

The crystal structure of ribose 5-phosphate isomerase has been determined to 1.72 Å resolution and is presented with a brief comparison to other known ribose 5-phosphate isomerase A structures.
Keywords: ribose 5-phosphate isomerase, in situ diffraction, Lactobacillus salivarius
Abstract
The structure of ribose 5-phosphate isomerase from the probiotic bacterium Lactobacillus salivarius UCC188 has been determined at 1.72 Å resolution. The structure was solved by molecular replacement, which identified the functional homodimer in the asymmetric unit. Despite only showing 57% sequence identity to its closest homologue, the structure adopted the typical α and β d-ribose 5-phosphate isomerase fold. Comparison to other related structures revealed high homology in the active site, allowing a model of the substrate-bound protein to be proposed. The determination of the structure was expedited by the use of in situ crystallization-plate screening on beamline I04-1 at Diamond Light Source to identify well diffracting protein crystals prior to routine cryocrystallography.
1. Introduction
Ribose 5-phosphate isomerase (EC 5.3.1.6; Rpi) is a key enzyme of the pentose phosphate pathway that catalyses the interconversion of ribose 5-phosphate (R5P) and ribulose 5-phosphate (Ru5P). It exists in two isoforms, RpiA and RpiB; although they catalyse the same reaction, they are evolutionarily distinct, sharing little sequence or structural homology. RpiA is widespread throughout all kingdoms of life, whereas RpiB is found in bacterial sources and some pathogenic eukaryotes. Rpi is involved in the synthesis of purine and pyrimidine nucleotides, NAD and amino acids, including histidine and tryptophan, by the production of R5P and in the synthesis of riboflavins via the precursor Ru5P (Hamada et al., 2003 ▶; Zhang, Andersson, Savchenko et al., 2003 ▶). In commensal bacteria such as lactobacilli, the pentose phosphate pathway provides an alternative to hexose metabolism, presumably because plant-derived ribose is common in the mammalian diet.
Structures of RpiA are known from a variety of organisms, including Escherichia coli (Rangarajan et al., 2002 ▶; Zhang, Andersson, Savchenko et al., 2003 ▶), Pyrococcus horikoshii (Ishikawa et al., 2002 ▶), Haemophilus influenzae (PDB entry 1m0s; Northeast Structural Genomics Consortium, unpublished work), Thermus thermophilus (Hamada et al., 2003 ▶), Saccharomyces cerevisiae (Graille et al., 2005 ▶), Plasmodium falciparum (Holmes et al., 2006 ▶), Bartonella henselae (PDB entry 3hhe; Seattle Structural Genomics Center for Infectious Disease, unpublished work), Vibrio vulnificus YJ016 (Kim et al., 2009 ▶), Methanocaldococcus jannaschii (MJ1603; Strange et al., 2009 ▶), Francisella tularensis (PDB entry 3kwm; Center for Structural Genomics of Infectious Diseases, unpublished work) and Burkholderia thailandensis (PDB entries 3uw1 and 3u7j; Seattle Structural Genomics Center for Infectious Disease, unpublished work). Fewer structures of RpiB are known, including those from Thermatoga maritima (Xu et al., 2004 ▶), E. coli (Zhang, Andersson, Skarina et al., 2003 ▶), M. tuberculosis (Roos et al., 2004 ▶, 2005 ▶), Clostridium thermocellum (Jung et al., 2011 ▶), Streptococcus mutans UA159 (PDB entry 3l7o; X.-X. Fan, K.-T. Wang & X.-D. Su, unpublished work), Trypanosoma cruzi (Stern et al., 2011 ▶), Coccidioides immitis (Edwards et al., 2011 ▶) and Giardia lamblia (PDB entry 3s5p; Seattle Structural Genomics Center for Infectious Disease, unpublished work).
Lactobacillus salivarius UC188 is a Gram-positive, probiotic, lactic acid bacterium which has been widely studied for its probiotic benefits as it forms part of the microbiota of the gastrointestinal tract of humans. Research to date has shown that L. salivarius can confer health benefits including, but not limited to, prevention or hindrance of intestinal infections, the elimination of foodborne pathogens and reduction in inflammation and food intolerance (Corr et al., 2007 ▶; Neville & O’Toole, 2010 ▶; O’Callaghan et al., 2012 ▶). Here, we present the high-resolution crystal structure of RpiA from L. salivarius, which was solved as part of a structural proteomics project to shed further light on how Lactobacilli colonize and adapt to the local environment in the complex endogenous microbiota of the human gut.
