyl or formyl tail end), the alkyl acid functionality was solely directed to position 3 of the indole product, whereas reagents 12 (ethylcarbonyl tail end) typically produce the alkyl acid functionality at position 2 (isolated products).26 Structure examples are given in Scheme 2 and Table 1.26 Elongation of the alkyl chain in between the two functional groups with respect to reagents 11 was tolerated and didn’t affect the selectivity (e.g. 9d). On the contrary, the inclusion of one or more extra methylene units into the terminal alkyl chain (beyond carbonyl) and/or extension of the alkyl spacer (between CO and COOH) of 12 continuously yielded isomeric mixtures (C2/C3). Such compounds are not part of the current work.
Poor regioselectivity is a general problem of Fischer indole reactions as evidenced in the use of unsymmetrical cyclic ketones. Interestingly, there have been a few examples for the selective cyclization of phenylhydrazones with e.g. 3-ketocyclo-hexanecarboxylic acid or 5-aryldihydro-3(2H)-thiophenones.27,28 To simplify matters in our present study we used symmetrical 4-ketocyclohexane-carboxylic acid to produce indole derivatives 15a (6′-methoxy, see above) and 15b (6′-fluoro) and to demonstrate the later availability for use of similar intermediates in subsequent N-acylation/ester cleavage reactions. In practice, indole derivative 15b could be isolated quantitatively after short reaction times using microwave-aided synthesis.
In order to demonstrate the feasibility of additional heteroatoms and to modify the hydrophilicity of the core skeletons, we also produced a 2-aza (10) and a 4-aza indole-3-acetic acid methyl ester analogue (17) (Schemes 2 and 4). The reaction for 10 was completed in 77% yield according to a patent method starting from 2-(1H-indazol-3-yl)acetic acid (14h reflux in methanol/H2SO4).29 In this case the originally attempted esterification with BOP-Cl/methanol did not succeed. Another literature method allowed for the synthesis of the 4-aza indole derivative 17.30,31 In contrast to the published procedure, we adopted the reaction under microwave irradiation with the respective methyl ester. Customized one-pot catalytic reduction/cyclization of a 3-cyano-3-(nitropyridin-2-yl)-methyl propionate precursor 16 in methanol using Pd/C and cyclohexadiene as hydrogen source (10 min at 120 °C) readily afforded 17 in 60% yield.
Scheme 4.

Synthesis of a 4-aza-indole-3-acetic acid ester analogue
2.1. 2. Substitution at nitrogen (N1)
N-substitution of indoles, 9 and 15, yielded a series of N-acyl-indole methyl alkanoates, 18 and 19, respectively (Table 2). Different benzoyl/aryl acid chlorides were chosen as acylating reagents. However, direct N-acylation of an indole nucleus is demanding and necessitates deprotonation of the N-H center, as neutral indoles are non nucleophilic at the nitrogen atom. Due to the low acidity of the indole-N1, full deprotonation to the reactive anion can be only realized under strong base catalysis in (polar) aprotic solvents.
For the small-scale one-pot reactions on indole 9 we first tried sodium hydride (powdered material) in DMF, though, with less satisfying results due to several handling issues (e.g. with compounds 18g, 18k or 18l). Then we switched to sodium tert-butoxide and used a 2M commercial solution in THF to produce a larger series of compounds 18–21 having different benzoyl (p-Cl, p-CH2Cl, p-F, p-Me, p-OMe, m/p-CF3) and other acyl moieties (Scheme 5, first step). Not only did we observe higher yields and reproducibility with this base (yields ranging from 20–60% for numerous analogues), but also total reaction processing and workup were cleaner than with NaH/DMF (pipetting volumes instead of weighing moisture-sensitive solids; phase separation after water quench). Reactions were run on a small scale (80–100 mg each) under argon on a parallel synthesis apparatus. N-Acylation with different acid chlorides occurred best at temperatures between 0 °C and 25 °C. Therefore, in a typical experiment, we pipetted the base to an ice-cold stirred solution of the indole in THF and after 20 min we added the acid chloride and let the mixture react at ambient temperature until the starting material was consumed or no further product formation could be detected (usually overnight). Progress of the reactions was monitored by LC-MS and TLC. In most cases, one major product was detected. By-products were predominantly unreacted or N-deacylated/hydrolyzed indole starting materials and the carboxyl counterparts of the applied benzoyl chlorides.
Scheme 5.

N-Acylation and ester cleavage reactions.
Isolated amounts of products with varied meta- and para-substituted N-benzoyl moieties appeared to be better (24–57%), when a 2-methyl group was present on the indole ring (18a-f and 18m-o) and were lower (13–29%) among the 2-des-methyl indole analogues (18g-k), except for 18q (42%). In compounds 18k and 18p the acyl substituent was ‘extended’ by an additional methylene unit, which further reduced the percent yields [18k: 13% (impure) and 18q: 4%, respectively]. A simple aliphatic N-propanoyl substituent was realized with indole acetic acid ester analogue 18l. C2/C3-cyclized indole intermediates, 12a and 12b could be substituted on their nitrogens with an m-CF3- and p-Cl-benzoyl substituent in 24 and 14% yield, respectively (19a, 19b). Acylation of an N1, C2, C3-unsubstituted 5-methoxy indole precursor using sodium tert-butoxide and p-Cl-benzoyl chloride afforded a mixture of three compounds, two of which were isomers with the benzoyl moiety on either position C3 (faint, not isolated) or N1 (20a, 25%). The other isolated product 20b (8%) was analyzed to be an N1/C3-dibenzoylated indole derivative. These reactions reflect the enhanced reactivity of the unsubstituted indole 3-position upon hydrogen abstraction at the nitrogen via the conjugated double-bond system (conjugate-like substitution) and in turn the facilitated N1-deprotonation reaction by the 3-acyl ring substituent.
The initial attempt to acylate a 2-propionic acid ester indole derivative (e.g., 11) with 4-chlorobenzoyl chloride using similar conditions failed and considerable amounts of starting material were detected beside some undefined products by LCMS. Nevertheless, N-substitution of a comparable indole derivative was successfully followed up later under larger scale and with optimized reaction conditions (compound 30, see below).
Lastly, compound 21 exemplifies an N1-benzoylated 2-aza-indole acetic acid methyl ester emerging in 37% yield from the parallel routine described above.
