

The CIDE-N domain of Fsp27 expressed in E. coli was purified as a monomer and identified by LC/MS/MS. Crystals of the Fsp27 CIDE-N domain were grown in sitting-drop mode and diffracted to 1.92 Å resolution.
Keywords: CIDEC, Fsp27, obesity, prokaryotic expression, unilocular lipid droplets
Abstract
Fsp27, a member of the CIDE protein family which is selectively expressed in adipocytes, has emerged as a novel regulator for unilocular lipid droplet (LD) formation, lipid metabolism, differentiation of adipocytes and insulin sensitivity. An LD is a subcellular compartment that is used by adipocytes for the efficient storage of fats. The CIDE-N domain of Fsp27 functions as a recruitment platform that induces the correct configuration of the Fsp27 CIDE-C domain to facilitate LD fusion. This study reports the high-yield expression of the mouse Fsp27 CIDE-N domain in Escherichia coli; a two-step purification protocol with high efficiency was established and crystallographic analysis was performed. The purity of the recombinant Fsp27 was >95% as assessed by SDS–PAGE. Crystals were obtained at 291 K using 28% polyethylene glycol 4000 as a precipitant. Diffraction data were collected to 1.92 Å resolution and the crystal belonged to space group P65, with unit-cell parameters a = b = 63.3, c = 37.4 Å, α = β = 90, γ = 120°. The components of the crystal were identified by ion-trap LC/MS/MS spectrometric analysis. The structure has been solved by molecular replacement and refinement is in progress.
1. Introduction
Obesity, diabetes and liver steatosis are a class of diseases that feature metabolic abnormalities and disturbed energy homeostasis; they have become the most common chronic disorders throughout the world. As has been demonstrated previously, adipose tissue plays critical roles in regulating systemic metabolic balance and glucose homeostasis in addition to its fundamental function in storing and utilizing triacylglycerol (TAG; Rajala & Scherer, 2003 ▶).
The CIDE family of proteins CIDEA, CIDEB and CIDEC (also designated Fsp27 in mice), which were originally identified based on homology to the N-terminus of DFF 40/45 (Liang et al., 2003 ▶; Inohara et al., 1998 ▶), are mainly expressed in brown adipose tissue (BAT, an energy-consumption organ), liver and white adipose tissue (WAT, an energy-storage organ) (Zhou et al., 2003 ▶; Li et al., 2007 ▶; Nishino et al., 2008 ▶; Toh et al., 2008 ▶; Gesta et al., 2007 ▶). CIDE protein deficiency-sensitized animals have a lean phenotype with higher energy expenditure and improved insulin sensitivity and are protected from a high-fat diet (Gong et al., 2009 ▶; Li et al., 2007 ▶; Zhou et al., 2003 ▶), demonstrating that this group of proteins play a key role in fatty-acid metabolism.
It has been shown that Fsp27 is highly and selectively expressed in WAT (and moderately in BAT; Danesch et al., 1992 ▶; Toh et al., 2008 ▶). In white adipocytes, excess energy is stored by the accumulation of TAG in the form of distinct large lipid droplets (LDs), which are often characterized as solitary cytoplasmic organelles. The unilocular LDs are composed of a core of neutral TAG and the core is covered by a phospholipid monolayer studded with proteins which regulate LD formation and mobilization (Wolins et al., 2006 ▶; Tauchi-Sato et al., 2002 ▶; Miura et al., 2002 ▶). Fsp27 is one of the LD-associated proteins and is essential for the formation and maintenance of unilocular LDs (Nishino et al., 2008 ▶). The sequestration of fat by large LDs also prevents fatty-acid overload, which impairs insulin signalling. The underlying mechanism responsible for unilocular LD formation in WAT remains elusive (Zweytick et al., 2000 ▶; Murphy & Vance, 1999 ▶). In in vitro studies, overexpression of Fsp27 in 3T3-L1 pre-adipocytes led to LDs of increased size (Matsusue et al., 2008 ▶), while depletion of Fsp27 in fully differentiated adipocytes stimulated lipolysis and resulted in uniformly dispersed LDs with smaller structures than those in control cells (Puri et al., 2007 ▶). Furthermore, it was reported that CIDEC expression was increased along with the differentiation of pre-adipocytes and that CIDEC knockdown in pre-adipocytes could block differentiation (Li et al., 2010 ▶). These facts demonstrate that Fsp27 acts as a novel regulator of the formation of unilocular LDs, lipid metabolism, adipocyte differentiation and insulin sensitivity.
