

The Xpln crystal structure provides structural insights into Rho GTPase binding.
Keywords: GEF proteins, DH-PH module structure
Abstract
Xpln is a guanine nucleotide-exchange factor (GEF) for Rho GTPases. A Dbl homology (DH) domain followed by a pleckstrin homology (PH) domain is a widely adopted GEF-domain architecture. The Xpln structure solely comprises these two domains. Xpln activates RhoA and RhoB, but not RhoC, although their GTPase sequences are highly conserved. The molecular mechanism of the selectivity of Xpln for Rho GTPases is still unclear. In this study, the crystal structure of the tandemly arranged DH-PH domains of mouse Xpln, with a single molecule in the asymmetric unit, was determined at 1.79 Å resolution by the multiwavelength anomalous dispersion method. The DH-PH domains of Xpln share high structural similarity with those from neuroepithelial cell-transforming gene 1 protein, PDZ-RhoGEF, leukaemia-associated RhoGEF and intersectins 1 and 2. The crystal structure indicated that the α4–α5 loop in the DH domain is flexible and that the DH and PH domains interact with each other intramolecularly, thus suggesting that PH-domain rearrangement occurs upon RhoA binding.
1. Introduction
The Rho-family GTPases, including Rho, Rac and Cdc42, function in cytoskeletal network reorganization and thereby regulate cell migration and cell–cell adhesion (Raftopoulou & Hall, 2004 ▶). The GTPases are activated by guanine nucleotide-exchange factors (GEFs), which promote GDP/GTP exchange (Rossman et al., 2005 ▶; Schmidt & Hall, 2002 ▶). The Dbl homology (DH) domain is responsible for the RhoGEF activity (Hart et al., 1994 ▶). A domain architecture consisting of a DH domain followed by a pleckstrin homology (PH) domain is widely adopted in the Dbl-family GEFs (Rossman et al., 2005 ▶). The Xpln protein is 531 amino acids in length and contains DH and PH domains with flexible N- and C-terminal regions. Among the Dbl-family GEFs, Xpln shares sequence similarity with the neuroepithelial cell-transforming gene 1 (Net1) protein (69% identity within the DH-PH module), which also possesses a DH-PH module with flexible terminal regions as in Xpln. Intersectins 1 and 2 are also closely aligned with Xpln in the phylogenetic tree (Rossman et al., 2005 ▶), although intersectin is the GEF for Cdc42.
Structural analyses have been intensively conducted for the DH-PH modules of GEFs and their complexes with GTPases (Snyder et al., 2002 ▶). The recognition of GTPases by GEFs is accomplished at the molecular surface, including α1, α5 and α6 in the DH domain. In many complex structures, the PH domain makes a minimal contribution to the GEF–GTPase interaction. However, it has also been reported that several residues in the PH domain are important for complex formation between Dbs and RhoA (Snyder et al., 2002 ▶).
The selectivity of GTPases by GEFs has been discussed in terms of the interactions among the residues on α1, α5 and α6 in the DH domain; for the Rho GTPases, the particularly important interactions for RhoA selectivity are the electrostatic interactions between the basic residues on the α4–α5 loop in the GEF and Asp45 and/or Glu54 in RhoA (Snyder et al., 2002 ▶). Xpln possesses a lysine at the corresponding position to the other RhoA-specific GEFs. In the present study, we have solved the crystal structure of the mouse Xpln DH-PH module at 1.79 Å resolution using the multi-wavelength anomalous dispersion (MAD) method. The crystal structure revealed the flexibility of the α4–α5 loop and the intramolecular interactions between the DH and PH domains, which overlap with the putative RhoA-binding site.
