

Crystals of S. scrofa quinolinate phosphoribosyltransferase purified from porcine kidney in complex with nicotinate mononucleotidewere obtained and diffraction data were collected and processed to 2.1 Å resolution.
Keywords: quinolinate phosphoribosyltransferase, NAD biosynthesis, nicotinate mononucleotide
Abstract
Quinolinate phosphoribosyltransferase (QAPRTase) is a key enzyme in NAD biosynthesis; it catalyzes the formation of nicotinate mononucleotide (NAMN) from quinolinate and 5-phosphoribosyl-1-pyrophosphate. In order to elucidate the mechanism of NAMN biosynthesis, crystals of Sus scrofa QAPRTase (Ss-QAPRTase) purified from porcine kidney in complex with NAMN were obtained and diffraction data were collected and processed to 2.1 Å resolution. The Ss-QAPRTase–NAMN cocrystals belonged to space group P321, with unit-cell parameters a = 119.1, b = 119.1, c = 93.7 Å, γ = 120.0°. The Matthews coefficient and the solvent content were estimated as 3.10 Å3 Da–1 and 60.3%, respectively, assuming the presence of two molecules in the asymmetric unit.
1. Introduction
Nicotinamide adenine dinucleotide (NAD) is a key component that is involved in numerous biological processes. NAD serves as a major hydrogen donor or acceptor in redox reactions in various metabolic pathways and also plays a key role in DNA repair, synthesis and recombination and protein post-translational modification such as poly- and mono-ADP-ribosylation and NAD-dependent protein deacetylation (Olivera & Lehman, 1967 ▶; Lehman, 1974 ▶; Chiarugi, 2002 ▶). Moreover, NAD can act as a precursor molecule of intracellular calcium-mobilizing agents, including cyclic ADP-ribose (cADPR) and nicotinate adenine dinucleotide phosphate (NAADP) (Lee, 2001 ▶). There are two pathways for the synthesis of NAD: a de novo pathway from intracellular tryptophan or nicotinate and a salvage pathway from nicotinamide as a precursor from dietary niacin. Quinolinate phosphoribosyltransferase (QAPRTase; EC 2.4.2.19) is an important intermediate enzyme for the de novo biosynthesis of NAD, especially from quinolinate (QUIN) using tryptophan catabolism, which is named the kynurenine pathway, in most eukaryotes including mammals and in the synthesis of the pyridine ring from other precursors as well as dihydroxylacetone phosphate and l-aspartate in prokaryotes (Foster & Moat, 1980 ▶). This enzyme catalyzes the production of nicotinic acid mononucleotide (NAMN), CO2 and pyrophosphate from QUIN and 5-phosphoribosyl-1-pyrophosphate (PRPP), which involves a decarboxylation reaction and phosphoribosyl transfer. Meanwhile, QUIN is a potent neuropathological agent that causes various neurodegenerative diseases via disturbing the neurotransmitter usage of intrinsic neurons and continuous activation of glutamate N-methyl-d-aspartate receptors (Foster et al., 1985 ▶; Foster & Schwarcz, 1985 ▶; Feldblum et al., 1988 ▶). Thus, the significance of QAPRTase is being magnified as a regulator of cerebral QUIN concentration.
As the result of extensive structural studies of QAPRTase, a series of structures have been determined from Homo sapiens (apo and a tartrate complex mimicking QUIN), Saccharomyces cerevisiae (apo, a PRPP complex, a QUIN complex and a phthalate complex as a QUIN analogue), Helicobacter pylori (a QUIN complex, an NAMN complex and a phthalate complex), Mycobacterium tuberculosis [apo and complexes with QUIN, NAMN and 5-phosphoribosyl-1-(β-methylene) pyrophosphate as a PRPP analogue], Salmonella typhimurium (a QUIN complex), Thermotoga maritima (apo) and Vibrio cholera (apo), which maintain an N-terminal four-stranded antiparallel β-sandwich domain and a C-terminal α/β-barrel domain that are well conserved from bacteria to human (Eads et al., 1997 ▶; Sharma et al., 1998 ▶; Schwarzenbacher et al., 2004 ▶; Kim et al., 2006 ▶; Liu et al., 2007 ▶; di Luccio & Wilson, 2008 ▶). The QAPRTases from H. sapiens and S. cerevisiae have hexameric structures in their biological assembly, which are consistent with experimental results on yeast, rat, porcine and human QAPRTases, which exist as hexamers in solution (Iwai & Taguchi, 1974 ▶; Okuno & Schwarcz, 1985 ▶; Okuno et al., 1988 ▶; Liu et al., 2007 ▶; di Luccio & Wilson, 2008 ▶). In contrast, most QAPRTases from prokaryotes, including those from S. typhimurium and T. maritima, contain dimers in their crystal structures; exceptions are those from H. pylori, M. tuberculosis and Thermus thermophilus, which have been reported to have hexameric structures similar to those of the eukaryotic QAPRTases.
