

The structure of ATP-dependent DNA ligase from Thermococcus sp. 1519 was determined at a resolution of 3.02 Å, showing a new relative arrangement of the OB-fold domain.
Keywords: ATP-dependent DNA ligase, OB fold, Thermococcus sp. 1519, DNA ligases
Abstract
DNA ligases join single-strand breaks in double-stranded DNA by catalyzing the formation of a phosphodiester bond between adjacent 5′-phosphate and 3′-hydroxyl termini. Their function is essential for maintaining genome integrity in the replication, recombination and repair of DNA. High flexibility is important for the function of DNA ligase molecules. Two types of overall conformations of archaeal DNA ligase that depend on the relative position of the OB-fold domain have previously been revealed: closed and open extended conformations. The structure of ATP-dependent DNA ligase from Thermococcus sp. 1519 (LigTh1519) in the crystalline state determined at a resolution of 3.02 Å shows a new relative arrangement of the OB-fold domain which is intermediate between the positions of this domain in the closed and the open extended conformations of previously determined archaeal DNA ligases. However, small-angle X-ray scattering (SAXS) measurements indicate that in solution the LigTh1519 molecule adopts either an open extended conformation or both an intermediate and an open extended conformation with the open extended conformation being dominant.
1. Introduction
DNA ligases catalyze the sealing of adjacent 5′-phosphate and 3′-hydroxyl termini at single-stranded breaks in double-stranded DNA. Their function is crucial in DNA replication, recombination and repair (Lehman, 1974 ▶). The reaction catalyzed by DNA ligases proceeds by three sequential nucleotidyl-transfer steps. In the first step, the active-site lysine is activated by the covalent attachment of AMP, which is accompanied by the release of pyrophosphate or nicotinamide mononucleotide from the cofactor (ATP or NAD+). AMP is then transferred to the 5′-end of the 5′-phosphate-terminated DNA strand to form DNA adenylate. In the final step, the phosphodiester bond is formed with concomitant release of AMP from the DNA-adenylate intermediate. DNA ligases are present in all living organisms and are conventionally divided into two families according to their cofactor specificity (Sriskanda et al., 2000 ▶). The first group contains ATP-dependent ligases (EC 6.5.1.1), which are found in eukaryotes, archaea, viruses and some bacteria. The second group comprises NAD+-dependent DNA ligases (EC 6.5.1.2); these are present in bacteria and some eukaryotic viruses.
DNA ligases from thermophilic and hyperthermophilic archaea have been used as model systems in structural and mechanistic studies of DNA ligation. A number of thermostable DNA ligases from archaea have been isolated and functionally characterized (Sriskanda et al., 2000 ▶; Lai et al., 2002 ▶; Keppetipola & Shuman, 2005 ▶). All of them utilized ATP as a cofactor and it was generally accepted that archaeal DNA ligases belong to the family of ATP-dependent ligases. However, it was subsequently found that some DNA ligases from hyperthermophilic euryarchaea of the order Thermococcales (Thermococcus kodakarensis, T. fumicolans, T. onnurineus and Pyrococcus abyssi) are also able to use NAD+ as a cofactor (Nakatani et al., 2000 ▶; Rolland et al., 2004 ▶; Kim et al., 2006 ▶). Therefore, it was suggested that the dual specificity of Thermococcales DNA ligases reflects an intermediate phase in the evolution of an ancestral enzyme into the cofactor-specific specialized present-day enzymes (Sun et al., 2008 ▶).
Only a few crystal structures of archaeal DNA ligases have been reported: those from the Euryarchaeota P. furiosus (PfuLig; Nishida et al., 2006 ▶), Archaeoglobus fulgidus (AfuLig; Kim et al., 2009 ▶) and T. sibiricus (TsibLig; Petrova et al., 2012 ▶) and the Crenarchaeota Sulfolobus solfataricus (SsoLig; Pascal et al., 2006 ▶) and Sulfophobococcus zilligii (Supangat et al., 2010 ▶). Previously, we identified and functionally characterized the ATP-dependent DNA ligase from the hyperthermophilic archaeon Thermococcus sp. 1519 from the order Thermococcales (LigTh1519; Smagin et al., 2008 ▶). We succeeded in the crystallization and structure determination of this enzyme at 2.9 Å resolution (Bezsudnova et al., 2009 ▶) and found a molecular-replacement solution for two domains only. We suggested that the third domain, the OB-fold domain, either adopted different positions in different cells of the crystal or was hydrolyzed during crystallization. Subsequently, we collected X-ray data from a crystal of LigTh1519 grown under different conditions. Here, we report the results of a crystallographic study of the ATP-dependent DNA ligase from Thermococcus sp. 1519 with all three domains. A comparative analysis of the structures of ATP-dependent DNA ligases may help in the identification of sites for directed mutagenesis aimed at changing the cofactor specificity of the enzyme from ATP to NAD.