2. Materials and methods
2.1. Protein expression and purification
The DH-PH module of mouse Xpln (residues 110–448 of Swiss-Prot entry Q91X46), with an N-terminal natural polyhistidine affinity (NHis) tag and a TEV protease cleavage site, was prepared by a two-step PCR method (Yabuki et al., 2007 ▶). The DNA fragment thus produced was cloned into the TA vector pCR2.1TOPO (Invitrogen). Selenomethionine-labelled mouse Xpln DH-PH module with an N-terminal NHis tag was produced using a cell-free expression system (Kigawa et al., 2002 ▶). The protein was loaded onto a HisTrap column (GE Healthcare) equilibrated with 20 mM Tris–HCl buffer pH 8.0 containing 1 M NaCl and 20 mM imidazole. The Xpln protein was eluted with a linear gradient of 20–500 mM imidazole. The peak fractions were pooled and applied onto a HiPrep desalting column (GE Healthcare). Tag cleavage was performed at 303 K for 60 min with TEV protease (46 µg ml−1). A six-linker sequence (SSGSSG) was inserted between the TEV protease recognition site (EHLYFQ↓G) and Cys110, in addition to a C-terminal insertion (SGPSSG). Consequently, seven residues (GSSGSSG) at the N-terminus and six residues at the C-terminus remained after tag cleavage using TEV protease. The tag-cleaved protein was applied onto a HisTrap column to remove the His-tag fragment and the buffer was exchanged to 20 mM Tris–HCl buffer pH 8.5 containing 50 mM NaCl and 5 mM β-mercaptoethanol on a desalting column. The sample solution was subsequently loaded onto a Mono Q column (GE Healthcare) equilibrated with 20 mM Tris–HCl buffer pH 8.5 containing 50 mM NaCl and 5 mM β-mercaptoethanol and eluted with a linear gradient of 0–1 M NaCl. Fractions containing the protein were pooled and subjected to gel filtration on a HiLoad Superdex 75 (GE Healthcare) column equilibrated with 20 mM Tris–HCl buffer pH 8.0 containing 150 mM NaCl and 2 mM DTT. The Xpln protein was finally concentrated to 18.0 mg ml−1. The protein concentration was determined by spectrophotometric measurements at 280 nm using an extinction coefficient of 29 910 l mol−1 cm−1.
2.2. Crystallization and data collection
Xpln was crystallized by the sitting-drop vapour-diffusion method at 293 K immediately after purification. The drops were composed of 1.0 µl protein solution and 1.0 µl reservoir solution (0.2 M MES buffer pH 6.5 containing 0.2 M ammonium sulfate and 30% polyethylene glycol monomethyl ether). The crystallization well contained 90 µl reservoir solution. The crystals grew to approximate dimensions of 0.2 × 0.05 × 0.05 mm in a week. Data collection was performed using crystals that had been transferred into the crystallization mother solution for 1 min before flash-cooling in a nitrogen stream at 110 K. The reflection data sets were collected at three wavelengths, 0.97888 Å (peak), 0.97964 Å (edge) and 0.96400 Å (remote), to 1.79 Å resolution on beamline BL-17A at the Photon Factory, Tsukuba, Japan. All diffraction data sets were integrated and scaled with the HKL-2000 program suite (Otwinowski & Minor, 1997 ▶). The Matthews coefficient was evaluated as V M = 2.51 Å3 Da−1, assuming the presence of one molecule in the asymmetric unit.
2.3. Structure determination and refinement
The crystal structure of Xpln revealed one protein molecule in the asymmetric unit and was determined using the MAD method. The determination of the selenium sites and the calculation of the MAD phases were accomplished with the program SOLVE (Terwilliger & Berendzen, 1999 ▶) using the fully measured data sets, and two selenium positions were identified. The resulting electron-density map was considerably improved by density modification with the program RESOLVE (Terwilliger & Berendzen, 1999 ▶). The protein model was built by ARP/wARP (Morris et al., 2004 ▶) and was modified manually into the electron-density map using the program O (Jones et al., 1991 ▶). The structure was refined with the program CNS (Brünger et al., 1998 ▶).
Since the N-terminal and C-terminal ends and the two loop regions [GSSGSSG plus Cys110–Asn112, Leu128–Leu140, Arg400–Gly401 and the C-terminal end (SGPSSG)] could not be identified in the electron-density map, these residues were excluded from the coordinates. The final model was assessed by PROCHECK in the CCP4 suite (Winn et al., 2011 ▶). The data-collection and refinement statistics are summarized in Table 1 ▶. The ribbon and molecular-surface models shown in the figures were produced using MolFeat (FiatLux, Tokyo, Japan). The atomic coordinates have been deposited in the Protein Data Bank with accession code 2z0q.