Homology analysis revealed that the members of the CIDE protein family contain two domains (designated CIDE-N and CIDE-C). The CIDE-N domains of CIDEA, CIDEB and CIDEC (Fsp27) share 39%, 29% and 38% amino-acid identity with the CAD domain in the N-terminus of Dffa/Dff45/ICAD, respectively. The CIDE-C domain is unique to the CIDE family; the CIDE-C domains of CIDEA and CIDEB share homology (54% and 53% identity, respectively) with that of CIDEC (Fsp27) (Inohara et al., 1998 ▶). Signature sequences of the CIDE-N and CIDE-C domains have been defined by sequence comparison: the unique penta-amino-acid signature RPXRV was observed within the evolutionarily conserved 37 amino-acid residues that surround the EDGT sequence in the CIDE-N domain, and a highly conserved XARXTFDXYXXNPXDXXGXLNKVATXYXXYSXSXD signature was identified in the CIDE-C domain (Wu et al., 2008 ▶). Previous reports suggested that the CIDE-N domains of CIDE proteins could regulate killing activity, perhaps by the formation of homophilic dimers (Inohara et al., 1998 ▶), while the CIDE-C domains were necessary and sufficient for apoptotic activity and LD localization (Inohara et al., 1998 ▶; Liu et al., 2009 ▶).
Recently, a new study demonstrated a novel molecular mechanism responsible for unilocular LD formation for the first time: Fsp27 is highly concentrated at LD–LD contact sites (LDCS) to mediate the formation of fusion pores through which net neutral lipids diffuse from the smaller to the larger LD and ultimately lead to a unique LD fusion. Furthermore, periliplin (Plin), another LD-associated protein, was identified as cooperating with Fsp27 in vivo through direct interaction, resulting in significant acceleration of Fsp27-dependent lipid diffusion and LD fusion. It was shown that the Fsp27 CIDE-N domain plays a modulatory role, functioning as a recruitment platform for the formation of an Fsp27 CIDE-N/CIDE-N homodimer or an Fsp27/Plin heterodimer (or both); the recruitment interactions can induce the correct configuration of the Fsp27 CIDE-C domain or stabilize it to enhance the formation/expansion of the fusion pore(s) and facilitate LD fusion (Gong et al., 2011 ▶). However, the detailed structural characteristics of the Fsp27 CIDE-N domain remained unknown.
In this study, the expression and purification of the CIDE-N domain of mouse Fsp27 in Escherichia coli are reported; crystallization and preliminary X-ray crystallographic analysis were carried out and the crystals obtained diffracted to high resolution (1.92 Å).
2. Materials and methods
2.1. Materials
The enzymes and the PCR amplification kit used for plasmid construction were all obtained from New England Biolabs. A mini plasmid kit and a DNA quick purify/recover kit were obtained from Omega Co. All other chemicals were of analytical grade.
Plasmid Fsp27-GFP was a generous gift from Professor Peng Li of Tsinghua University.
2.2. Construction of plasmid pRSFDuet-1-Fsp27 39–119
The primers used for the Fsp27 CIDE-N domain (residues 39–119) were designed based on the published nucleotide sequence of Mus musculus CIDEC (GenBank Accession No. BC099676.1). FseI and AscI sites (shown in bold), which provide convenience for subcloning into the pRSFDuet-1 vector, were introduced into the 5′ ends of the primers. The forward and reverse primers were 5′-GGCCGGCCACCGAGGGCCAGGCC-3′ and 5′-GGCGCGCCTCAGGGCTTCCACTTCTGC-3′, respectively.
PCR amplification was performed with a DNA Thermal Cycler (Eppendorf) using standard PCR settings with the plasmid Fsp27-GFP as template.
The PCR product Fsp27 39–119 was isolated on a gel, purified using a DNA recover kit and subjected to FseI/AscI digestion for 5 h. The cleaved Fsp27 39–119 coding sequence was subcloned into a modified pRSFDuet-1 vector (Novagen) containing an N-terminal 6×His tag and FseI and AscI restriction-enzyme sites.
2.3. Expression and purification of the Fsp27 CIDE-N domain
Positive colonies transformed with the plasmid pRSFDuet-1-Fsp27 39–119 were grown in LB medium containing 100 µg ml−1 ampicillin at 310 K; when the culture density approached an OD600 of ∼0.8, the temperature of the shaker-incubator was reset to 289 K to allow gradual cooling of the cultures. After IPTG induction (0.5 mM), the cultures were incubated in the shaker-incubator for a further 12–16 h and collected by centrifugation. The cell pellets were resuspended in lysis buffer and lysed by sonication (Table 1 ▶). The homogenized product was clarified by centrifugation (15 000g, 45 min, 277 K).