2. Materials and methods
Expression and purification have previously been described by Bezsudnova et al. (2009 ▶).
2.1. Crystallization
Freshly prepared recombinant LigTh1519 (22.1 mg ml−1) in 50 mM Tris–HCl pH 7.5 containing 100 mM NaCl and 0.5 mM DTT was used. Initial crystallization screening of LigTh1519 was performed at 291 K by the hanging-drop vapour-diffusion method using Crystal Screen, Index and Crystal Screen Cryo kits (Hampton Research). A single crystal was obtained after six months using the gel-tube counter-diffusion method at 291 K. The truncated square bipyramidal crystal had dimensions of about 200–300 µm. The reservoir solution consisted of 0.1 M sodium cacodylate pH 6.5, 0.2 M MgCl2, 20% glycerol, 16% PEG 8000, 100 mM NaCl, 50 mM Tris–HCl pH 7.5.
2.2. X-ray data collection and treatment
X-ray diffraction data were collected on beamline BL41XU at the Spring-8 synchrotron using a MAR Mosaic 225 mm CCD detector. A crystal was flash-cooled in a stream of cold nitrogen gas at 100 K. Data collection was performed using the HKL-2000 software package (Otwinowski & Minor, 1997 ▶). Experimental details are summarized in Table 1 ▶. X-ray diffraction images were indexed, integrated and subsequently scaled using HKL-2000. Data reduction was performed using the CCP4 package (Winn et al., 2011 ▶). Data-collection statistics are given in Table 1 ▶.
Table 1. Experimental setup and statistics of data collection and processing.
Values in parentheses are for the highest resolution shell.
| Space group | P212121 |
| Radiation source | National Research Center ‘Kurchatov Institute’ |
| Unit-cell parameters (Å) | a = 76.950, b = 85.600, c = 105.960 |
| Temperature (K) | 100 |
| Wavelength (Å) | 0.800 |
| Crystal-to-detector distance (mm) | 410 |
| Oscillation range (°) | 0.5 |
| Mosaicity (°) | 0.20 |
| No. of frames | 720 |
| Resolution limit (Å) | 29.26–3.02 (3.20–3.02) |
| Total reflections | 68411 (10505) |
| Molecules per asymmetric unit | 1 |
| Solvent content (%) | 55 |
| Independent reflections | 13339 (2121) |
| Average I/σ(I) | 14.19 (1.75) |
| Completeness (%) | 93.3 (94.0) |
| R merge | 0.10 (0.66) |
SAXS measurements were performed at a wavelength of 0.1542 nm using an AMUR-K diffractometer with an OD3M linear position-sensitive detector and a graphite monochromator. The Kratky-type geometry was used with a sample-to-detector distance of 700 mm and a sample slit width of 0.2 mm to cover the range of momentum transfer 0.12 < s < 12.0 nm−1 (s = 4πsinθ/λ, where 2θ is the scattering angle). Measurements were carried out at room temperature for 2 h using sample solutions placed in 1 mm quartz capillaries. The data were normalized to the intensity of the incident beam and corrected for X-ray absoption and collimation distortions using standard procedures (Feigin & Svergun, 1987 ▶).
2.3. Structure determination and refinement
The structure of LigTh1519 was solved using BALBES (Long et al., 2008 ▶). The best solution found by BALBES was obtained using the PfuLig model (PDB entry 2cfm; Nishida et al., 2006 ▶) as a starting model for molecular replacement and refinement. The model was further refined using phenix.refine (Afonine et al., 2005 ▶). The refined parameters were the coordinates for each atom and the B factors for groups of atoms. The relatively small number of observations did not allow us to refine B factors for each atom. Two B factors were refined for each residue: one for the atoms of the main chain and the other for the side-chain atoms. Along with a set of standard stereochemical restraints, secondary-structure restraints and Ramachandran restraints were applied during refinement. Examination of density maps and the manual rebuilding of the model were performed using the program Coot (Emsley & Cowtan, 2004 ▶).
In addition to a LigTh1519 molecule, the final model contained four water molecules and one Mg2+ ion. There was no interpretable electron density for 11 N-terminal residues, including six purification-tag residues, and six C-terminal residues. The geometry of the final model was inspected by PROCHECK (Laskowski et al., 1993 ▶). A summary of the refinement statistics and model quality is given in Table 2 ▶. Atomic coordinates and experimental structure factors were deposited in the Protein Data Bank and are accessible as entry 3rr5.