Table 1. Crystal parameters and data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Peak | Edge | Remote | |
|---|---|---|---|
| Crystal characteristics | |||
| Space group | P212121 | ||
| Unit-cell parameters (Å) | a = 51.6, b = 62.7, c = 115.3 | ||
| Molecules in asymmetric unit | 1 | ||
| Protein molecular weight (Da) | 40202.6 | ||
| MAD data | |||
| Wavelength (Å) | 0.97888 | 0.97964 | 0.96400 |
| Resolution range (Å) | 42.4–1.79 (1.85–1.79) | 42.5–1.79 (1.85–1.79) | 42.5–1.79 (1.85–1.79) |
| Observed reflections | 251364 | 250794 | 251684 |
| Unique reflections | 35822 (3441) | 35888 (3450) | 35883 (3463) |
| Multiplicity | 7.0 (6.5) | 7.0 (6.5) | 7.0 (6.5) |
| Completeness (%) | 99.4 (96.4) | 99.4 (96.4) | 99.4 (96.8) |
| 〈I/σ(I)〉 | 28.2 (4.7) | 28.4 (3.2) | 26.3 (2.7) |
| R merge † | 0.059 (0.443) | 0.063 (0.610) | 0.069 (0.737) |
| f′′/f′ | 3.83/−4.84 | 1.83/−7.58 | 3.6/−3.2 |
| Wilson B factor (Å2) | 23.8 | 24.8 | 24.6 |
| Figure of merit (FOM) | |||
| Before solvent modification | 0.33 | ||
| After solvent modification | 0.59 | ||
| Refinement statistics | |||
| Resolution range (Å) | 42.4–1.79 | ||
| No. of reflections used | 35760 | ||
| R factor‡ | 0.203 (0.244) | ||
| Free R factor‡ | 0.243 (0.286) | ||
| No. of protein atoms | 2614 | ||
| No. of ion atoms§ | 4 | ||
| No. of water molecules | 360 | ||
| R.m.s. deviation from ideal geometry | |||
| Bond lengths (Å) | 0.013 | ||
| Bond angles (°) | 1.50 | ||
| Average isotropic B value (Å2) | |||
| Protein atoms | 27.9 | ||
| Nonprotein atoms | 34.8 | ||
| Ramachandran plot | |||
| Most favoured regions (%) | 95.5 | ||
| Allowed regions (%) | 4.5 | ||
R
merge = 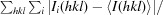
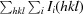 .
.
R = 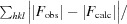
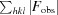 . The free R factor was calculated using 5% of reflections omitted from refinement.
. The free R factor was calculated using 5% of reflections omitted from refinement.
Sulfate ions.
3. Results and discussion
3.1. DH-PH module structure
The structure of the Xpln DH-PH module (residues 110–448) was solved by the MAD method at 1.79 Å resolution. The DH domain is composed of elongated helical bundles, including six major helical segments (α1–α6; Fig. 1 ▶ a). The α1 and α2 segments are further divided into two helices. The α6 segment forms a long helix consisting of 34 residues. The loop region after the short helix α4 was not modelled owing to disorder, as mentioned above. This loop is located near the solvent region in the crystal and makes no interactions in the crystal packing. The PH domain is composed of seven β-strands flanked by an α-helix (αC), forming an antiparallel β-sandwich structure, as described in other PH-domain structures (Lemmon, 2003 ▶). The PH domain is located adjacent to the DH domain and is stabilized by four hydrogen bonds (Leu369 O⋯Arg315 Nη1, Tyr371 N⋯Arg315 O, Tyr371 O⋯Leu317 N and Leu373 N⋯Leu317 O) as well as van der Waals contacts between α3 and α6 in the DH domain (Ala222, His226, Arg315 and Leu317) and the β3–β4 loop (His365, Gln368, Leu369 and Tyr371) in the PH domain (Fig. 1 ▶ b).
Figure 1.

Structure of Xpln. (a) Ribbon representation of the Xpln structure. The secondary structures are coloured blue (helices) and yellow (strands). (b) Interdomain interactions between the DH and PH domains. (c) Superimposed structures of Xpln and LARG (PDB entry 1txd). The ribbon models are coloured pink (Xpln) and cyan (LARG).