Table 1. Buffers used for Fsp27 CIDE-N domain purification.
| Lysis buffer | 25 mM Tris–HCl pH 7.7, 500 mM NaCl, 20 mM imidazole, 1 mM PMSF |
| Buffer A | 25 mM Tris–HCl pH 7.7, 500 mM NaCl, 20 mM imidazole |
| Buffer B | 25 mM Tris–HCl pH 7.7, 500 mM NaCl, 500 mM imidazole |
| Buffer C | 10 mM Tris–HCl pH 7.7, 150 mM NaCl, 2 mM DTT |
All of the following procedures for Fsp 39–119 purification were carried out at 277 K. The clarified lysate was loaded onto a 5 ml HisTrap HP column (GE Healthcare) coupled to an ÄKTApurifier system (GE Healthcare) and the contaminant proteins were washed out with wash buffer (buffer A; Table 1 ▶). The His-tagged recombinant Fsp27 was eluted with a 90 ml linear gradient of 20–500 mM imidazole (buffer A to buffer B; see Table 1 ▶). Fractions containing the recombinant protein were collected and concentrated using an Ultrafree 10K filter (Millipore) before being applied onto a Superdex 75 column (GE Healthcare) using buffer C (see Table 1 ▶). The purity of the recombinant Fsp27 was assessed by SDS–PAGE.
2.4. Crystallization
The purified Fsp27 CIDE-N domain was concentrated to 30 mg ml−1. Protein concentrations were measured by UV–Vis spectroscopy with a NanoVue Plus spectrophotometer (GE Healthcare). The protein solution was clarified by centrifugation at 13 000g for 20 min at 277 K prior to searching for initial crystallization conditions. The screening process was carried out in 24-well plates at 291 K by the hanging-drop vapour-diffusion technique using Index, Crystal Screen and Crystal Screen 2 reagent kits from Hampton Research as described previously (Zhang et al., 2010 ▶; Jancarik et al., 1991 ▶). The initial conditions were optimized by changing the PEG molecular weight, buffer pH and protein concentration as well as by the use of additives.
2.5. Ion-trap LC/MS/MS spectrometry analysis
Several crystals were washed with reservoir solution, submitted to SDS–PAGE and stained with Coomassie Brilliant Blue, allowing the visualization of a single 14 kDa band. We excised the protein band, chopped it into smaller portions and digested them in-gel using sequencing-grade trypsin at 310 K overnight (Promega). The sample was centrifuged and the supernatant was concentrated for ion-trap LC/MS/MS spectrometric analysis with an Agilent 6300 Ion Trap LC/MS System (Agilent Technologies); the peptide masses were submitted to the database-analysis software Spectrum Mill (Agilent Technologies).
2.6. X-ray crystallographic studies
To prepare for X-ray analysis, crystals were incubated with a cryoprotectant solution (crystallization solution plus 15% glycerol), mounted in a nylon-fibre loop and cryocooled at 100 K in a liquid-nitrogen gas stream. Diffraction data collection was performed with single Fsp27 crystals on the BL17U beamline (wavelength 0.9792 Å) of the Shanghai Synchrotron Radiation Facility (SSRF) using a MAR CCD 225 detector. The exposure time was 0.8 s per frame, with a 0.5° oscillation angle; the crystal-to-detector distance was 150 mm. The HKL-2000 programs DENZO and SCALEPACK were employed to index, integrate and scale the intensity data (Otwinowski & Minor, 1997 ▶).
3. Results and discussion
3.1. Cloning of the CIDE-N domain of Fsp27
Using PCR amplification, a DNA fragment of 260 bp containing the coding sequence for the Fsp27 CIDE-N domain was obtained and subjected to FseI/AscI digestion. The digested product was subcloned into the modified pRSFDuet-1 vector cleaved with FseI and AscI at 289 K for 3–5 h. The ligation product was transformed into the Rossetta strain of E. coli. The positive colony containing the plasmid pRSFDuet-1-Fsp27 39–119 was identified by PCR and FseI/AscI digestion and was verified by nucleotide sequencing.