2. Materials and methods
2.1. Protein production and crystallization
The full-length coding sequence for L. salivarius Rpi (LSL_1806) was cloned into pOPINF using the InFusion method described previously (Bird, 2011 ▶; Berrow et al., 2007 ▶). The protein was produced in E. coli strain Rosetta pLysS (DE3) using the auto-induction method (Studier, 2005 ▶). The cells were harvested by centrifugation and frozen at 193 K (the yield of pure protein was 3 mg per litre). The purification protocol followed that described previously (Nichols et al., 2009 ▶); briefly, defrosted cells were lysed and the soluble fraction was purified via nickel-chelation chromatography followed by gel-filtration chromatography. Protein-containing fractions were pooled and the N-terminal His6 tag was removed using rhinovirus 3C protease followed by reverse purification using nickel-chelation chromatography. Purified protein was concentrated to 20 mg ml−1 in 20 mM Tris pH 7.5, 200 mM NaCl prior to crystallization.
Crystallization screening was carried out as published elsewhere (Walter et al., 2005 ▶) and several conditions were identified, with crystals appearing after between 78 and 128 d incubation at 294 K.
2.2. In situ crystal screening and data collection
Ten crystal leads were identified in 96-well sparse-matrix screens from Emerald BioSystems (Wizard I and II) and Molecular Dimensions (Morpheus; Gorrec, 2009 ▶). X-ray diffraction of these initial crystal hits was screened using in situ plate screening on beamline I04-1 at Diamond Light Source. The setup currently in place at the beamline provides for up to four plates to be hosted in a plate hotel adjacent to the robot dewar in the experimental hutch. These plates were loaded by the CATS robotic arm (Ohana et al., 2004 ▶) and inserted to intersect the X-ray beam at the standard sample position. The additional area required to sample the plate area of any standard Society for Biomolecular Sciences (SBS) 96-well plate in the beamline sample environment was generated by modification of the standard xyz alignment stage of the Maatel MD2 microdiffractometer. Specifically, the space envelope at the sample position in x and y has been augmented by increasing the translation range of the single ω axis by 100 mm and through changes under the goniometer to reclaim 50 mm.
Here, ten drops from two plates were screened using a 1 s exposure with a 50 µm2 beam with an incident flux of 1.7 × 1011 photons s−1. A rotation width of 4° was used to allow unequivocal distinction between salt and protein crystals. Diffraction data were collected on a PILATUS 2M detector. The total time for screening was of the order of an hour. The best crystal identified from the screening was obtained from condition No. 35 of the Wizard II screen from Emerald BioSystems, which consists of 800 mM sodium phosphate monobasic, 1.2 M potassium phosphate dibasic as the precipitant mixture in 0.1 M sodium acetate buffer. The crystals were cryoprotected directly in the crystallization drop by the addition of crystallization buffer containing 30% glycerol prior to flash-cooling in liquid nitrogen and the collection of diffraction data to a resolution of 1.72 Å. The data were processed automatically using xia2 (Winter, 2010 ▶; Evans, 2006 ▶; Leslie, 2006 ▶; Sauter et al., 2004 ▶; Zhang et al., 2006 ▶). Data-collection and reduction statistics are summarized in Table 1 ▶.
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the outermost shell.
| Data collection | |
| X-ray source | I04-1, Diamond Light Source |
| Wavelength () | 0.917 |
| Space group | C2 |
| Unit-cell parameters () | a = 129.32, b = 63.74, c = 59.84, = 90, = 107.6, = 90 |
| Resolution () | 221.72 (1.761.72) |
| R merge † | 0.066 (0.704) |
| I/(I) | 17.3 (2.8) |
| Mosaicity () | 0.2 |
| Completeness (%) | 99.2 (99.8) |
| Multiplicity | 6.8 (6.8) |
| Refinement | |
| No. of reflections | 331889 (24579) |
| No. of unique reflections | 48927 (3613) |
| R cryst ‡ | 0.17 |
| R free ‡ | 0.206 |
| No. of atoms | |
| Protein | 3574 |
| Water | 260 |
| No. of phosphate ions | 3 |
| No. of potassium ions | 2 |
| Average B factors (2) | |
| Protein | 23.2 |
| Phosphate (PO4 3) | 35.5 |
| Potassium (K+) | 13.7 |
| Waters | 31.6 |
| R.m.s. deviations | |
| Bond lengths () | 0.018 |
| Bond angles () | 1.93 |
| Ramachandran statistics (%) | |
| Most favoured | 98.9 |
| Generously allowed | 1.1 |
| Disallowed | 0 |
| MolProbity all-atom clashscore | 10.48 |
R
merge = 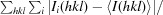
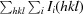 .