Most structures of prokaryotic QAPRTases have been determined from bacterial pathogens such as H. pylori, M. tuberculosis and S. typhimurium, thus allowing the design of novel antibacterial drugs that target NAD biosynthesis in pathogenic bacteria (Eads et al., 1997 ▶; Sharma et al., 1998 ▶; Kim et al., 2006 ▶). Structural information on human QAPRTase is also important for the development of structure-based antibacterial inhibitors which specifically bind to bacterial targets and avoid host QAPRTase. Currently, structures of eukaryotic QAPRTases have been solved in complexes with PRPP, QUIN, tartrate or phthalate molecules, which mimic QUIN in part and are substrates of QAPRTases. Thus, it is essential to determine the three-dimensional structure of QAPRTase in complex with its product in order to elucidate the phosphoribosyl-transfer and decarboxylation mechanisms of intermediate NAD biosynthesis in eukaryotes. Here, we present the crystallization of QAPRTase purified from porcine kidney, which is highly homologous to the human enzyme (amino-acid sequence identity of ∼90% and similarity of ∼94% for porcine Met1–Glu288 and human Met1–Lys288), in complex with NAMN and its preliminary X-ray crystallographic characterization as the first step in providing structural information and exploring the interaction of mammalian QAPRTase with its product at atomic resolution.
2. Materials and methods
2.1. Purification of Sus scrofa QAPRTase (Ss-QAPRTase)
Ss-QAPRTase was purified from porcine kidney as described previously (Shibata & Iwai, 1980 ▶). Briefly, frozen porcine kidney was homogenized in 50 mM potassium phosphate buffer pH 7.0 containing 10 mM β-mercaptoethanol (standard buffer). The crude extract was centrifuged followed by ammonium sulfate fractionation. The precipitated protein in 40% ammonium sulfate was dissolved in standard buffer and dialyzed for 24 h. The dialyzed solution was loaded onto a DEAE Sephadex A-50 column and eluted with 50–500 mM potassium phosphate pH 7.0, 10 mM β-mercaptoethanol. Fractions containing Ss-QAPRTase were pooled, precipitated using 60% ammonium sulfate and dissolved using standard buffer. The solution was dialyzed for one week with 50 mM Tris–HCl pH 8.5 containing 130 mM sodium citrate and loaded onto a Superdex 200 16/60 column (Pharmacia) equilibrated with 20 mM HEPES–NaOH pH 7.5, 100 mM KCl. The protein eluted as a hexamer. Fractions containing pure Ss-QAPRTase were pooled and concentrated to 15 mg ml−1.
2.2. Crystallization and data collection
Ss-QAPRTase was cocrystallized with NAMN at room temperature (294 ± 1 K) using the hanging-drop vapour-diffusion method. Ss-QAPRTase (15 mg ml−1) in 20 mM HEPES–NaOH pH 7.5, 100 mM KCl was mixed with a half volume of reservoir solution. The reservoir solution consisted of 100 mM Tris–HCl pH 8.0, 16–24%(w/v) polyethylene glycol (PEG) 8000, 150–200 mM ammonium acetate, 5 mM NAMN. Rod-like single crystals grew to maximal dimensions of 0.3 × 0.1 × 0.1 mm over a week (Fig. 1 ▶). For data collection, Ss-QAPRTase crystals were transferred to cryoprotectant solution consisting of 100 mM Tris–HCl pH 8.0, 16–24% PEG 8000, 150–200 mM ammonium acetate, 20%(v/v) ethylene glycol and flash-cooled in liquid nitrogen at 95 K. Native data were collected from the Ss-QAPRTase–NAMN cocrystal on the BL-18B beamline at the Photon Factory, Tsukuba, Japan. The data set was processed and scaled with HKL-2000 (Otwinowski & Minor, 1997 ▶). Data-collection statistics are listed in Table 1 ▶.