Table 2. Refinement statistics.
| R work/R free | 0.233/0.310 |
| R.m.s. deviations from ideal geometry | |
| Bond lengths (Å) | 0.009 |
| Bond angles (°) | 1.319 |
| Average B factor (Å2) | |
| Main-chain atoms | 84.3 |
| Side-chain atoms | 84.2 |
| Ramachandran plot (%) | |
| Most favourable | 95.8 |
| Allowed | 3.8 |
| Outliers | 0.4 |
3. Results and discussion
3.1. Overall conformation of the LigTh1519 molecule
Like previously determined molecules of ATP-dependent DNA ligases, the LigTh1519 molecule comprises three domains: an N-terminal DNA-binding domain (DBD), an adenylation domain (AdD) and a C-terminal OB-fold domain (OBD) (Fig. 1 ▶ a). Previously, the following conformations of ATP-dependent DNA ligases have been determined depending on the relative position of the OBD.
(i) Closed conformation. This conformation was revealed for PfuLig (Nishida et al., 2006 ▶; PDB entry 2cfm; Fig. 1b ▶), AfuLig (Kim et al., 2009 ▶; PDB entry 3gde) and TsibLig (Petrova et al., 2012 ▶; PDB entry 4eq5).
(ii) Open extended conformation. This conformation was displayed by SsoLig (Pascal et al., 2006 ▶; PDB entries 2hiv and 2hix; Fig. 1 ▶ c).
(iii) Open conformation. This conformation was displayed by human DNA ligase I bound to DNA (Pascal et al., 2004 ▶; PDB entry 1x9n) and human DNA ligase III bound to DNA (Cotner-Gohara et al., 2010 ▶; PDB entry 3l2p).
Thus, only two conformations have previously been revealed for archaeal DNA ligases: the closed and open extended conformations.
Figure 1.

Different conformations of archaeal DNA ligases. (a) A ribbon diagram of LigTh1519, which shows a new position of the OBD; (b) a ribbon diagram of PfuLig, which shows a closed conformation; (c) a ribbon diagram of SsoLig, which shows an open extended conformation.
The LigTh1519 molecule in the crystalline state shows a new relative arrangement of the OBD which is intermediate between the positions of this domain in the open extended and closed conformations of DNA ligases (Fig. 2 ▶). To derive this new intermediate position from that corresponding to the open extended conformation, the OBD should be rotated in the anticlockwise direction by approximately 90° around the AdD (Fig. 2 ▶ a). Further rotation of the OBD around a slightly different axis by about 120° in the anticlockwise direction leads to the position that corresponds to the closed conformation (Fig. 2 ▶ b). The position of the OBD in LigTh1519 rather resembles the position of the OBD in the two-domain DNA ligases from Chlorella virus (PDB entry 1p8l; Odell et al., 2003 ▶) and from bacteriophage T7 (PDB entry 1a0i; Subramanya et al., 1996 ▶) (Figs. 2 ▶ c and 2 ▶ d). Note that while the relative position of the OBD differs significantly in the open extended, closed and intermediate conformations, the relative positions of the DBD and AdD are very similar. The r.m.s.d.s calculated over Cα atoms of the first two domains, DBD and AdD, for superimposed molecules of LigTh1519 and PfuLig and of LigTh1519 and SsoLig are 1.3 and 2.2 Å, respectively.
Figure 2.

Superposition of LigTh1519 (green) with (a) SsoLig (pink), (b) PfuLig (blue), (c) Clorella virus DNA ligase (magenta) and (d) DNA ligase from bacteriophage T7 (violet).
Interestingly, unlike other DNA ligases from archaea, the OBD in LigTh1519 is not connected to the other two domains through hydrogen bonds and salt bridges; the only link between the OBD and the rest of the molecule is a covalent bond. It appears as if the OBD is floating around the AdD. In the crystal, this position of the OBD is fixed by eight hydrogen bonds and three salt bridges to the AdD of a symmetry-related molecule (Figs. 3 ▶ a and 3 ▶ b). Note that the residues involved in these interactions form the only contact region between the OBD and the other domains and fix the position of the OBD in the crystal structure. The side chains of these residues are easily visible in the electron-density map. It seems that the position of the OBD is stabilized by these interactions.
Figure 3.