.
R
cryst = 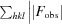
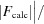
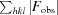 , where F
obs and F
calc are the observed and calculated structure-factor amplitudes, respectively. R
free is calculated as for R
cryst but using a random 5% subset of the data excluded from the refinement.
, where F
obs and F
calc are the observed and calculated structure-factor amplitudes, respectively. R
free is calculated as for R
cryst but using a random 5% subset of the data excluded from the refinement.
2.3. Structure determination and refinement
The crystal structure of L. salivarius Rpi was solved by molecular replacement using MrBUMP (Keegan & Winn, 2007 ▶; Murzin et al., 1995 ▶; Pearson & Lipman, 1988 ▶). A solution was identified using MOLREP (Vagin & Teplyakov, 2010 ▶) with chain A of PDB entry 3enw (Kim et al., 2009 ▶) prepared by CHAINSAW (Stein, 2008 ▶) as a model. The initial R factor of 52.3% was refined to R cryst and R free values of 36.8% and 41.6%, respectively, after 30 cycles of REFMAC5 (Murshudov et al., 2011 ▶). The ARP/wARP web server (Evrard et al., 2007 ▶) was used to autobuild the structure, which resulted in an almost completed model with R cryst and R free values of 19.8% and 24.3%, respectively. Subsequent manual model building and refinement with Coot (Emsley & Cowtan, 2004 ▶) and REFMAC5, respectively, which included the modelling of 21 amino-acid side chains with dual conformations, resulted in a final model with R cryst and R free values of 17.0% and 20.6%, respectively. The final model was validated by MolProbity (Chen et al., 2010 ▶) and the RCSB Validation Server (Berman et al., 2000 ▶, 2003 ▶). Refinement statistics are detailed in Table 1 ▶.
3. Results and discussion
3.1. Benefits of in situ crystal screening
In situ diffraction experiments were first reported in 2004 (Jacquamet et al., 2004 ▶) and clearly illustrated the potential of the method for accelerating the crystal-to-structure turnaround time. However, these initial experiments were compromised somewhat by the crystallization-plate geometry and the significant scattering from the plate plastic. Although an integrated X-ray system for the home laboratory (the PX scanner from Agilent) has been available for some time, the true potential of the method is now being realised, with a number of synchrotron beamlines enabling the method to be routinely accessed by users, notably at the Swiss Light Source, where a dedicated facility has been implemented (Bingel-Erlenmeyer et al., 2011 ▶), BM30 at the ESRF (Jacquamet et al., 2009 ▶) and Diamond (Axford et al., 2012 ▶). Moreover, the development of low X-ray scattering SBS-format crystallization plates (CrystalQuickX, Greiner Bio-One, Germany) has significantly increased the tractability of performing in situ data collections (le Maire et al., 2011 ▶; Wang et al., 2012 ▶) and the development of other crystallization devices developed with in situ capabilities in mind should have an impact on the method (see Hargreaves, 2012 ▶ and references therein).
The recently commissioned in situ diffraction capability available on beamline I04-1 at Diamond Light Source provides an ideal facility for the rapid screening of crystal quality and subsequent standard data collection after crystal harvesting. Use of the CATS robot for handling the SBS standard plates allows the method to be integrated into the standard beamline setup, thus allowing the user to swap quickly between an in situ screening experiment and a standard data collection with a cryogenically cooled loop-mounted crystal using the Maatel MD2 diffractometer. The experiment workflow is accommodated within the beamline software control (GDA at Diamond; http://opengda.org), which provides an interface for the user to easily navigate to the crystallization wells containing crystal hits; the viewing system at low zoom levels is able to display a clear view of the crystallization well, allowing the user to quickly centre on the crystal of interest in the drop.