2.1.3. Methyl Ester Cleavage
In order to obtain the free acid derivatives 2, 22, 23, and 24 (Table 3) of the N-acylated indole compounds 18, 19, and 21, the methyl ester groups had to be hydrolyzed. Different standard ‘saponification’ methods exist to achieve this task, however common drawbacks include elimination reactions induced by the aqueous-basic conditions.32 This circumstance became particularly significant, as the N-acyl bonds of our indole derivatives were perceptibly fragile toward basic (and strong acidic) treatment and easily hydrolyzed to liberate N-unsubstituted indole alkanoic acids. This was true for numerous 2-des-methyl indole analogues and also for the sterically more demanding 2-propionic acid derivatives (see below), notwithstanding a limited number of literature instances for successful ester saponification toward N-acyl 2-methyl indole acetic acids.20
We attempted a recently reported non-aqueous metal catalytic method for the mild and selective hydrolysis of esters with trimethyltin hydroxide.24 It had been illustrated for many (complex) aromatic and aliphatic systems that methyl, allyl and benzyl esters are hydrolyzed in quantitative yields using this reagent. For our current study, we once more transferred the stated settings to microwave conditions (for detailed parameters see experimental part) and consistently applied 5 equivalents of Me3SnOH beside our starting methyl esters in 1,2-dichloroethane for the expedited ester hydrolysis. Reactions normally ran to completion within 20 to 30 min (IPC by TLC and LC-MS) and workup was quite simple, as products could be extracted into dichloromethane after addition of 50% acetic acid. Residual Me3SnOH was removed with water or brine; the organic layer was dried over Na2SO4, filtered and concentrated. Flash chromatography on silica gel afforded the pure N-acyl indole alkanoic acids 2, 22 and 23 quantitatively. As shown in Table 3, this technique proved truly valuable in getting the selective and high-yielding (76–99%) hydrolysis of methyl esters within the sensitive substrates 18 and 19. Unfortunately, this procedure failed with the 2-aza derivative 21 to obtain the corresponding free acid derivative 24. The latter compound repeatedly decomposed into several unidentified products, regardless of whether the reaction was conducted with microwaves or under conventional heating conditions (60–80 °C).
To extend this strategy to the large-scale synthesis of individual N-acyl indole derivatives, we further optimized the reaction conditions as described below, particularly with a view to improving capacities of the N-acylation step.
2.2. Large-scale synthesis
New synthetic methodology was adopted for the scale-up to multi-gram scale. Beside the efforts with N-substitution, the major problematic step in the synthesis remained the final hydrolysis of the ester in the presence of the N-acyl functional group. As mentioned above, the N-benzoyl bond is labile under basic conditions, so it was hydrolyzed easily at normal hydrolysis condition for the ester hydrolysis step.32 Therefore we considered using allyl,33,34 benzyl,35,36 and tert-butyl ester37 to overcome the side reaction and allyl ester 26 was evaluated.
A large quantity of 2 was prepared from the commercially available 2-(5-methoxy-1H-indol-3-yl)acetic acid 7b in three steps starting with esterification with allyl bromide to afford allylester 25 in 75% yield (Scheme 6). Efficient acylation of 25 followed by treatment with triethylamine and DMAP then gave N-benzoate 26 (92% yield). Subsequently, cleavage of the allyl ester was examined. Optimal conditions employed tetrakis(triphenylphosphine) palladium in combination with morpholine in THF to quantitatively provide 2 without unwanted hydrolysis of the N-acyl group.38
Scheme 6.

Large-scale synthesis of compound 2.
From the result of the synthesis for 2, allyl ester was projected as an advanced intermediate in our synthesis so it was applied to another large-scale synthesis of a 2-propionic acid derivative 30 en route to a more complex sulfonimide derivative (COOH-bioisostere) 31.
Indole 28 was not commercially available and therefore should be prepared from succinyl chloride using Boger’s synthesis.39 Succinyl chloride was treated with ethyldichloroaluminum and allyl alcohol in dichloromethane to provide allyl-4-oxohexanoate 27. Fischer indolization of 27 with 4-methoxyphenyl hydrazine followed by treatment with acetic acid then gave indole compound 28 in 71%. Acylation (87%) and ester cleavage (79%) produced carboxylic acid, 30.15 Finally compound 31 was obtained by amide coupling between methylsulfonylamine and acid 30 using CDI coupling reagent.
3. Conclusions
In summary, we have found that different INDO analogues, including 2′-des-methyl INDO 2 are efficiently prepared in a parallel approach via their N-unsubstituted indole methyl-/allylalkanoate intermediates starting from commercially available arylhydrazine hydrochlorides and a variety of ketoacids/esters. Substituted phenylhydrazine precursors were subjected to Fischer cyclization in a one-pot procedure to provide respective indole products. The N-unsubstituted indole alkanoic acid esters (optimally methyl or allyl esters) were deprotonated and further elaborated by N-acylation followed by aqueous-free ester cleavage in the presence of different metal-based catalyst systems. Concomitant N-deacylation was not observed with the applied technique. The synthetic strategy enabled assembly and variation of all relevant substituents within only three reaction steps. Target compounds were readily formed under optimized settings and could be isolated in good to high overall yields and in acceptable analytical quality. The procedure also allowed for scale-up to afford large amounts of selected compounds of interest for further derivatization reactions and/or direct use in in vitro and in vivo experiments. The range of indole analogs will be evaluated for activity against peroxisome proliferator activated receptor γ, induction of tumor cell apoptosis, and inhibition of aldo-keto reductase enyzmes.10,40
4. Experimental part
All commercial reagents, solvents and other materials were used as received without further purification. Flash chromatography was conducted on a Biotage SP1 automated flash chromatography system equipped with a fixed wavelength UV detector (λ = 254 nm) using prefabricated ‘Flash KP-SIL’ columns (size according to requirements). Thin-layer chromatography was performed on precoated fluorescent silica gel 60 F254 plates (250 um) from Whatman (Partisil® LK6D, Cat. No. 4865-821). Spots were visualized under natural light, and UV illumination at λ = 254 and 365 nm. Microwave reactions were performed in an automated Biotage Initiator Eight Synthesizer. NMR spectra were recorded with a Bruker AV-400 instrument with sample changer (BACS 60) (400 MHz for 1H, 100 MHz for 13C) or a Bruker AV-300 system (282 MHz for 19F) and calibrated with DMSO-d6 as solvent and TMS as internal standard signal. Chemical shifts (δ) are reported in parts per million (ppm). Coupling constants (J) are given in Hz. Low resolution mass spectra (LCMS) were obtained on an Agilent 1200 LCMS system with electrospray ionization. High resolution mass spectra (HRMS) were recorded on a Waters QTof-API-US plus Acquity system with ES as the ion source. Analytical high pressure liquid chromatography (HPLC) was performed on an Agilent 1200 analytical LCMS with UV detection at 214 nm and 254 nm along with ELSD detection. General methods are specified in the Supporting Information. Compounds 9a, 9b, 10 and 17 were prepared according to literature procedures.25,31,41,30
Synthesis of exemplified intermediates and target compounds:
4.1. Methyl 2-(5-fluoro-1H-indol-3-yl)acetate 9e
According to general procedure B_variant2 (conventional heating), (4-fluorophenyl)hydrazine hydrochloride (100 mg, 0.62 mmol), 4-oxobutanoic acid (79 mg, 0.68 mmol) and 160 μL H2SO4 in 2 mL methanol were refluxed for 3 h under argon to afford 110 mg (86%) of 9e as brownish viscous mass. C11H10FNO2, Mr = 207.20; 1H NMR (400 MHz, DMSO-d6) δ: 3.60 (s, 3H), 3.72 (s, 2H), 6.91 (td, J=2.6/9.2 Hz, 1H), 7.22 (dd, J=2.4/10.0 Hz, 1H), 7.31–7.35 (m, 2H); 19F NMR (282 MHz, DMSO-d6) δ: −123.52 (5′-F); LCMS (ESI) tR: 2.16 min (>99%, ELSD), m/z: 208.2 [M+H]+; HRMS (TOF, ES+) C11H10FNO2 [M+H]+ calc. mass 208.0774, found 208.0772.