3.2. Structural comparison with other RhoGEFs
A structure-similarity search for Xpln was performed using the DALI server (Holm & Rosenström, 2010 ▶). The highest Z-score (22.9) was assigned to the RhoA-specific GEF protein Net1 (PDB entry 3eo2; Structural Genomics Consortium, unpublished work). Other Rho-specific GEFs, including PDZ-RhoGEF (Bielnicki et al., 2011 ▶; Derewenda et al., 2004 ▶; Chen et al., 2010 ▶), leukaemia-associated RhoGEF (LARG; Kristelly et al., 2004 ▶) and p115-RhoGEF (Chen et al., 2011 ▶), shared structural homology with Xpln with high Z-scores (higher than 20). Intersectin also shared a high structural similarity with Xpln, although it is a GEF for Cdc42 (Snyder et al., 2002 ▶; Kapp et al., 2012 ▶; Ahmad & Lim, 2010 ▶). Superimposition of Xpln with the other GEF structures revealed that the relative position of the Xpln PH domain is quite different from those in the other GEFs. Superimposed structures of Xpln and the DH-PH module of LARG (PDB entry 1txd; Kristelly et al., 2004 ▶) are shown in Fig. 1 ▶(c). In the other GEFs interdomain interactions were not observed within the molecule, although differences in the relative orientations between the DH and PH domains in the DH-PH module structures are commonly observed (Kristelly et al., 2004 ▶). Crystal-packing contacts occur between the PH domain and α5, α6 and αC of the neighbouring molecules. Although the packing contacts might influence the relative orientation of the DH-PH module, its orientation in the current crystal structure of Xpln seems to be a unique variation.
In the Xpln structure, the α4–α5 loop was not identified owing to disorder. On the other hand, in the DH-domain structure of Net1, the top homologue of Xpln, the corresponding loop was included in the refined structure. In the Net1 structure, α4 is longer than in Xpln and is flanked by the C-terminal end of α1. Nevertheless, the sequence of the α4–α5 loop is fully conserved between Xpln and Net1. The crystal packing revealed that the α4–α5 loop of Net1 forms intermolecular contacts with α1 and the α2–α3 loop of the neighbouring molecule in the crystal. The α4–α5 loop in Xpln/Net1 may be flexible in solution and become folded upon contacting the other molecule. A secondary-structure prediction by PSIPred (Buchan et al., 2010 ▶) suggested that α4 of Xpln has the potential to form a longer helix.
3.3. RhoA-binding model
The relative orientation between the DH and PH domains in the Xpln structure was distinct from those in the structures of other DH-PH modules. In the modelled structure with RhoA, the current PH-domain position definitely conflicts with RhoA (Fig. 2 ▶ b). Therefore, the DH-PH domain arrangement in the RhoA–Xpln complex will be different from that in the current crystal structure. Although it is difficult to predict the position of the PH domain in the complex, the relatively long β3–β4 loop (nine residues in Xpln; six residues in PDZRhoGEF and LARG) seems to be similar to that in the RhoA–Dbs complex (Snyder et al., 2002 ▶), in which the long β3–β4 loop (11 residues) is involved in RhoA recognition. Several factors may be involved in the selectivity of Rho GTPases by Xpln. The interactions between the PH domain and RhoA must be considered to be one of the possible determinants of selectivity. The crystal structure of Xpln complexed with a Rho GTPase will clarify the molecular mechanism of the selectivity and preparation of this complex is currently under way.
Figure 2.

Model of the RhoA–Xpln complex. (a) The ribbon models are coloured pink (Xpln) and orange (RhoA). The loop region between α4 and α5 is depicted by a dashed line. The PH domain of Xpln is omitted from the figure for clarity. (b) Structural conflict in the model. RhoA is shown in a surface representation. The arrow indicates the overlapping structural regions between the Xpln PH domain and RhoA.