For the RpiA experiment, crystals were ranked based on the in situ diffraction experiment; ten crystallization conditions with crystal hits were screened directly in situ, which showed that although nine of the ten conditions contained protein crystals, only that in one condition (Fig. 1 ▶ a) gave strong diffraction to beyond 3 Å resolution (Table 2 ▶). This crystal was then harvested and used to collect a 1.72 Å resolution data set immediately after the in situ experiment (Fig. 1 ▶ b). As the crystallization condition for these crystals only yielded crystals after between 78 and 128 d, reproducing the crystals proved to be challenging. In this case crystal hits were effectively ranked by in situ screening, enabling the most effective use of the allocated beamtime, with data being collected immediately following the screening session on the beamline from the best identified crystal. The changeover time from in situ screening to the standard data-collection setup at I04-1 is currently under 30 min. The process has been successfully repeated for two other projects and is proving to be highly effective for making the best use of the synchrotron beamtime available.
Figure 1.

(a) Snapshot of the RpiA crystal in the 96-well plate before in situ screening. The plates were stored at 293 K and imaged using a Formulatrix Rock Imager. (b) An image captured from the I04-1 on-axis viewing system showing the same crystal mounted and cryoprotected at the beamline before data collection.
Table 2. Results of in situ screening of RpiA crystallization hit conditions.
| Crystal screen | Well | Well solution | Outcome |
|---|---|---|---|
| Wizard I and II | A6 | 20% PEG 3000, 0.1M citrate pH 5.5 | Salt crystals |
| A8 | 2.0M ammonium sulfate, 0.1M citrate pH 5.5 | No diffraction | |
| G11 | 0.8M sodium phosphate/1.2M potassium phosphate, 0.1M acetate pH 4.5 | Strong diffraction to 2.7 resolution, easily indexed and subsequently used for data collection | |
| Morpheus | A5 | 0.06M Divalents Mix, 0.1M Buffer System 2 pH 7.5, 30% PEG550MME_P20K Mix | No diffraction |
| C1 | 0.09M NPS Mix, 0.1M Buffer System 1 pH 6.5, 30% PEG550MME_P20K Mix | Diffraction spots observed to 6 resolution | |
| D1 | 0.12M Alcohols Mix, 0.1M Buffer System 1 pH 6.5, 30% PEG550MME_P20K Mix | Diffraction spots observed to 6 resolution | |
| E1 | 0.12M Ethylene Glycols Mix, 0.1M Buffer System 1 pH 6.5, 30% PEG550MME_P20K Mix | Diffraction spots observed to 6.5 resolution | |
| G4 | 0.10M Carboxylic Acids Mix, 0.1M Buffer System 1 pH 6.5, 37.5% MPD_P1K_P3350 Mix | Highly mosaic diffraction with spots observed to 3 resolution (harvesting of crystals unsuccessful, no further characterization) | |
| H4 | 0.10M Amino Acids Mix, 0.1M Buffer System 1 pH 6.5, 37.5% MPD_P1K_P3350 Mix | Diffraction spots observed to 6 resolution | |
| H5 | 0.10M Amino Acids Mix, 0.1M Buffer System 2 pH 7.5, 30% PEG550MME_P20K Mix | Diffraction spots observed to 9 resolution |
3.2. RpiA structure and comparison with the d-ribose-5-phosphate isomerase family
The overall topology of L. salivarius RpiA (Figs. 2 ▶ and 3 ▶) is broadly similar to other known RpiA structures. The N-terminal domain consists of α-helices 1–5 and β-strands 1–3, 6, 12 and 13. The C-terminal domain consists of α-helices 6 and 7 and β-strands 7–10. The remaining three β-strands form the interface between the two domains. In RpiA structures the domain interface typically consists of four β-strands. From a structural superposition (not shown) we see that the fourth strand, which would have been made by residues 124–126, is in fact an extended loop in the L. salivarius structure. While there is this very subtle change in topology, the extended loop occupies broadly the same position as the short β-strand that it replaces. The L. salivarius structure fits well into the SCOP α and β family of d-ribose-5-phosphate isomerase (RpiA) catalytic domains, as would be expected.
Figure 2.