4.2. Methyl 2-(1H-indazol-3-yl)acetate 10
According to general procedure A_variant2, 2-(1H-indazol-3-yl)acetic acid (300 mg, 1.70 mmol) was refluxed in methanol (15 mL) for 14 h. The reaction mixture was worked up as described to afford 250 mg (77%) of 10. C10H10N2O2, Mr = 190.20; 1H NMR (400 MHz, DMSO-d6) δ: 3.62 (s, 3H), 4.01 (s, 2H), 7.09 (td, J=0.8/7.4 Hz, 1H), 7.33 (td, J=1.0/7.7 Hz, 1H), 7.48 (d, J=8.4 Hz, 1H), 7.69 (d, J=8.4 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ: 33.04 (s, -CH2-), 52.17 (s, -OCH3), 110.49 (s, C7′), 120.29 (s), 120.43 (s), 122.20 (s, C4a′), 126.40 (s, C6′), 138.83 (s, C7a′), 141.20 (s, C3′), 171.03 (s, >C=O); LCMS (ESI) tR: 1.65 min (>99%, UV254), m/z: 191.2 [M+H]+; HRMS (TOF, ES+) C10H10N2O2 [M+H]+ calc. mass 191.0821, found 191.0821.
4.3. Methyl 3-(5-fluoro-3-methyl-1H-indol-2-yl)propanoate 14
According to general procedure B_variant2, the title compound was obtained from (4-fluorophenyl)hydrazine hydrochloride (100 mg, 0.62 mmol), 4-oxohexanoic acid (88.8 g, 0.68 mmol) and 80 μL H2SO4 in 3 mL methanol after 10 min at 120 °C in 46% yield (67 mg) as brownish viscous mass. C13H14FNO2, Mr = 235.25; 1H NMR (400 MHz, DMSO-d6) δ: 2.12 (s, 3H), 2.66 (t, J=8.0 Hz, 2H), 2.93 (t, J=7.6 Hz, 2H), 3.58 (s, 3H), 6.80 (td, J=2.8/9.2 Hz, 1H), 7.10 (dd, J=2.4/10.2 Hz, 1H), 7.20 (dd, J=4.6/8.6 Hz, 1H), 10.74 (bs, 1H); 19F NMR (282 MHz, DMSO-d6) δ: −124.03 (d, 5′-F); LCMS (ESI) tR: 2.58 min (>99%, UV220), m/z: 236.2 [M+H]+; HRMS (TOF, ES+) C13H14FNO2 [M+H]+ calc. mass 236.1087, found 236.1085.
4.4. Methyl 6-methoxy-2,3,4,9-tetrahydro-1H-carbazole-3-carboxylate 15a
According to general procedure B_variant1, the title compound was obtained from (4-Methoxyphenyl)hydrazine hydrochloride (80.0 mg, 0.46 mmol) and methyl 4-oxocyclohexanecarboxylate (85.9 mg, 0.55 mmol) in 0.5 mL glacial acetic acid after 3 h at 80 °C in 90% yield (107 mg) as an off-white solid. C15H17NO3, Mr = 259.30; 1H NMR (400 MHz, DMSO-d6) δ: 1.81–1.91 (m, 1H), 2.15–2.19 (m, 1H), 2.66–2.84 (m, 4H), 2.90 (dd, J=4.6/14.2 Hz, 1H), 3.65 (s, 3H), 3.72 (s, 3H), 6.61 (dd, J=2.4/8.8 Hz, 1H), 6.86 (d, J=2.4 Hz, 1H), 7.11 (d, J=8.8 Hz, 1H), 10.51 (s, 1H); LCMS (ESI) tR: 2.27 min (>99%, ELSD), m/z: 260.2 [M+H]+; HRMS (TOF, ES+) C15H17NO3 [M+H]+ calc. mass 260.1287, found 260.1285.
4.5. Methyl 2-(5-methoxy-1H-pyrrolo[3,2-b]pyridin-3-yl)acetate 17
A 2 mL microwave process vial with a stir bar was charged with crude methyl 3-cyano-3-(6-methoxy-3-nitropyridin-2-yl)propanoate 16 (50 mg, 0.19 mmol), 10% Pd/C (5 Mol%, 20 mg, 0.01 mmol), and methanol (1.5 mL). An excess of 1,4-cyclohexadiene (91 mg, 1.13 mmol) was added and the vessel flooded with argon, capped and heated under microwave conditions at 120 °C for 5 min. The reaction was filtered through Celite® and the solvent was evaporated in vacuo. The crude material was purified by flash chromatography (SiO2, ethyl acetate/hexane gradient) to yield the product as greenish oil (25 mg, 60%). C11H12N2O3, Mr = 220.22; 1H NMR (400 MHz, DMSO-d6) δ: 3.61 (s, 3H), 3.72 (s, 2H), 3.83 (s, 3H), 6.53 (d, J=8.8 Hz, 1H), 7.38 (d, J=2.8 Hz, 1H), 7.66 (d, J=8.4 Hz, 1H), 11.01 (bs, 1H); 13C NMR (100 MHz, DMSO-d6) δ: 29.41 (s, -CH2-), 51.86 (s, -C(O)OCH3), 52.83 (s, -OCH3), 104.86 (s, C6′), 107.19 (s, C3′), 122.78 (s, C2′), 124.60 (s, C7a′), 126.80 (s, C7′), 141.29 (s, C3a′), 159.16 (s, C5′), 172.47 (s, >C=O); LCMS (ESI) tR: 0.69 min (>97%, UV220, ELSD), m/z: 221.2 [M+H]+; HRMS (TOF, ES+) C11H12N2O3 [M+H]+ calc. mass 221.0926, found 221.0926.
4.6. Methyl 2-(5-methoxy-2-methyl-1-(4-methylbenzoyl)-1H-indol-3-yl)acetate 18d
According to general procedure C_variant2, the title compound was obtained from from methyl 2-(5-methoxy-2-methyl-1H-indol-3-yl)acetate (80 mg, 0.34 mmol), 4-methylbenzoyl chloride (63.6 mg, 0.41 mmol) and tBuONa (2 M in THF, 206 μL, 0.41 mmol) in anhydrous THF (2.5 mL). The crude residue was subjected to flash chromatography (SiO2, ethyl acetate/hexane gradient) to afford the pure title compound in 57% yield (68.5 mg). C21H21NO4, Mr = 351.40; 1H NMR (400 MHz, DMSO-d6) δ: 2.22 (s, 3H), 2.42 (s, 3H), 3.62 (s, 3H), 3.74 (s, 3H), 3.77 (s, 2H), 6.68 (dd, J=2.6/9.0 Hz, 1H), 6.85 (d, J=8.8 Hz, 1H), 7.01 (d, J=2.4 Hz, 1H), 7.37 (d, J=8.0 Hz, 2H), 7.54 (pseudo-d, J=8.4 Hz, 2H); LCMS (ESI) tR: 2.74 min (>95%, ELSD), m/z: 352.2 [M+H]+; HRMS (TOF, ES+) C21H21NO4 [M+H]+ calc. mass 352.1549, found 352.1548.