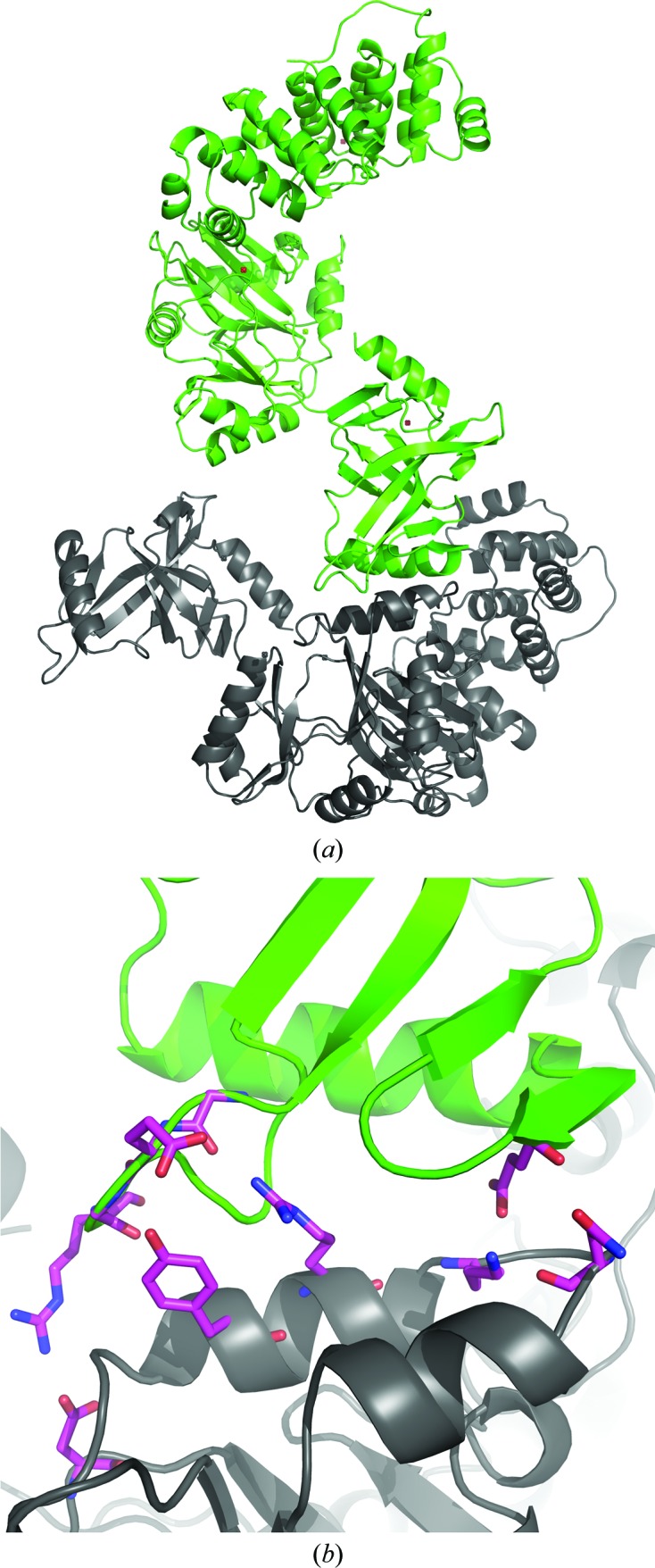
The OBD of LigTh1519 (green) and a symmetry-related molecule (grey) in the crystal. (a) The OBD of LigTh1519 is only covalently bound to the rest of the molecule. In the crystal, the position of the OBD is fixed by a symmetry-related molecule. (b) An enlarged view of the region between the OBD and the AdD of a symmetry-related molecule. Residues participating in hydrogen bonds between the OBD and the AdD of a symmetry-related molecule are shown in stick representation.
The final model of LigTh1519 did not contain an ATP molecule. Although ATP was not externally added to the crystallization solution, it might have copurified with the ligase (as in PfuLig and TsibLig). Therefore, the final structure may be a complex of the enzyme with endogenous ATP. The relatively poor electron density did not allow unambiguous determination of whether or not an ATP molecule was present in the active site. In addition, Arg280, the conformation of which was determined unambiguously, would be too close to the possible position of the cofactor (Fig. 4 ▶)
Figure 4.