Supplementary Material
PDB reference: Xpln, 2z0q
Acknowledgments
We thank Yuki Kamewari-Hayami, Machiko Yamaguchi-Hirafuji and Aishan Tuerxun for assistance with protein preparation and crystallization. We thank Dr Takanori Kigawa for the preparation of PCR fragments. We are grateful to Dr Yoshihide Hayashizaki (RIKEN OSC) for the FANTOM clone (E430019N10). The synchrotron-radiation experiments were performed at the Photon Factory, Tsukuba, Japan. We would like to thank the beamline staff at BL17A of the Photon Factory for assistance during data collection. This work was supported by the RIKEN Structural Genomics/Proteomics Initiative (RSGI), the National Project on Protein Structural and Functional Analyses and Special Coordination Funds for Promoting Science and Technology through the Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
- Ahmad, K. F. & Lim, W. A. (2010). PLoS One, 5, e11291. [DOI] [PMC free article] [PubMed]
- Bielnicki, J. A., Shkumatov, A. V., Derewenda, U., Somlyo, A. V., Svergun, D. I. & Derewenda, Z. S. (2011). J. Biol. Chem. 286, 35163–35175. [DOI] [PMC free article] [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Buchan, D. W., Ward, S. M., Lobley, A. E., Nugent, T. C., Bryson, K. & Jones, D. T. (2010). Nucleic Acids Res. 38, W563–W568. [DOI] [PMC free article] [PubMed]
- Chen, Z., Guo, L., Sprang, S. R. & Sternweis, P. C. (2011). Protein Sci. 20, 107–117. [DOI] [PMC free article] [PubMed]
- Chen, Z., Medina, F., Liu, M., Thomas, C., Sprang, S. R. & Sternweis, P. C. (2010). J. Biol. Chem. 285, 21070–21081. [DOI] [PMC free article] [PubMed]
- Derewenda, U., Oleksy, A., Stevenson, A. S., Korczynska, J., Dauter, Z., Somlyo, A. P., Otlewski, J., Somlyo, A. V. & Derewenda, Z. S. (2004). Structure, 12, 1955–1965. [DOI] [PubMed]
- Hart, M. J., Eva, A., Zangrilli, D., Aaronson, S. A., Evans, T., Cerione, R. A. & Zheng, Y. (1994). J. Biol. Chem. 269, 62–65. [PubMed]
- Holm, L. & Rosenström, P. (2010). Nucleic Acids Res. 38, W545–W549. [DOI] [PMC free article] [PubMed]
- Jones, T. A., Zou, J.-Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed]
- Kapp, G. T., Liu, S., Stein, A., Wong, D. T., Reményi, A., Yeh, B. J., Fraser, J. S., Taunton, J., Lim, W. A. & Kortemme, T. (2012). Proc. Natl Acad. Sci. USA, 109, 5277–5282. [DOI] [PMC free article] [PubMed]
- Kigawa, T., Yamaguchi-Nunokawa, E., Kodama, K., Matsuda, T., Yabuki, T., Matsuda, N., Ishitani, R., Nureki, O. & Yokoyama, S. (2002). J. Struct. Funct. Genomics, 2, 29–35. [DOI] [PubMed]
- Kristelly, R., Gao, G. & Tesmer, J. J. G. (2004). J. Biol. Chem. 279, 47352–47362. [DOI] [PubMed]
- Lemmon, M. A. (2003). Traffic, 4, 201–213. [DOI] [PubMed]
- Morris, R. J., Zwart, P. H., Cohen, S., Fernandez, F. J., Kakaris, M., Kirillova, O., Vonrhein, C., Perrakis, A. & Lamzin, V. S. (2004). J. Synchrotron Rad. 11, 56–59. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Raftopoulou, M. & Hall, A. (2004). Dev. Biol. 265, 23–32. [DOI] [PubMed]
- Rossman, K. L., Der, C. J. & Sondek, J. (2005). Nature Rev. Mol. Cell Biol. 6, 167–180. [DOI] [PubMed]
- Schmidt, A. & Hall, A. (2002). Genes Dev. 16, 1587–1609. [DOI] [PubMed]
- Snyder, J. T., Worthylake, D. K., Rossman, K. L., Betts, L., Pruitt, W. M., Siderovski, D. P., Der, C. J. & Sondek, J. (2002). Nature Struct. Biol. 9, 468–475. [DOI] [PubMed]
- Terwilliger, T. C. & Berendzen, J. (1999). Acta Cryst. D55, 849–861. [DOI] [PMC free article] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Yabuki, T., Motoda, Y., Hanada, K., Nunokawa, E., Saito, M., Seki, E., Inoue, M., Kigawa, T. & Yokoyama, S. (2007). J. Struct. Funct. Genomics, 8, 173–191. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: Xpln, 2z0q