4.7. Methyl 3-(1-(4-chlorobenzoyl)-5-fluoro-2-methyl-1H-indol-3-yl)propanoate 18o
According to general procedure C_variant2, the title compound was obtained from methyl 3-(5-fluoro-2-methyl-1H-indol-3-yl)propanoate (80 mg, 0.34 mmol), 4-chlorobenzoyl chloride (71.4 mg, 0.41 mmol) and tBuONa (2 M in THF, 204 μL, 0.41 mmol) in anhydrous THF (2.5 mL). The crude residue was subjected to flash chromatography (SiO2, ethyl acetate/hexane gradient) to afford the pure title compound as yellow oil in 49% yield (62 mg). C20H17ClFNO3, Mr = 373.81; 1H NMR (400 MHz, DMSO-d6) δ: 2.19 (s, 3H), 2.59 (t, J=7.4 Hz, 2H), 2.92 (t, J=7.6 Hz, 2H), 3.56 (s, 3H), 6.94 (td, J=2.4/9.2 Hz, 1H), 7.08 (dd, J=4.6/9.0 Hz, 1H), 7.39 (dd, J=2.4/9.2 Hz, 1H), 7.63–7.68 (m, 4H); LCMS (ESI) tR: 3.21 min (>95%, ELSD), m/z: 374.3 [M+H]+; HRMS (TOF, ES+) C20H17ClFNO3 [M+H]+ calc. mass 374.0959, found 374.0962.
4.8. Methyl 6-methoxy-9-(3-(trifluoromethyl)benzoyl)-2,3,4,9-tetrahydro-1H-carbazole-3-carboxylate 19a
According to general procedure C_variant2, the title compound was obtained from from methyl 6-methoxy-2,3,4,9-tetrahydro-1H-carbazole-3-carboxylate (80 mg, 0.31 mmol), 3-(trifluoromethyl) benzoyl chloride (77.2 mg, 0.37 mmol) and tBuONa (2 M in THF, 185 μL, 0.37 mmol) in anhydrous THF (2.5 mL). The crude residue was subjected to flash chromatography (SiO2, ethyl acetate/hexane gradient) to afford the pure title compound in 24% yield (32 mg). C23H20F3NO4, Mr = 431.40; 1H NMR (400 MHz, DMSO-d6) δ: 1.70–1.80 (m, 1H), 2.03–2.07 (m, 1H), 2.47 (m, 2H, partly overlayed by DMSO signal), 2.73–2.79 (m, 1H), 2.82–2.89 (m, 1H), 2.94 (dd, J=5.0/15.8 Hz, 1H), 3.65 (s, 3H), 3.77 (s, 3H), 6.73 (dd, J=2.6/9.0 Hz, 1H), 7.04 (d, J=2.4 Hz, 1H), 7.08 (d, J=9.2 Hz, 1H), 7.79 (t, J=8.0 Hz, 1H), 7.93 (d, J=8.0 Hz, 1H), 7.99 (s, 1H), 8.04 (d, J=8.0 Hz, 1H); 19F NMR (282 MHz, DMSO-d6) δ: −59.41 (m-CF3); LCMS (ESI) tR: 0.95 min (>95%, UV254), m/z: 432.0 [M+H]+; HRMS (TOF, ES+) C23H20F3NO4 [M+H]+ calc. mass 432.1423, found 432.1422.
4.9. Methyl 2-(1-(4-chlorobenzoyl)-1H-indazol-3-yl)acetate 21
According to general procedure C_variant2, methyl 2-(1H-indazol-3-yl)acetate (80 mg, 0.42 mmol) was subjected to reaction with 4-chlorobenzoyl chloride (88.3 mg, 0.50 mmol) (252 μL tBuONa). The crude product was purified by flash chromatography (SiO2, ethyl acetate/hexane gradient) to afford 13.5 mg (52%) of the title compound as bright yellow solid. The compound permanently crystallized upon drying at high vacuum and storage at −20 °C. Yield: 51 mg (37%). C17H13ClN2O3, Mr = 328.75; 1H NMR (400 MHz, DMSO-d6) δ: 3.65 (s, 3H), 4.16 (s, 2H), 7.50 (td, J=0.8/7.6 Hz, 1H), 7.63–7.66 (m, 2H), 7.71 (td, J=1.0/7.6 Hz, 1H), 7.91 (d, J=8.0 Hz, 1H), 8.00–8.03 (m, 2H), 8.41 (d, J=8.4 Hz, 1H); LCMS (ESI) tR: 2.80 min (>99%, ELSD), m/z: 329.0 [M+H]+; HRMS (TOF, ES+) C17H13ClN2O3 [M+H]+ calc. mass 329.0693, found 329.0693.
4.10. 2-(1-(4-(chloromethyl)benzoyl)-5-methoxy-2-methyl-1H-indol-3-yl)acetic acid 22e
According to general procedure D, methyl 2-(1-(4-(chloromethyl)benzoyl)-5-methoxy-2-methyl-1H-indol-3-yl)acetate (15 mg, 0.039 mmol) was subjected to reaction with trimethyltin hydroxide (35 mg, 0.194 mmol) in 1,2-DCE (1 mL). The crude product was chromatographed on a short silica gel column using 1:1 ethyl acetate/hexane (0.5% AcOH) mixtures of increasing polarity as eluants to afford the title compound quantitatively as off-white solid. Yield: 14.1 mg (98%). C20H18ClNO4, Mr = 371.81; 1H NMR (400 MHz, DMSO-d6) δ: 2.19 (s, 3H), 3.65 (bs, 2H), 3.75 (s, 3H), 4.88 (s, 2H), 6.68 (dd, J=2.4/9.2 Hz, 1H), 6.90 (d, J=8.8 Hz, 1H), 7.02 (d, J=2.4 Hz, 1H), 7.61–7.67 (m, 4H); 13C NMR (125 MHz, DMSO-d6) δ: 13.34 (s), 29.93 (s), 45.23 (s), 55.72 (s), 101.14 (s), 111.68 (s), 115.08 (s), 128.79 (s, 2C), 130.14 (s, 2C), 130.41 (s), 130.85 (s), 135.38 (s), 136.27 (s), 142.26 (s), 156.02 (s), 168.79 (s), 176.3 (s), one signal invisible; LCMS (ESI) tR: 2.48 min (>99%, UV220, UV254), m/z: 372.0 [M+H]+; HRMS (TOF, ES+) C20H18ClNO4 [M+H]+ calc. mass 372.1003, found 372.1003.