The overall dimeric structure of RpiA from L. salivarius, with each chain coloured by secondary structure (helices in red, β-strands in blue and random coils in teal). The two K+ ions located at the dimer interface are shown as purple spheres and the three phosphate (PO4 3−) ions are shown in stick representation: two in the active site of chain A and one in the active site of chain B (yellow sticks).
Figure 3.

(a) Structure-based sequence alignment of the L. salivarius structure with the published structures with PDB codes 3l7o, 3enq (Kim et al., 2009 ▶), 1uj4 (Hamada et al., 2003 ▶), 3ixq (Strange et al., 2009 ▶) and 1xtz (Graille et al., 2005 ▶). (b) Superposed structures of one protomer from the above members of the RpiA family coloured as in Fig. 2 ▶. The L. salivarius structure is shown in cartoon representation. Molecular-graphics figures were all prepared using PyMOL (Schrödinger LLC). The sequence alignment was prepared in PROMALS3D (Pei et al., 2008 ▶).
The structure presented contains a dimer in the asymmetric unit (Fig. 2 ▶). The two protomers in the asymmetric unit are highly similar, with a global r.m.s difference of 0.99 Å on superposition using proSMART (Nicholls, 2011 ▶). The presence of a dimer correlates with two observations on the protein in solution. Firstly, during gel-filtration chromatography to purify RpiA the protein eluted at a point corresponding to approximately the molecular weight of a dimer. Secondly, the purified protein was assessed using a Viscotek OmniSEC Tetra detector, which gave a molecular mass in solution of 44.388 kDa (dimer molecular weight of 49.74 kDa; data not shown). Analysis of the dimer interaction site using the PISA web server (Krissinel & Henrick, 2007 ▶; http://www.ebi.ac.uk/pdbe/prot_int/pistart.html) indicated that the dimer interface involves 33 amino acids from each protomer and buries an interface area of approximately 2167 Å2. While there are no covalent bonds or salt bridges in the dimer interface, there are seven hydrogen bonds (involving residues Thr54, Asp74, Lys112, Tyr141, Ser143, Gly144 and Thr145 from both protomers) and numerous hydrophobic interactions stabilizing dimerization. This dimer interface is replicated across the RpiA family, where the biologically relevant dimer interfaces are comprised of predominantly hydrophobic interactions with a few key hydrogen bonds, although the dimer interface is only slightly more conserved than the surface residues. The role of the dimer in biological activity is unclear; each subunit presents an active site and there is no evidence for the dimer interface having a role in allosteric regulation (Zhang, Andersson, Savchenko et al., 2003 ▶). Two crystallographic potassium ions complexed from the crystallization buffer have been modelled in the dimer interface. Each is coordinated by three protein residues, Ile195, Gln197 and Val200, and by three water molecules in extended water networks.
The active site is located in a shallow groove at the interface between the N-terminal and the C-terminal domains and is made up of two regions of conserved residues (Figs. 2 ▶ and 3 ▶ a). The conserved sequence GXG(T/S)GST that is known to contain the phosphate-recognition sequence is 25GLGTGST31 in L. salivarius. The second conserved region, DGADE(X)8KGXG, which contains the sugar-recognition sequence and the catalytic residues, is 84DGADE(ISSDFQGI)KGGG100 in L. salivarius. The crystals were grown in the absence of substrate, product or analogues. In some structures, the crystallization of apo RpiA has given rise to apo structures, including the S. mutans structure 3l7o, the V. vulnificus structure 3enq and the E. coli structure 1ks2. However, the structure presented here contains three phosphates, which are clearly visible in the electron density. Phosphate 1 is bound to the sugar-recognition sequence of protomer A, whilst phosphates 2 and 3 are bound to the phosphate-recognition pockets of protomer A and B, respectively (Fig. 4 ▶).
Figure 4.

(a) The active site of L. salivarius RpiA showing the two phosphate (PO4 3−) ions bound in the phosphate- and sugar-binding pockets. (b) Model of Ru5P bound to L. salivarius RpiA using the bound phosphate ions as a reference based on the structure of Ru5P bound to V. vulnificus, showing that the mode of binding is likely to be essentially identical.