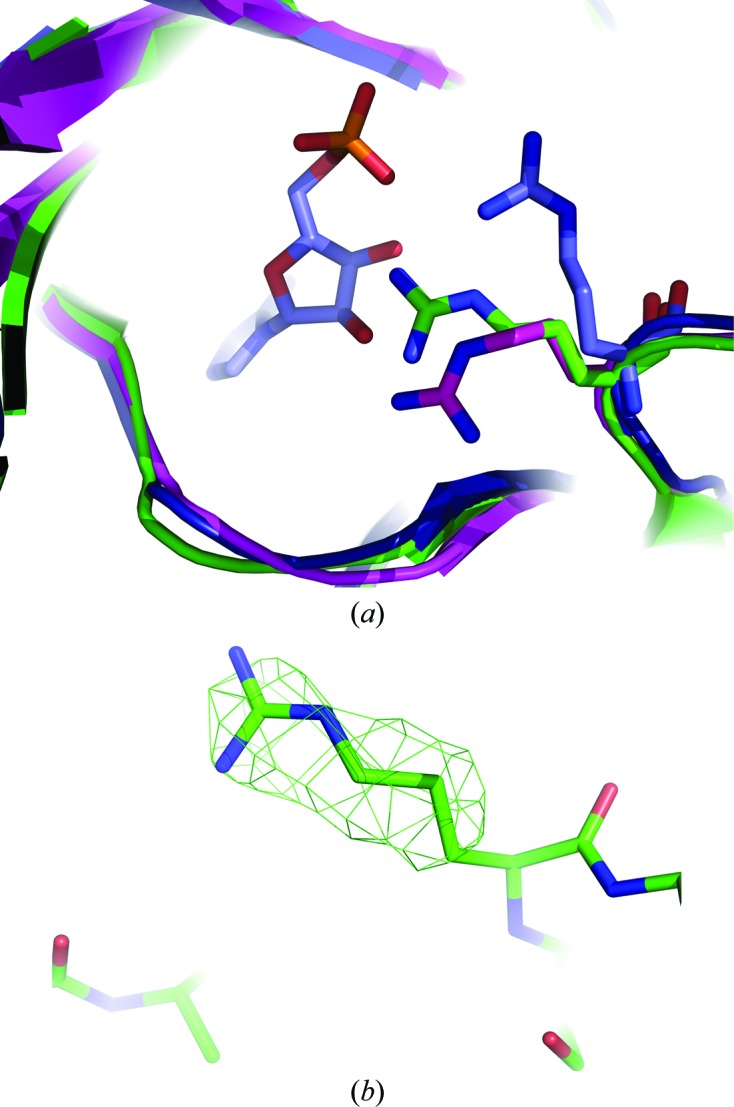
Superposition of the active sites of LigTh1519 (green), PfuLig (blue) and SsoLig (magenta). (a) The conformation of Arg280 in LigTh1519 is significantly different from the conformation of Arg269 in PfuLig, which is in the closed conformation and contains an AMP molecule in the active site. The conformations of Arg280 in LigTh1519 and in SsoLig, which is in the open extended conformation, are different, although not significantly. (b) An F obs − F calc OMIT map of the side chain of Arg280 confirms the position of the side chain of Arg280. The map is shown in green and contoured at the 2.5σ level.
In all three DNA ligases that adopt the closed conformation, PfuLig, TsibLig and AfuLig, the conformation of the corresponding Arg residue differs from that observed in the LigTh1519 structure. Note that PfuLig and TsibLig contain an ATP molecule and AfuLig contains phosphate ions in the active site. The position of the corresponding Arg residue in SsoLig, which adopts the open extended conformation and contains no cofactor in the active site, is different and is much closer to the conformation of Arg280 in LigTh1519 (Fig. 4 ▶).
3.2. Comparison of the OBD arrangement in PfuLig and LigTh1519
PfuLig has very high sequence similarity (79%) to LigTh1519; the similarity between their OBDs is 85%. However, the relative positions of the OBDs in the crystal structures differ significantly. Using the PISA server at the European Bioinformatics Institute (http://www.ebi.ac.uk/pdbe/prot_int/pistart.html; Krissinel & Henrick, 2007 ▶), we revealed those amino-acid residues in the structure of PfuLig that participate in hydrogen-bond contacts and salt bridges between the OBD and the other two domains. Alignment of the PfuLig and the LigTh1519 sequences shows that most of the residues are present in the same positions in the structure of LigTh1519 (Table 3 ▶). A few residues (shown in italics in Table 3 ▶) at equivalent positions in LigTh1519 and PfuLig are homologues with similar properties (Glu457→Asp468, Gln229→Asn240, Asp235→Glu246 and Asp532→Glu543).
Table 3. Hydrogen bonds and salt bridges in PfuLig.
(a).
Hydrogen bonds and salt bridges between the AdD and the OBD.
| No. | OBD residue atom | Distance (Å) | AdB residue atom |
|---|---|---|---|
| 1 | Glu426 N | 3.08 | Gln229 OE1 |
| 2 | Lys554 NZ | 3.89 | Asp235 OD2 |
| 3 | Thr424 O | 2.93 | Gln229 NE2 |
| 4 | Glu426 OE1 | 2.56 | Lys420 NZ |
| 5 | Glu426 OE2 | 3.88 | Lys249 NZ |
| 6 | Val528 O | 3.78 | Gln313 NE2 |
| 7 | Asp532 O | 3.02 | Arg270 NH1 |
| 8 | Asp532 OD1 | 2.84 | Arg320 NH1 |
| 9 | Asp533 O | 2.84 | Arg270 N |
| 10 | Lys534 O | 3.55 | Arg270 NH1 |
| 11 | Asp540 OD1 | 3.53 | Arg414 NH2 |
| 12 | Asp532 OD2 | 3.81 | Arg320 NE |
| 13 | Asp532 OD2 | 3.21 | Arg320 NH1 |
(b).