4.11. 2-(1-(4-chlorobenzoyl)-5-fluoro-1H-indol-3-yl)acetic acid 22n
According to general procedure D, methyl 2-(1-(4-chlorobenzoyl)-5-fluoro-1H-indol-3-yl)acetate (15 mg, 0.043 mmol) was subjected to reaction with trimethyltin hydroxide (39 mg, 0.216 mmol) in 1,2-DCE (1 mL). The crude product was chromatographed on a short silica gel column using 1:1 ethyl acetate/hexane (0.5% AcOH) mixtures of increasing polarity as eluants to afford the title compound quantitatively as off-white solid. Yield: 11.2 mg (78%). C17H11ClFNO3, Mr = 331.73; 1H NMR (400 MHz, DMSO-d6) δ: 3.66 (bs, 2H), 7.23 (td, J=2.6/9.2 Hz, 1H), 7.40–7.43 (m, 2H), 7.65–7.69 (m, 2H), 7.75–7.79 (m, 2H), 8.28 (dd, J=4.8/9.2 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ: 30.62 (s), 104.99 (d, J=24.3 Hz, 1C), 113.34 (d, J=24.9 Hz, 1C), 113.92 (s), 117.76 (d, J=8.9 Hz, 1C), 127.35 (s), 129.08 (s, 2C), 130.57 (s, 2C), 131.38 (d, J=9.7 Hz, 1C), 132.37 (s), 132.47 (s), 138.58 (s), 159.96 (d, J=242 Hz, 1C), 167.09 (s), 175.85 (s); 19F NMR (282 MHz, DMSO-d6) δ: −116.72 (5′-F); LCMS (ESI) tR: 2.54 min (>99%, UV215, UV254, ELSD), m/z: 332.0 [M+H]+; HRMS (TOF, ES+) C17H11ClFNO3 [M+H]+ calc. mass 332.0490, found 332.0490.
4.12. 6-methoxy-9-(3-(trifluoromethyl)benzoyl)-2,3,4,9-tetrahydro-1H-carbazole-3-carboxylic acid 23
According to general procedure D, methyl 6-methoxy-9-(3-(trifluoromethyl)benzoyl)-2,3,4,9-tetrahydro-1H-carbazole-3-carboxylate (15 mg, 0.035 mmol; ~60% pure) was subjected to reaction with trimethyltin hydroxide (31 mg, 0.174 mmol) in 1,2-DCE (1 mL). The crude product was chromatographed on a short silica gel column using 1:1 ethyl acetate/hexane (0.5% AcOH) mixtures of increasing polarity as eluants to afford the title compound quantitatively as bright yellow solid. Yield: 8.5 mg (98%). C22H18F3NO4, Mr = 417.38; 1H NMR (400 MHz, DMSO-d6) δ: 1.69–1.75 (m, 1H), 2.02–2.06 (m, 1H), ~2.47 (m, 2H, partially overlaid by DMSO signal), 2.71–2.74 (m, 2H), 2.89–2.92 (m, 1H), 3.78 (s, 3H), 6.73 (dd, J=2.6/9.0 Hz, 1H), 7.03 (d, J=2.4 Hz, 1H), 7.10 (d, J=9.2 Hz, 1H), 7.78 (t, J=7.8 Hz, 1H), 7.93 (d, J=7.6 Hz, 1H), 7.99 (s, 1H), 8.03 (d, J=7.6 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ: 23.51 (s), 24.73 (s), 25.81 (s), 38.74 (s), 55.68 (s), 101.09 (s), 111.96 (s), 115.49 (s), 116.78 (s), 123.48 (d, J=272 Hz, 1C), 126.18 (s), 128.81 (s), 129.34 (s), 130.63 (s), 130.95 (s), 131.37 (d, J=29.0 Hz, 1C), 132.42 (s), 135.55 (s), 136.53 (s), 156.35 (s), 167.29 (s), 179.92 (s); 19F NMR (282 MHz, DMSO-d6) δ: −59.38 (m-CF3); LCMS (ESI) tR: 2.74 min (>99%, UV254, ELSD), m/z: 418.0 [M+H]+; HRMS (TOF, ES+) C22H18F3NO4 [M+H]+ calc. mass 418.1266, found 418.1267.
4.13. Allyl 2-(5-methoxy-1H-indol-3-yl)acetate 25
To a solution of 2-(5-methoxy-1H-indol-3-yl)acetic acid 7b (15.25 g, 74.3 mmol) in 250 mL of acetone was added 29 g of cesium carbonate (89.2 mmol) and allyl bromide (7.1 mL, 81.74 mmol). The reaction mixture was stirred at room temperature for 12 h. The residual cesium carbonate was removed by filtration and solvent was concentrated in vacuo. The residue was purified by column chromatography using Hex/EtOAc (gradient: 0 to 30% EtOAc) to afford allyl 2-(5-methoxy-1H-indol-3-yl)acetate 25 (13.6 g, 75 %). C14H15NO3, Mr = 245.27; 1H NMR (400 MHz, CDCl3) δ 3.8 (s, 2H), 3.89 (s, 3H), 4.64 (dt, J=1.2, 5.7, 2H), 5.2 (dd, J=1.2, 10.4, 1H), 5.32 (dd, J=1.5, 17.2, 1H), 6.0-5.90 (m, 1H), 6.89 (dd, J=2.4, 8.8 Hz, 1H), 7.09 (d, J=2.4 Hz, 1H), 7.17 (s, 1H), 7.26 (d, J=8.8 Hz, 1H).
4.14. Allyl 2-(1-(4-chlorobenzoyl)-5-methoxy-1H-indol-3-yl)acetate 26
To a solution of allyl 2-(5-methoxy-1H-indol-3-yl)acetate 25 (10 g, 40.8 mmol) in dichloromethane (150 mL) was added triethylamine (17.1 mL, 122. 4 mmol) and DMAP (4.9 g, 40.8 mmol) at 0°C. After stirring 30 min, 4-chlorobenzoyl chloride (7.9 mL, 61.2 mmol) was added to the reaction mixture, which was allowed to warm to room temperature and stirred for 12 h. The reaction mixture was then quenched with saturated ammonium chloride (200 mL), extracted with dichloromethane (3 × 150 mL), dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography using Hex/EtOAc (gradient: 0 to 30% EtOAc) to afford a yellow solid of allyl 2-(1-(4-chlorobenzoyl)-5-methoxy-1H-indol-3-yl)acetate 26 (14.3 g, 92 %). C21H18ClNO4, Mr = 383.82; 1H NMR (400 MHz, CDCl3) δ 3.69 (s, 2H), 3.88 (s, 3H), 4.62 (d, J=6.0 Hz, 2H), 5.23 (dd, J=1.2, 10.4 Hz, 1H), 5.29 (dd, J=1.2, 17.2 Hz, 1H), 5.95-5.84 (m, 1H), 7.01 (d, J=7.6 Hz, 2H), 7.25 (s, 1H), 7.50 (d, J=8.4 Hz, 2H), 7.67 (d, J=8.4 Hz, 2H), 8.28 (d, J=10.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ: 30.97 (s), 55.70 (s), 65.74 (s), 101.93 (s), 113.76 (s), 114.68 (s), 117.39 (s), 118.69 (s), 126.31 (s), 128.92 (s, 2C), 130.50 (s, 2C), 130.73 (s), 131.43 (s), 131.78 (s), 132.85 (s), 138.15 (s), 156.89 (s), 166.96 (s), 170.25 (s); LCMS (ESI), single peak, tR: 0.92 min (>99%, UV, ELSD), m/z:, 384.0 [M+H]+; HRMS (TOF, ES+) C21H18ClNO4 [M+H]+ calc. mass 384.1003, found 384.1003.