A close inspection of the active site of RpiA has been made focusing on three species: L. salivarius, S. mutans and V. vulnificus. S. mutans is phylogenetically close to the lactobacilli; it encodes the closest sequence homologue (57%) to the L. salivarius enzyme and is an apo structure. The enzyme from the more phylogenetically distant Gram-negative marine organism V. vulnificus has only 44% sequence identity but has been solved as an apo structure, with the substrate Ru5P bound and with the inhibitor arabinose 5-phosphate bound. The structures from these three species superpose very closely, with only two minor side-chain differences in the active site. Firstly, at position 100 in L. salivarius and S. mutans there is a glycine residue which makes a hydrogen bond to phosphate 1. In V. vulnificus this is replaced by an alanine residue, which causes a change in the backbone conformation. Secondly, in the phosphate-binding region the L. salivarius structure has a hydrogen bond to the phosphate from Thr31. In the Ru5P-bound V. vulnificus structure this threonine occupies a different geometry and the hydrogen bond is made by Lys97 (Fig. 4 ▶). These two changes slightly extend the substrate-binding pocket, allowing it to easily accommodate the linear substrate molecule. The family of V. vulnificus structures demonstrate that binding of the substrate causes no perturbation of the active site. The substrate-bound V. vulnificus structure provides an excellent model for substrate binding in L. salivarius (Fig. 4 ▶ b). In contrast, the inhibitor is predominantly solvent-exposed, with the sugar ring binding the phosphate-binding pocket and the phosphate group directed towards the solvent.
L. salivarius RpiA was compared with 20 known RpiA structures using proSMART, a structure-alignment tool that uses the conservation of local structure to produce a conformation-independent structural comparison of protein chains (Nicholls, 2011 ▶). This analysis showed that despite low sequence identity (26–57%) the global r.m.s. difference is indicative of close structural homology (0.7–2.0 Å; abbreviated data are given in Table 3 ▶ and Fig. 3 ▶). While the domain structure of RpiA is largely identical, conformational differences can be observed in the domain interface, causing subtle changes in the angle between the N-terminal and C-terminal domains. The other area in which the structures differ is in the extended loop connecting β-strands 8 and 9 (residues 160–175), where some flexibility can be inferred from the deviations in modelled positions across the 20 structures.
Table 3. Structural superposition of RpiA structures.
Each chain was independently aligned and superposed on both chain A and chain B of the L. salivarius RpiA structure using proSMART. Results are shown for the comparison of chain A. Ru5P, ribulose 5-phosphate; A5P, arabinose-5-phosphate.
| PDB code | Chain | Species | Ligands | Sequence identity (%) | Global r.m.s.d.† () |
|---|---|---|---|---|---|
| 4gmk | A | L. salivarius UCC118 | PO4 3, K+ | 100.0 | |
| B | 100.0 | 0.99 | |||
| 3l7o | A | S. mutans UA159 | Apo | 56.6 | 1.4 |
| B | 57.4 | 0.7 | |||
| 3enq | A | V. vulnificus YJ016 | Apo | 44.4 | 1.6 |
| B | 44.4 | 1.8 | |||
| 3env | A | V. vulnificus YJ016 | A5P | 44.4 | 1.8 |
| B | 44.4 | 1.7 | |||
| 3enw | A | V. vulnificus YJ016 | Ru5P | 44.4 | 1.6 |
| B | 44.4 | 1.7 |
Global r.m.s.d. of main-chain atoms after superposition of all aligned residues based on aligned residues.
Supplementary Material
PDB reference: ribose 5-phosphate isomerase, 4gmk
Acknowledgments
The authors thank Matthew Jenions for the Viscotek OmniSEC Tetra detector results. Initial work on this project was supported by a joint MRC/BBSRC grant to MAW. Work in the laboratory of PWO’T is supported by a Science Foundation Ireland grant to the Alimentary Pharmabiotic Centre and a Principal Investigator award. We thank Alimentary Health Ltd for providing strain UCC118.