Figure 1.
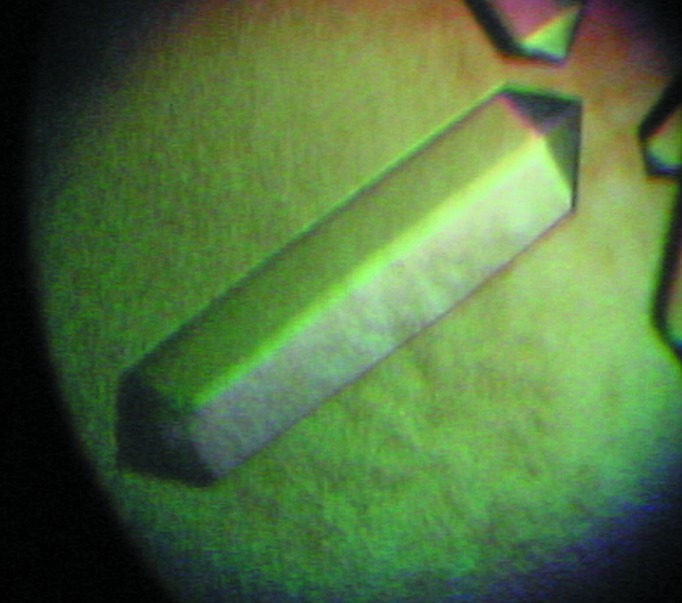
Crystal of Ss-QAPRTase in complex with NAMN grown for 7 d in 100 mM Tris–HCl pH 8.0, 16–24%(w/v) PEG 8K, 150–200 mM ammonium acetate, 5 mM NAMN. The crystal dimensions are approximately 0.3 × 0.1 × 0.1 mm.
Table 1. Data-collection statistics for the Ss-QAPRTase–NAMN complex.
Values in parentheses are for the highest resolution shell.
| Wavelength (Å) | 1.0000 |
| Space group | P321 |
| Unit-cell parameters (Å, °) | a = 119.1, b = 119.1, c = 93.7, γ = 120.0 |
| Resolution range (Å) | 50–2.1 (2.14–2.10) |
| Observed reflections | 264166 |
| Unique reflections | 86365 |
| Multiplicity | 3.1 (3.0) |
| Completeness (%) | 99.5 (100.0) |
| R merge † (%) | 6.4 (36.1) |
| 〈I/σ(I)〉 | 15.5 (3.3) |
R
merge = 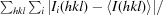
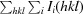 , where Ii(hkl) is the intensity of the ith observation of reflection hkl and 〈I(hkl)〉 is the average intensity of reflection hkl.
, where Ii(hkl) is the intensity of the ith observation of reflection hkl and 〈I(hkl)〉 is the average intensity of reflection hkl.
3. Results and discussion
The Ss-QAPRTase–NAMN cocrystals belonged to space group P321, with unit-cell parameters a = 119.1, b = 119.1, c = 93.7 Å, γ = 120.0°, and diffracted to 2.1 Å resolution. Assuming the presence of two molecules in the asymmetric unit, the Matthews coefficient was calculated to be 3.10 Å3 Da−1, corresponding to a solvent content of 60.3% (Matthews, 1968 ▶). The program Phaser (McCoy et al., 2007 ▶) was employed to calculate phase information in the resolution range 45–2.1 Å using the structure of human QAPRTase (PDB entry 2jbm; Liu et al., 2007 ▶) as the search model. The molecular-replacement solution had a log-likelihood gain of 3230. Although the asymmetric unit contained two subunits of porcine QAPRTase, a hexameric architecture was formed by generation of crystallographic symmetry-related molecules (Fig. 2 ▶). Furthermore, the hexameric structure of porcine QAPRTase is consistent with those from eukaryotes such as yeast and human (PDB entries 3c2e and 2jbm; di Luccio & Wilson, 2008 ▶; Liu et al., 2007 ▶). The resultant electron-density map (R work and R free of 29.3 and 34.7%, respectively) at an early stage of refinement using REFMAC5 (Murshudov et al., 2011 ▶) showed the NAMN structure to fit near the active site, similar to the binding sites of substrates including QUIN and PRPP (Fig. 3 ▶). Model building and further refinement are now in progress.