4.15. 2-(1-(4-chlorobenzoyl)-5-methoxy-1H-indol-3-yl)acetic acid 2
To a solution of allyl 2-(1-(4-chlorobenzoyl)-5-methoxy-1H-indol-3-yl)acetate 3 (14.3 g, 37.3 mmol) in THF (200 mL) was added morpholine (32.5 mL, 373 mmol) and Pd(PPh3)4 (2.3 g, 1.9 mmol) under argon at room temperature. Stirring was continued until LCMS analysis indicated starting material has disappeared. The mixture was filtered and concentrated in vacuo. The residue was redissolved in dichloromethane and acidified with 2N HCl (50 mL) to afford white solid 2 (10 g, 79 %). C18H14ClNO4, Mr = 343.76; 1H NMR (400 MHz, DMSO-d6) δ: 3.67 (s, 2H), 3.81 (s, 3H), 6.99 (dd, J=2.4/9.2 Hz, 1H), 7.12 (d, J=2.4 Hz, 1H), 7.32 (s, 1H), 7.65–7.77 (m, 4H), 8.17 (d, J=9.2 Hz, 1H); ); 13C NMR (100 MHz, DMSO-d6) δ: 30.15 (s), 55.47 (s), 102.70 (s), 113.05 (s), 115.70 (s), 116.70 (s), 126.84 (s), 128.82 (s, 2C), 129.99 (s), 130.69 (s, 2C), 131.87 (s), 133.00 (s), 136.57 (s), 156.30 (s), 166.60 (s), 171.97 (s); LCMS (ESI) tR: 2.60 min (>99%, ELSD), m/z: 344.0 [M+H]+; HRMS (TOF, ES+) C18H14ClNO4 [M+H]+ calc. mass 344.0690, found 344.0687.
4.16. Allyl 4-oxohexanoate 27
Succinyl dichloride 4 (4.6 mL, 41.6 mmol) in dichloromethane (200 mL) was cooled to −40 °C and ethylaluminum dichloride (50 mL, 50 mmol) was added at −40 °C. The reaction mixture was stirred at −40 °C for 3.5 h and quenched with the addition of anhydrous allyl alcohol (100 mL). The reaction mixture was concentrated to dryness and redissolved in dichloromethane (100 mL). The organic layer was washed with aqueous sodium potassium tartrate (2 × 50 mL), water (50 mL) and saturated aqueous NaCl (50 mL). The organic layer was dried over MgSO4, filtered and concentrated to provide pure allyl 4-oxohexanoate 27 (5.2 g, 73 %). C9H14O3, Mr = 170.21; 1H NMR (400 MHz, CDCl3) δ 1.07 (t, J=7.2 Hz, 3H), 2.47 (q, J=7.2 Hz, 2H), 2.62 (t, J=6.4 Hz, 2H), 2.73 (t, J=6.4 Hz, 2H), 4.57 (d, J=5.6 Hz, 2H), 5.23 (dd, J=1.2, 10.4 Hz, 1H), 5.31 (dd, J=1.2, 18.0 Hz, 1H), 5.96–5.84 (m, 1H).
4.17. Allyl 3-(5-methoxy-3-methyl-1H-indol-2-yl)propanoate 28
To a mixture of (4-methoxyphenyl)hydrazine (10.4 g, 59.7 mmol) and allyl 4-oxohexanoate 5 (10.2 g, 59.7 mmol) in 1,4-dioxane (60 mL) was added acetic acid (30 mL) at room temperature. The reaction was heated at 80 °C for 6 h then quenched with saturated NaHCO3 (150 mL), extracted with EtOAc (3 × 60 mL), and dried over MgSO4. The residue was purified by column chromatography using Hex/EtOAc (gradient: 0 to 40 % EtOAc) to afford brown oil allyl 3-(5-methoxy-3-methyl-1H-indol-2-yl)propanoate 28 (11.6 g, 71 %). C16H19NO3, Mr = 273.33; 1H NMR (400 MHz, CDCl3) δ 2.21 (s, 3H), 2.68 (t, J=6.4 Hz, 2H), 3.03 (t, J=6.4 Hz, 2H), 3.86 (s, 3H), 4.60 (d, J=7.2 Hz, 2H), 5.23 (dd, J=1.2, 10.4 Hz, 1H), 5.29 (dd, J=1.2, 17.2 Hz, 1H), 5.95-5.83 (m, 1H), 6.78 (dd, J=2.4, 8.8 Hz, 1H), 6.93 (d, J=2.4 Hz, 1H), 7.16 (d, J = 8.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ: 8.43 (s), 20.91 (s), 34.09 (s), 55.95 (s), 65.47 (s), 100.50 (s), 106.91 (s), 111.04 (s), 111.08 (s), 118.54 (s), 129.32 (s), 130.39 (s), 131.85 (s), 134.45 (s), 153.73 (s), 173.45 (s); LCMS (ESI), single peak, tR: 0.79 min, m/z: 274.0 [M+H]+; HRMS (TOF, ES+) C16H19NO3 [M+H]+ calc. mass 274.1443, found 274.1441.
4.18. Allyl 3-(1-(4-chlorobenzoyl)-5-methoxy-3-methyl-1H-indol-2-yl)propanoate 29
To a solution of allyl 3-(5-methoxy-3-methyl-1H-indol-2-yl)propanoate 28 (16 g, 61.8 mmol) in 1,2-dichloroethane (200 mL) was added triethylamine (34.5 mL, 247.2 mmol) and DMAP (7.55 g, 61.8 mmol) at 0°C. After stirring for 30 min, 4-chlorobenzoyl chloride (19.7 mL, 154.6 mmol) was added to the reaction mixture, which was allowed to stir for 12 h under reflux condition. The reaction mixture was cooled down, quenched with saturated ammonium chloride (200 mL), extracted with dichloromethane (3 × 150 mL), dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography using Hex/EtOAc (gradient: 0 to 30% EtOAc) to afford brown oil of allyl 3-(1-(4-chlorobenzoyl)-5-methoxy-3-methyl-1H-indol-2-yl)propanoate 29 (22.2 g, 87 %). C23H22ClNO4, Mr = 411.88; 1H NMR (400 MHz, CDCl3) δ 2.24 (s, 3H), 2.69 (t, J=7.6 Hz, 2H), 3.29 (t, J=7.6 Hz, 2H), 3.83 (s, 3H), 4.53 (d, J=6.0 Hz, 2H), 5.18 (dd, J=1.2, 10.4 Hz, 1H), 5.25 (dd, J=1.2, 17.2 Hz, 1H), 5.93-5.78 (m, 1H), 6.44 (d, J=9.2 Hz, 1H), 6.59 (dd, J=2.8, 8.4 Hz, 1H), 6.89 (d, J=2.8 Hz, 1H), 7.46 (d, J=8.4 Hz, 2H), 7.65 (d, J=8.4 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 8.67 (s), 21.67 (s), 34.14 (s), 55.65 (s), 65.18 (s), 101.38 (s), 111.48 (s), 114.78 (s), 116.38 (s), 118.28 (s), 129.10 (s, 2C), 129.33 (s), 130.76 (s), 131.15 (s, 2C), 131.66 (s), 131.84 (s), 132.06 (s), 133.88 (s), 136.71 (s), 139.15 (s), 155.77 (s), 168.13 (s), 172.23 (s); LCMS (ESI), single peak, tR: 0.99 min, m/z, 412.0 [M+H]+; HRMS (TOF, ES+) C23H22ClNO4 [M+H]+ calc. mass 412.1316, found 412.1320.