References
- Axford, D. et al. (2012). Acta Cryst. D68, 592–600. [DOI] [PMC free article] [PubMed]
- Berman, H., Henrick, K. & Nakamura, H. (2003). Nature Struct. Biol. 10, 980. [DOI] [PubMed]
- Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N. & Bourne, P. E. (2000). Nucleic Acids Res. 28, 235–242. [DOI] [PMC free article] [PubMed]
- Berrow, N. S., Alderton, D., Sainsbury, S., Nettleship, J., Assenberg, R., Rahman, N., Stuart, D. I. & Owens, R. J. (2007). Nucleic Acids Res. 35, e45. [DOI] [PMC free article] [PubMed]
- Bingel-Erlenmeyer, R., Olieric, V., Grimshaw, J. P. A., Gabadinho, J., Wang, X., Ebner, S. G., Isenegger, A., Schneider, R., Schneider, J., Glettig, W., Pradervand, C., Panepucci, E. H., Tomizaki, T., Wang, M. & Schulze-Briese, C. (2011). Cryst. Growth Des. 11, 916–923.
- Bird, L. E. (2011). Methods, 55, 29–37. [DOI] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Corr, S. C., Li, Y., Riedel, C. U., O’Toole, P. W., Hill, C. & Gahan, C. G. (2007). Proc. Natl Acad. Sci. USA, 104, 7617–7621. [DOI] [PMC free article] [PubMed]
- Edwards, T. E., Abramov, A. B., Smith, E. R., Baydo, R. O., Leonard, J. T., Leibly, D. J., Thompkins, K. B., Clifton, M. C., Gardberg, A. S., Staker, B. L., Van Voorhis, W. C., Myler, P. J. & Stewart, L. J. (2011). BMC Struct. Biol. 11, 39. [DOI] [PMC free article] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Evrard, G. X., Langer, G. G., Perrakis, A. & Lamzin, V. S. (2007). Acta Cryst. D63, 108–117. [DOI] [PMC free article] [PubMed]
- Gorrec, F. (2009). J. Appl. Cryst. 42, 1035–1042. [DOI] [PMC free article] [PubMed]
- Graille, M., Meyer, P., Leulliot, N., Sorel, I., Janin, J., Van Tilbeurgh, H. & Quevillon-Cheruel, S. (2005). Biochimie, 87, 763–769. [DOI] [PubMed]
- Hamada, K., Ago, H., Sugahara, M., Nodake, Y., Kuramitsu, S. & Miyano, M. (2003). J. Biol. Chem. 278, 49183–49190. [DOI] [PubMed]
- Hargreaves, D. (2012). J. Appl. Cryst. 45, 138–140. [DOI] [PMC free article] [PubMed]
- Holmes, M. A. et al. (2006). Acta Cryst. F62, 427–431.
- Ishikawa, K., Matsui, I., Payan, F., Cambillau, C., Ishida, H., Kawarabayasi, Y., Kikuchi, H. & Roussel, A. (2002). Structure, 10, 877–886. [DOI] [PubMed]
- Jacquamet, L., Joly, J., Bertoni, A., Charrault, P., Pirocchi, M., Vernede, X., Bouis, F., Borel, F., Périn, J.-P., Denis, T., Rechatin, J.-L. & Ferrer, J.-L. (2009). J. Synchrotron Rad. 16, 14–21. [DOI] [PubMed]
- Jacquamet, L., Ohana, J., Joly, J., Borel, F., Pirocchi, M., Charrault, P., Bertoni, A., Israel-Gouy, P., Carpentier, P., Kozielski, F., Blot, D. & Ferrer, J.-L. (2004). Structure, 12, 1219–1225. [DOI] [PubMed]
- Jung, J., Kim, J.-K., Yeom, S.-J., Ahn, Y.-J., Oh, D.-K. & Kang, L.-W. (2011). Appl. Microbiol. Biotechnol. 90, 517–527. [DOI] [PubMed]
- Keegan, R. M. & Winn, M. D. (2007). Acta Cryst. D63, 447–457. [DOI] [PubMed]
- Kim, T. G., Kwon, T. H., Min, K., Dong, M.-S., Park, Y. I. & Ban, C. (2009). Mol. Cells, 27, 99–103. [DOI] [PubMed]
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol. 372, 774–797. [DOI] [PubMed]
- Leslie, A. G. W. (2006). Acta Cryst. D62, 48–57. [DOI] [PubMed]
- Maire, A. le, Gelin, M., Pochet, S., Hoh, F., Pirocchi, M., Guichou, J.-F., Ferrer, J.-L. & Labesse, G. (2011). Acta Cryst. D67, 747–755. [DOI] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Murzin, A. G., Brenner, S. E., Hubbard, T. & Chothia, C. (1995). J. Mol. Biol. 247, 536–540. [DOI] [PubMed]
- Neville, B. A. & O’Toole, P. W. (2010). Future Microbiol. 5, 759–774. [DOI] [PubMed]
- Nicholls, R. A. (2011). PhD thesis. University of York.