Hydrogen bonds and salt bridges between the DBD and the OBD.
| No. | OBD residue atom | Distance (Å) | DdB residue atom |
|---|---|---|---|
| 1 | Tyr454 OH | 2.55 | Glu168 OE2 |
| 2 | Glu457 OE1 | 2.53 | Arg172 NH2 |
| 3 | Glu457 OE2 | 3.26 | Arg172 NH1 |
| 4 | Glu457 OE1 | 3.03 | Arg172 NH1 |
The PfuLig molecule contains a C-terminal extension helix (residues 541–561) which passes through the cleft between the AdD and the OBD and contributes, mainly through ionic interactions, to the link between these two domains. Nishida et al. (2006 ▶) proposed that the C-terminal extension helix in the PfuLig structure plays a crucial role in stabilizing the closed conformation. In the C-terminal parts of PfuLig and LigTh1519 only two residues differ. In the structure of LigTh1519 seven terminal residues are disordered and, as a result, the terminal helix is shorter than that in the PfuLig structure. In fact, the superposition of the OBDs of PfuLig and LigTh1519 shows that the terminal part of the C-terminal extension helix would overlap with residues of the AdD of LigTh1519 in the vicinity of the active site.
Thus, the difference between the sequences of PfuLig and LigTh1519 cannot explain the difference in the arrangement of the OBD. In other words, it seems that there is nothing that prevents the LigTh1519 molecule from adopting the closed conformation.
3.3. Comparison of the OBD arrangement in SsoLig and LigTh1519
The sequence similarity between SsoLig and LigTh1519 is considerably lower (40%) than that between PfuLig and LigTh1519. Alignment of the SsoLig and the LigTh1519 sequences reveals that the hinge between the AdD and the OBD in the SsoLig structure comprises eight amino acids, while in the LigTh1519, PfuLig and AfuLig structures the hinge involves only six amino acids. The larger hinge might favour the open extended conformation in the crystal structure of SsoLig and may explain why this conformation is not observed in the crystal structures of PfuLig and LigTh1519. In addition, the arrangement of the OBD in the structure of SsoLig is fixed by numerous hydrogen bonds and salt bridges between the OBD and the AdD (Table 4 ▶). With the exception of Arg564 (shown in bold in Table 4 ▶), the enumerated residues or their homologues are present at the same positions in LigTh1519.
Table 4. Hydrogen bonds and salt bridges between the AdD and the OBD in SsoLig.
Residues that are present in LigTh1519 are shown in plain text. Ser398, Ile404 and Ser405 (shown in italics) are not present in LigTh1519; residues with similar properties are present at the equivalent positions. Arg564 (shown in bold) is not present in LigTh1519; Ala540 is at the equivalent position. Note that residues in equivalent positions in the models of SsoLig and LigTh1519 have different numbers.
| No. | OBD residue atom | Distance (Å) | AdB residue atom |
|---|---|---|---|
| 1 | Arg561 NH1 | 3.24 | Glu394 OE1 |
| 2 | Arg561 NH2 | 2.71 | Glu394 OE2 |
| 3 | Arg561 NH1 | 3.65 | Ser398 OG |
| 4 | Arg561 NH2 | 3.18 | Glu394 OE1 |
| 5 | Arg564 NH2 | 3.84 | Ile404 O |
| 6 | Arg564 NH2 | 2.66 | Ser405 O |
| 7 | Gly488 O | 2.86 | Arg402 NH1 |
| 8 | Gln493 OE1 | 2.72 | Arg402 NH1 |
3.4. SAXS experiment
An SAXS experiment was performed in order to reveal the conformation of LigTh1519 in solution.
The scattering intensity from known PDB structures was calculated using CRYSOL (Svergun et al., 1995 ▶), which takes the scattering from the excluded volume and hydration shell into account. The scattering curve calculated from the SsoLig structure is in better agreement with the experimental intensities than the scattering curve calculated from LigTh1519 (Fig. 5 ▶ a, Table 5 ▶).
Figure 5.

Results of the SAXS experiment. (a) SAXS intensity profiles for the experimental data (circles) and calculated from models of SsoLig (continuous line) and LigTh1519 (dashed line) and a mixture of the SsoLig (81%) and LigTh1519 (19%) models (dashed/dotted line). The scattering profile for the mixture is shifted vertically by 0.05. The experimental profile is shown twice; one of the copies is also shifted vertically by 0.05 for comparison with the profile for the mixture. (b) Normalized pair-distance distribution functions calculated from experimental SAXS data (circles) and from models of SsoLig (continuous line) and LigTh1519 (dashed line) in the crystalline state.
Table 5. Structural parameters derived from SAXS experimental data and calculated from the models of SsoLig and of LigTh1519 in the crystalline state and a mixture of the SsoLig and LigTh1519 models.