4.19. 3-(1-(4-chlorobenzoyl)-5-methoxy-3-methyl-1H-indol-2-yl)propanoic acid 30
To a solution of allyl 3-(1-(4-chlorobenzoyl)-5-methoxy-3-methyl-1H-indol-2-yl)propanoate 29 (11.1 g, 26.9 mmol) in THF (200 mL) was added morpholine (23.0 mL, 269 mmol) and Pd(PPh3)4 (1.67 g, 1.35 mmol) under argon at room temperature. Stirring was continued until LCMS analysis indicated starting material has disappeared. The mixture was filtered and concentrated in vacuo. The residue was redissolved in dichloromethane, acidified with 2N HCl (50 mL), concentrated in vacuo, and purified by column chromatography using Hex/EtOAc (gradient: 0 to 100 % EtOAc) to afford yellow solid of 3-(1-(4-chlorobenzoyl)-5-methoxy-3-methyl-1H-indol-2-yl)propanoic acid 30 (7.9 g, 79%). C20H18ClNO4, Mr = 371.81; 1H NMR (400 MHz, CDCl3,) δ 2.24 (s, 3H), 2.72 (t, J=7.6 Hz, 2H), 3.29 (t, J=7.6 Hz, 2H), 3.83 (s, 3H), 6.42 (d, J=9.2 Hz, 1H), 6.59 (dd, J=2.8, 9.2 Hz, 1H), 6.90 (d, J=2.8 Hz, 1H), 7.45 (d, J=8.4 Hz, 2H), 7.64 (d, J=8.4 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ: 8.40 (s), 21.24 (s), 33.74 (s), 55.42 (s), 101.55 (s), 111.58 (s), 114.38 (s), 115.74 (s), 129.15 (s, 2C), 130.26 (s), 131.24 (s, 2C), 134.04 (s), 136.72 (s), 137.78 (s), 155.46 (s), 167.77 (s), 173.40 (s), one signal invisible; LCMS (ESI), single peak, tR: 0.82 min, m/z:, 372.0 [M+H]+; HRMS (TOF, ES+) C20H18ClNO4 [M+H]+ calc. mass 372.1003, found 372.1005.
4.20. 3-(1-(4-chlorobenzoyl)-5-methoxy-3-methyl-1H-indol-2-yl)-N-(methylsulfonyl)propanamide 31
The reaction mixture of 3-(1-(4-chlorobenzoyl)-5-methoxy-3-methyl-1H-indol-2-yl)propanoic acid 30 (7 g, 18.8 mmol) and methanesulfonamide (1.97 g, 20.7 mmol) in dichloromethane (94 mL) was treated with 1,1′-carbonyldiimidazole (3.05 g, 18.8 mmol) and DBU (3.4 mL, 22.6 mmol) at 0°C. The reaction mixture was stirred at room temperature for 4h then quenched with acetic acid (5 mL) and washed with brine (3 × 40 mL). The organic layer was then dried over MgSO4 and the solvent was removed in vacuo. The yellow solid 31 (5.6 g, 61 %) was obtained after column chromatography purification using Hex/EtOAc (gradient: 0 to 100 % EtOAc with 0.5 % of acetic acid). C21H21ClN2O5S, Mr = 170.21; 1H NMR (400 MHz, CDCl3) δ 2.20 (s, 3H), 2.54 (dd, J=7.2, 7.6 Hz, 2H), 3.10 (dd, J=7.2, 7.6 Hz, 2H), 3.14 (s, 3H), 3.75 (s, 3H), 6.42 (d, J=9.2 Hz, 1H), 7.70-7.64 (m, 4H), 6.63 (dd, J=2.4, 9.2 Hz, 1H), 7.01 (d, J=2.4 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ: 8.42 (s), 20.57 (s), 35.30 (s), 40.96 (s), 55.42 (s), 101.59 (s), 111.66 (s), 114.45 (s), 116.01 (s), 129.13 (s, 2C), 130.25 (s), 131.20 (s), 131.27 (s, 2C), 134.00 (s), 136.31 (s), 137.77 (s), 155.48 (s), 167.74 (s), 171.42 (s); LCMS (ESI), single peak, 0.81 min, m/z, 449.08 [M+H]+; HRMS (TOF, ES+) C21H21ClN2O5S [M+H]+ calc. mass 449.0938, found 449.0941.
Supplementary Material
Scheme 7.

Large-scale synthesis of compounds 30 (free acid) and 31 (COOH bioisostere).
Acknowledgments
The authors are grateful to the National Institutes of Health (CA89450) and the National Foundation for Cancer Research for financial support. We thank Dr. Matthew Mulder (Craig Lindsley Group) for recording all high-resolution mass spectra and Christian Wallen (2011 Vanderbilt REU Trainee) for the assistance with the parallel syntheses of the fluorinated indole precursors. A.J.L. also thanks the German Research Foundation (DFG) for obtaining a Research Stipend (LI2019/1-1, Einzelprojekt).