- Nichols, C. E., Sainsbury, S., Ren, J., Walter, T. S., Verma, A., Stammers, D. K., Saunders, N. J. & Owens, R. J. (2009). Acta Cryst. F65, 204–209. [DOI] [PMC free article] [PubMed]
- O’Callaghan, J., Buttó, L. F., MacSharry, J., Nally, K. & O’Toole, P. W. (2012). Appl. Environ. Microbiol. 78, 5196–5203. [DOI] [PMC free article] [PubMed]
- Ohana, J., Jacquamet, L., Joly, J., Bertoni, A., Taunier, P., Michel, L., Charrault, P., Pirocchi, M., Carpentier, P., Borel, F., Kahn, R. & Ferrer, J.-L. (2004). J. Appl. Cryst. 37, 72–77.
- Pearson, W. R. & Lipman, D. J. (1988). Proc. Natl Acad. Sci. USA, 85, 2444–2448. [DOI] [PMC free article] [PubMed]
- Pei, J., Kim, B.-H. & Grishin, N. V. (2008). Nucleic Acids Res. 36, 2295–2300. [DOI] [PMC free article] [PubMed]
- Rangarajan, E. S., Sivaraman, J., Matte, A. & Cygler, M. (2002). Proteins, 48, 737–740. [DOI] [PubMed]
- Roos, A. K., Andersson, C. E., Bergfors, T., Jacobsson, M., Karlén, A., Unge, T., Jones, T. A. & Mowbray, S. L. (2004). J. Mol. Biol. 335, 799–809. [DOI] [PubMed]
- Roos, A. K., Burgos, E., Ericsson, D. J., Salmon, L. & Mowbray, S. L. (2005). J. Biol. Chem. 280, 6416–6422. [DOI] [PubMed]
- Sauter, N. K., Grosse-Kunstleve, R. W. & Adams, P. D. (2004). J. Appl. Cryst. 37, 399–409. [DOI] [PMC free article] [PubMed]
- Stein, N. (2008). J. Appl. Cryst. 41, 641–643.
- Stern, A. L., Naworyta, A., Cazzulo, J. J. & Mowbray, S. L. (2011). FEBS J. 278, 793–808. [DOI] [PubMed]
- Strange, R. W., Antonyuk, S. V., Ellis, M. J., Bessho, Y., Kuramitsu, S., Yokoyama, S. & Hasnain, S. S. (2009). Acta Cryst. F65, 1214–1217. [DOI] [PMC free article] [PubMed]
- Studier, F. W. (2005). Protein Expr. Purif. 41, 207–234. [DOI] [PubMed]
- Vagin, A. & Teplyakov, A. (2010). Acta Cryst. D66, 22–25. [DOI] [PubMed]
- Walter, T. S. et al. (2005). Acta Cryst. D61, 651–657. [DOI] [PMC free article] [PubMed]
- Wang, X. et al. (2012). Nature Struct. Mol. Biol. 19, 424–429. [DOI] [PMC free article] [PubMed]
- Winter, G. (2010). J. Appl. Cryst. 43, 186–190.
- Xu, Q. et al. (2004). Proteins, 56, 171–175. [DOI] [PubMed]
- Zhang, R., Andersson, C. E., Savchenko, A., Skarina, T., Evdokimova, E., Beasley, S., Arrowsmith, C. H., Edwards, A. M., Joachimiak, A. & Mowbray, S. L. (2003). Structure, 11, 31–42. [DOI] [PMC free article] [PubMed]
- Zhang, R. G., Andersson, C. E., Skarina, T., Evdokimova, E., Edwards, A. M., Joachimiak, A., Savchenko, A. & Mowbray, S. L. (2003). J. Mol. Biol. 332, 1083–1094. [DOI] [PMC free article] [PubMed]
- Zhang, Z., Sauter, N. K., van den Bedem, H., Snell, G. & Deacon, A. M. (2006). J. Appl. Cryst. 39, 112–119.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: ribose 5-phosphate isomerase, 4gmk