R g is the radius of gyration and D max is the maximum interatomic distance.
| R g (nm) | D max (nm) | χ2 experiment–model | |
|---|---|---|---|
| SAXS experimental data | 3.4 ± 0.05 | 11.5 ± 0.5 | — |
| SsoLig | 3.3 ± 0.01 | 10.8 ± 0.2 | 0.84 |
| LigTh1519 | 3.1 ± 0.01 | 9.2 ± 0.2 | 1.1 |
| Mixture of the SsoLig (81 ± 3%) and LigTh1519 (19 ± 3%) models | 3.28 ± 0.01 (averaged value) | — | 0.78 |
The pair-distance distribution functions p(r) were calculated from experimental intensities using GNOM (Svergun et al., 1988 ▶). The radius of gyration R g was calculated using the Guinier approximation (Guinier, 1939 ▶) at s R g < 1.5,
and the relation (International Tables for Crystallography, 1992 ▶)
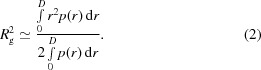 |
Estimate (2) is much less influenced by aggregation effects, because relatively large aggregates significantly increase the scattering intensity only at small angles, thus increasing estimate (1). The approximately equal values, 3.40 ± 0.05 and 3.41 ± 0.05 nm, obtained from (1) and (2), respectively, indicate that aggregation was negligibly small. The maximal size D max of dissolved molecules (or the maximal interatomic distance) was estimated from the profile of the p(r) function.
The graph for the normalized distance-distribution function and structural parameters calculated from SAXS experimental data are much closer to those calculated for the SsoLig model than to those of LigTh1519 (Fig. 5 ▶ b and Table 5 ▶). The right shoulder on the experimental distribution curve indicates the presence of relatively large distances intrinsic to the open extended conformation of the protein. Thus, we concluded that the LigTh1519 molecule adopts an open extended conformation in solution, but not the conformation that it shows in the crystalline state. We also checked whether the experimental data can be fitted to the data calculated for a mixture of SsoLig and LigTh1519 form factors using the program OLIGOMER (Konarev et al., 2003 ▶). The results showed (see Table 5 ▶ and Fig. 5 ▶ a) that a somewhat better fit to the experimental data was attained for the mixture of SsoLig (81%) and LigTh1519 (19%). Thus, LigTh1519 molecules might exist in both conformations in solution, with scattering from the molecule in the open extended conformation being dominant.
3.5. A new intermediate conformation of ATP-dependent DNA ligase
The structure of LigTh1519 was studied using X-ray crystallography in combination with SAXS and comparative structure analysis with known archaeal ATP-dependent DNA ligases. A new conformation of LigTh1519 was revealed which is intermediate between the open extended and closed conformations. Taking the absence of bound or trapped ATP or phosphate ions in the LigTh1519 structure into account, in contrast to PfuLig, TsibLig or AfuLig, we might consider this intermediate conformation of LigTh1519 as a structural variation of the open extended conformation. In the LigTh1519 crystal the absence of stabilizing hydrogen bonds between the OBD and the AdD is partially compensated for by hydrogen bonds and salt bridges between the OBD and the neighbouring symmetry-related molecules; the OBD is additionally fixed by eight intermolecular hydrogen bonds and three intermolecular salt bridges.
Previously, based on the strikingly different positions of the OBD in different DNA ligases, a conclusion was made about high OBD flexibility, which is essential for the function of DNA ligases. Moreover, if the respective movement of the AdD and the OBD is restricted, the efficiency of adenylation will be reduced (Ochi et al., 2012 ▶). Here, we report further experimental evidence for this high flexibility. The amino-acid composition, the structural arrangement of the catalytic domains (AdD and OBD) and the presence of ligands do not explain the advantage of the open, closed or intermediate conformations for DNA-free enzymes in crystals. However, the SAXS experiment showed that the LigTh1519 molecule in solution predominantly adopts the open extended conformation, thereby fixing the OBD and providing additional stabilization of the whole structure in an aqueous environment.
Supplementary Material
PDB reference: LigTh1519, 3rr5
Acknowledgments
This work was supported by the Ministry of Education and Sciences of Russia (contract 02.740.11.0765 and grant NS 7200.2012.4).
References
- Afonine, P. V., Grosse-Kunstleve, R. W. & Adams, P. D. (2005). CCP4 Newsl. 42, contribution 8.
- Bezsudnova, E. Y., Kovalchuk, M. V., Mardanov, A. V., Poliakov, K. M., Popov, V. O., Ravin, N. V., Skryabin, K. G., Smagin, V. A., Stekhanova, T. N. & Tikhonova, T. V. (2009). Acta Cryst. F65, 368–371. [DOI] [PMC free article] [PubMed]
- Cotner-Gohara, E., Kim, I.-K., Hammel, M., Tainer, J. A., Tomkinson, A. E. & Ellenberger, T. (2010). Biochemistry, 49, 6165–6176. [DOI] [PMC free article] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Feigin, L. A. & Svergun, D. I. (1987). Structure Analysis by Small-Angle X-ray and Neutron Scattering, p. 363. New York: Plenum Press.