Footnotes
Detailed synthetic procedures, routine spectroscopic and spectrometric data and HPLC data, as well as exemplified 1H and 13C NMR spectra of intermediates and final compounds are available.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Bartolini W, Cali BM, Chen B, Chien Y-T, Currie MG, Milne GT, Pearson JP, Talley JJ, Zimmerman C. Application: US US. USA: 2005. p. 118. Cont -in-part of U S Ser No 883, 900. [Google Scholar]
- 2.Munoz B, Prasit P, Stock NS. Application: WO WO. Merck & Co., Inc; USA: 2004. p. 40. [Google Scholar]
- 3.Torisu K, Kobayashi K, Iwahashi M, Egashira H, Nakai Y, Okada Y, Nanbu F, Ohuchida S, Nakai H, Toda M. Bioorganic & Medicinal Chemistry Letters. 2004;14:4557–4562. doi: 10.1016/j.bmcl.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 4.Torisu K, Kobayashi K, Iwahashi M, Egashira H, Nakai Y, Okada Y, Nanbu F, Ohuchida S, Nakai H, Toda M. European Journal of Medicinal Chemistry. 2005;40:505–519. doi: 10.1016/j.ejmech.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 5.Wey SJ, Augustyniak ME, Cochran ED, Ellis JL, Fang X, Garvey DS, Janero DR, Letts LG, Martino AM, Melim TL, Murty MG, Richardson SK, Schroeder JD, Selig WM, Trocha AM, Wexler RS, Young DV, Zemtseva IS, Zifcak BM. Journal of Medicinal Chemistry. 2007;50:6367–6382. doi: 10.1021/jm0611861. [DOI] [PubMed] [Google Scholar]
- 6.Blobaum AL, Marnett LJ. Journal of Medicinal Chemistry. 2007;50:1425–1441. doi: 10.1021/jm0613166. [DOI] [PubMed] [Google Scholar]
- 7.Rainsford KD. Sub-cellular biochemistry. 2007;42:3–27. doi: 10.1007/1-4020-5688-5_1. [DOI] [PubMed] [Google Scholar]
- 8.Wallace JL, McKnight W, Reuter BK, Vergnolle N. Gastroenterology. 2000;119:706–714. doi: 10.1053/gast.2000.16510. [DOI] [PubMed] [Google Scholar]
- 9.Sandham DA, Adcock C, Bala K, Barker L, Brown Z, Dubois G, Budd D, Cox B, Fairhurst RA, Furegati M, Leblanc C, Manini J, Profit R, Reilly J, Stringer R, Schmidt A, Turner KL, Watson SJ, Willis J, Williams G, Wilson C. Bioorg Med Chem Lett. 2009;19:4794–8. doi: 10.1016/j.bmcl.2009.06.042. [DOI] [PubMed] [Google Scholar]
- 10.Felts AS, Ji C, Stafford JB, Crews BC, Kingsley PJ, Rouzer CA, Washington MK, Subbaramaiah K, Siegel BS, Young SM, Dannenberg AJ, Marnett LJ. ACS Chemical Biology. 2007;2:479–483. doi: 10.1021/cb700077z. [DOI] [PubMed] [Google Scholar]
- 11.Byrns MC, Steckelbroeck S, Penning TM. Biochemical Pharmacology. 2008;75:484–493. doi: 10.1016/j.bcp.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Piazza GA, Keeton AB, Tinsley HN, Whitt JD, Gary BD, Mathew B, Singh R, Grizzle WE, Reynolds RC. Pharmaceuticals. 2010;3:1652–1667. doi: 10.3390/ph3051652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schuligoi R, Sturm E, Luschnig P, Konya V, Philipose S, Sedej M, Waldhoer M, Peskar BA, Heinemann A. Pharmacology. 2010;85:372–82. doi: 10.1159/000313836. [DOI] [PubMed] [Google Scholar]
- 14.Hata AN, Lybrand TP, Marnett LJ, Breyer RM. Mol Pharmacol. 2005;67:640–7. doi: 10.1124/mol.104.007971. [DOI] [PubMed] [Google Scholar]
- 15.Marnett LJ, Prusakiewicz JJ, Felts AS, Ji C. Application: US US. Vanderbilt University; USA: 2005. p. 63. [Google Scholar]
- 16.Prusakiewicz JJ, Felts AS, Mackenzie BS, Marnett LJ. Biochemistry. 2004;43:15439–15445. doi: 10.1021/bi048534q. [DOI] [PubMed] [Google Scholar]
- 17.Mukai C, Takahashi Y. Organic Letters. 2005;7:5793–5796. doi: 10.1021/ol052179u. [DOI] [PubMed] [Google Scholar]
- 18.Maki BE, Scheidt KA. Organic Letters. 2009;11:1651–1654. doi: 10.1021/ol900306v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Campos KR, Woo JCS, Lee S, Tillyer RD. Organic Letters. 2004;6:79–82. doi: 10.1021/ol036113f. [DOI] [PubMed] [Google Scholar]
- 20.Takahashi H, Wakamatsu S, Tabata H, Oshitari T, Harada A, Inoue K, Natsugari H. Organic Letters. 2011;13:760–763. doi: 10.1021/ol103008d. [DOI] [PubMed] [Google Scholar]
- 21.Yamamoto H. Journal of Organic Chemistry. 1967;32:3693–5. [Google Scholar]
- 22.Yamamoto H. Chem Pharm Bull (Tokyo) 1968;16:17–9. doi: 10.1248/cpb.16.17. [DOI] [PubMed] [Google Scholar]
- 23.Maguire AR, Plunkett SJ, Papot S, Clynes M, O’Connor R, Touhey S. Bioorganic & Medicinal Chemistry. 2001;9:745–762. doi: 10.1016/s0968-0896(00)00292-3. [DOI] [PubMed] [Google Scholar]
- 24.Nicolaou KC, Estrada AA, Zak M, Lee SH, Safina BS. Angewandte Chemie, International Edition. 2005;44:1378–1382. doi: 10.1002/anie.200462207. [DOI] [PubMed] [Google Scholar]
- 25.Kalgutkar AS, Crews BC, Saleh S, Prudhomme D, Marnett LJ. Bioorganic & Medicinal Chemistry. 2005;13:6810–6822. doi: 10.1016/j.bmc.2005.07.073. [DOI] [PubMed] [Google Scholar]
- 26.Conn RSE, Douglas AW, Karady S, Corley EG, Lovell AV, Shinkai I. Journal of Organic Chemistry. 1990;55:2908–13. [Google Scholar]
- 27.Allen GR., Jr J Heterocycl Chem. 1970;7:239–41. [Google Scholar]
- 28.Karthikeyan SV, Perumal S, Shetty KA, Yogeeswari P, Sriram D. Bioorganic & Medicinal Chemistry Letters. 2009;19:3006–3009. doi: 10.1016/j.bmcl.2009.04.029. [DOI] [PubMed] [Google Scholar]
- 29.Vasudevan A, Souers AJ, Freeman JC, Verzal MK, Gao J, Mulhern MM, Wodka D, Lynch JK, Engstrom KM, Wagaw SH, Brodjian S, Dayton B, Falls DH, Bush E, Brune M, Shapiro RD, Marsh KC, Hernandez LE, Collins CA, Kym PR. Bioorganic & Medicinal Chemistry Letters. 2005;15:5293–5297. doi: 10.1016/j.bmcl.2005.08.049. [DOI] [PubMed] [Google Scholar]
- 30.Jeanty M, Suzenet F, Guillaumet G. Journal of Organic Chemistry. 2008;73:7390–7393. doi: 10.1021/jo8011623. [DOI] [PubMed] [Google Scholar]
- 31.Mylari BL, Zembrowski WJ. Application: EP EP. Pfizer Inc; USA: 1989. p. 15. [Google Scholar]
- 32.Honba K, Yamada T, Yoneda M, Ohkawa Y, Nagai H. Iyakuhin Kenkyu. 1982;13:323–33. [Google Scholar]
- 33.Guibe F. Tetrahedron. 1998;54:2967–3042. [Google Scholar]
- 34.Tsuji J, Mandai T. Synthesis. 1996:1–24. [Google Scholar]
- 35.Goossen LJ, Doehring A. Synlett. 2004:263–266. [Google Scholar]
- 36.Lee JC, Oh YS, Cho SH, Lee J., II Org Prep Proced Int. 1996;28:480–483. [Google Scholar]
- 37.Anderson GW, Callahan FM. J Am Chem Soc. 1960;82:3359–63. [Google Scholar]
- 38.Kunz H, Waldmann H. Helv Chim Acta. 1985;68:618–22. [Google Scholar]
- 39.Choi Y, Ishikawa H, Velcicky J, Elliott GI, Miller MM, Boger DL. Organic Letters. 2005;7:4539–4542. doi: 10.1021/ol051975x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adeniji AO, Twenter BM, Byrns MC, Jin Y, Chen M, Winkler JD, Penning TM. J Med Chem. 2012;55:2311–23. doi: 10.1021/jm201547v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mylari BL, Zembrowski WJ. Application: US US. Pfizer Inc; USA: 1993. p. 8. Cont of U S Ser No 499, 279, abandoned. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.