Figure 2.

The Ss-QAPRTase dimer in the asymmetric unit is shown in yellow and orange. The hexameric architecture is formed by a crystallographic threefold symmetry operation. The noncrystallographic twofold axes and the crystallographic threefold axis are indicated by arrows and a triangle, respectively.
Figure 3.

OMIT map of the Ss-QAPRTase–NAMN complex including NAMN and neighbouring residues contoured at 1.0σ. The NAMN molecule is represented as a stick model; C, O, N and P atoms are coloured grey, red, blue and orange, respectively.
Acknowledgments
This work was supported by the GIST Systems Biology Infrastructure Establishment Grant, the Korea Healthcare Technology R&D Project (A092006) and NRF grants (20120000771 and 20110016226).
References
- Chiarugi, A. (2002). Trends Pharmacol. Sci. 23, 122–129. [DOI] [PubMed]
- di Luccio, E. & Wilson, D. K. (2008). Biochemistry, 47, 4039–4050. [DOI] [PubMed]
- Eads, J. C., Ozturk, D., Wexler, T. B., Grubmeyer, C. & Sacchettini, J. C. (1997). Structure, 5, 47–58. [DOI] [PubMed]
- Feldblum, S., Rougier, A., Loiseau, H., Loiseau, P., Cohadon, F., Morselli, P. L. & Lloyd, K. G. (1988). Epilepsia, 29, 523–529. [DOI] [PubMed]
- Foster, J. W. & Moat, A. G. (1980). Microbiol. Rev. 44, 83–105. [DOI] [PMC free article] [PubMed]
- Foster, A. C. & Schwarcz, R. (1985). J. Neurochem. 45, 199–205. [DOI] [PubMed]
- Foster, A. C., Whetsell, W. O., Bird, E. D. & Schwarcz, R. (1985). Brain Res. 336, 207–214. [DOI] [PubMed]
- Iwai, K. & Taguchi, H. (1974). Biochem. Biophys. Res. Commun. 56, 884–891. [DOI] [PubMed]
- Kim, M., Im, Y. J., Lee, J. H. & Eom, S. H. (2006). Proteins, 63, 252–255. [DOI] [PubMed]
- Lee, H. C. (2001). Annu. Rev. Pharmacol. Toxicol. 41, 317–345. [DOI] [PubMed]
- Lehman, I. R. (1974). Science, 186, 790–797. [DOI] [PubMed]
- Liu, H., Woznica, K., Catton, G., Crawford, A., Botting, N. & Naismith, J. H. (2007). J. Mol. Biol. 373, 755–763. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Okuno, E. & Schwarcz, R. (1985). Biochem. Biophys. Acta, 841, 112–119. [DOI] [PubMed]
- Okuno, E., White, R. J. & Schwarcz, R. (1988). J. Biochem. 103, 1054–1059. [DOI] [PubMed]
- Olivera, B. M. & Lehman, I. R. (1967). Proc. Natl Acad. Sci. USA, 57, 1700–1704. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Schwarzenbacher, R. et al. (2004). Proteins, 55, 768–771. [DOI] [PubMed]
- Sharma, V., Grubmeyer, C. & Sacchettini, J. C. (1998). Structure, 6, 1587–1599. [DOI] [PubMed]
- Shibata, K. & Iwai, K. (1980). Biochim. Biophys. Acta, 611, 280–288. [DOI] [PubMed]