- Guinier, A. (1939). Ann. Phys. (Paris), 12, 161–236.
- International Tables for Crystallography (1992). Vol. C, Mathematical, Physical and Chemical Tables, 1st ed., edited by A. J. C. Wilson, p. 91. Dordrecht/Boston/London: Kluwer Academic Publishers.
- Keppetipola, N. & Shuman, S. (2005). J. Bacteriol. 187, 6902–6908. [DOI] [PMC free article] [PubMed]
- Kim, D. J., Kim, O., Kim, H.-W., Kim, H. S., Lee, S. J. & Suh, S. W. (2009). Acta Cryst. F65, 544–550. [DOI] [PMC free article] [PubMed]
- Kim, Y. J., Lee, H. S., Bae, S. S., Jeon, J. H., Yang, S. H., Lim, J. K., Kang, S. G., Kwon, S.-T. & Lee, J.-H. (2006). Biotechnol. Lett. 28, 401–407. [DOI] [PubMed]
- Konarev, P. V., Volkov, V. V., Sokolova, A. V., Koch, M. H. J. & Svergun, D. I. (2003). J. Appl. Cryst. 36, 1277–1282.
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol. 372, 774–797. [DOI] [PubMed]
- Lai, X., Shao, H., Hao, F. & Huang, L. (2002). Extremophiles, 6, 469–477. [DOI] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst. 26, 283–291.
- Lehman, I. R. (1974). Science, 186, 790–797. [DOI] [PubMed]
- Long, F., Vagin, A. A., Young, P. & Murshudov, G. N. (2008). Acta Cryst. D64, 125–132. [DOI] [PMC free article] [PubMed]
- Nakatani, M., Ezaki, S., Atomi, H. & Imanaka, T. (2000). J. Bacteriol. 182, 6424–6433. [DOI] [PMC free article] [PubMed]
- Nishida, H., Kiyonari, S., Ishino, Y. & Morikawa, K. (2006). J. Mol. Biol. 360, 956–967. [DOI] [PubMed]
- Ochi, T., Wu, Q., Chirgadze, D. Y., Grossmann, J. G., Bolanos-Garcia, V. M. & Blundell, T. L. (2012). Structure, 20, 1212–1222. [DOI] [PMC free article] [PubMed]
- Odell, M., Malinina, L., Sriskanda, V., Teplova, M. & Shuman, S. (2003). Nucleic Acids Res. 31, 5090–5100. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Pascal, J. M., O’Brien, P. J., Tomkinson, A. E. & Ellenberger, T. (2004). Nature (London), 432, 473–478. [DOI] [PubMed]
- Pascal, J. M., Tsodikov, O. V., Hura, G. L., Song, W., Cotner, E. A., Classen, S., Tomkinson, A. E., Tainer, J. A. & Ellenberger, T. (2006). Mol. Cell, 24, 279–291. [DOI] [PubMed]
- Petrova, T. E., Bezsudnova, E. Y., Dorokhov, B. D., Slutskaya, E. S., Polyakov, K. M., Dorovatovskiy, P. V., Ravin, N. V., Skryabin, K. G., Kovalchuk, M. V. & Popov, V. O. (2012). Acta Cryst. F68, 163–165. [DOI] [PMC free article] [PubMed]
- Rolland, J.-L., Gueguen, Y., Persillon, C., Masson, J.-M. & Dietrich, J. (2004). FEMS Microbiol. Lett. 236, 267–273. [DOI] [PubMed]
- Smagin, V. A., Mardanov, A. V., Bonch-Osmolovskaia, E. A. & Ravin, N. V. (2008). Prikl. Biokhim. Mikrobiol. 44, 523–528. [PubMed]
- Sriskanda, V., Kelman, Z., Hurwitz, J. & Shuman, S. (2000). Nucleic Acids Res. 28, 2221–2228. [DOI] [PMC free article] [PubMed]
- Subramanya, H. S., Doherty, A. J., Ashford, S. R. & Wigley, D. B. (1996). Cell, 85, 607–615. [DOI] [PubMed]
- Sun, Y., Seo, M. S., Kim, J. H., Kim, Y. J., Kim, G. A., Lee, J. I., Lee, J.-H. & Kwon, S.-T. (2008). Environ. Microbiol. 10, 3212–3224. [DOI] [PubMed]
- Supangat, S., An, Y. J., Sun, Y., Kwon, S.-T. & Cha, S.-S. (2010). Acta Cryst. F66, 1583–1585. [DOI] [PMC free article] [PubMed]
- Svergun, D., Barberato, C. & Koch, M. H. J. (1995). J. Appl. Cryst. 28, 768–773.
- Svergun, D. I., Semenyuk, A. V. & Feigin, L. A. (1988). Acta Cryst. A44, 244–250.
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: LigTh1519, 3rr5