3.2. Efficient expression and purification of the Fsp27 CIDE-N domain
The CIDE-N domain of Fsp27 N-terminally fused with a 6×His tag was solubly expressed in E. coli with a yield of more than 6 mg from 1 l culture medium. The molecular weight of Fsp27 39–119 was calculated to be approximately 11.9 kDa using the ProtParam characterization tool on the ExPASy server. After a two-step purification consisting of affinity and gel-filtration chromatography (see Table 2 ▶ and Fig. 1 ▶), the purity of the protein was more than 95% as assessed by SDS–PAGE visualized by Coomassie R-250 staining (Fig. 1 ▶). The molecular mass of the purified product identified by size-exclusion chromatography followed by static laser light-scattering (data not shown) showed that Fsp27 may form a monomer.
Table 2. Purification summary of His-Fsp27 CIDE-N domain.
| Purification step | Volume (ml) | Protein concentration (mg ml−1) | Total protein (mg) | Yield (%) |
|---|---|---|---|---|
| Soluble cell fraction | 60 | 22.28 | 1336.8 | 100 |
| Ni-affinity | 26 | 0.81 | 20.96 | 1.57 |
| Superdex 75 | 11 | 1.64 | 18.04 | 1.35 |
Figure 1.

Elution profile of the CIDE-N domain of Fsp27 on Superdex 75 and SDS–PAGE (inset) of the eluted fractions. The left lane contains molecular-mass markers (labelled in kDa).
3.3. Mass-spectrometric analysis of the Fsp27 CIDE-N domain
The ion-trap LC/MS/MS spectra of the gel-excised band are shown in Fig. 2 ▶(b). The list of peptide masses was submitted to the data-analysis software Spectrum Mill (Agilent Technologies) and the identity of the protein was verified with a coverage of 27.62% (amino acids 56–112; Fig. 2 ▶ a) and a score of 1421.30. Thus, the components of the crystal were fully identified.
Figure 2.

Characterization of the components of the Fsp27 crystals by ion-trap LC/MS/MS spectrometric analysis. (a) Matched peptides are indicated in light grey; the identity of the protein was verified with a sequence coverage of 27.62% (amino acids 56–112). (b) Ion-trap LC/MS/MS spectra arranged according to the peptide score. (1) LC/MS/MS spectrum of the m/z 991.2 ion corresponding to the peptide with amino-acid residues 78–103 of Fsp27; (2) LC/MS/MS spectrum of the m/z 565.3 ion corresponding to amino-acid residues 104–112; (3) LC/MS/MS spectrum of the m/z 720.9 ion corresponding to amino-acid residues 57–69; (4) LC/MS/MS spectrum of the m/z 784.9 ion corresponding to amino-acid residues 56–69; (5) LC/MS/MS spectrum of the m/z 1071.6 ion corresponding to amino-acid residues 76–103; (6) LC/MS/MS spectrum of the m/z 715.5 ion corresponding to amino-acid positions 70–75.
3.4. Crystallization and data collection of single Fsp27 crystals
Small prolate crystals were observed after one week in several conditions from the Crystal Screen and Index kits (Hampton Research), all of which contained PEG as a precipitant. Optimizations of these initial conditions were carried out as described above, rendering crystals of larger size that diffracted to high resolution (Fig. 3 ▶). The best crystals were obtained at 291 K in sitting-drop mode by dispensing droplets comprising 1.5 µl protein solution (20 mg ml−1) and 1.5 µl crystallization buffer (0.2 M sodium acetate trihydrate, 28% PEG 4000, 0.1 M Tris–HCl pH 8.5). The largest crystals were immersed in an optimized cryoprotectant (crystallization buffer plus 15% glycerol) for data collection.
Figure 3.

Typical crystals of the CIDE-N domain of Fsp27. (a) An initial crystal. (b) The best crystal grown using the optimized conditions.
A complete 1.92 Å resolution data set was obtained from a crystal belonging to space group P65, with unit-cell parameters a = b = 63.3, c = 47.4 Å (see Fig. 4 ▶ and Table 3 ▶). The Matthews coefficient was 2.41 Å3 Da−1 (solvent content of 48.9%), with one Fsp27 molecule per asymmetric unit. Our gel-filtration results also indicate that Fsp27 39–119 may exist as a monomer in solution.
Figure 4.

Diffraction image of the CIDE-N domain of Fsp27. The crystal diffracted to 1.92 Å resolution.
Table 3. Data-collection and processing statistics.
| Wavelength (Å) | 0.9792 |
| Space group | P65 |
| Unit-cell parameters (Å, °) | a = b = 63.3, c = 37.4, α = β = 90, γ = 120 |
| Solvent content (%) | 48.9 |
| V M (Å3 Da−1) | 2.41 |
| Resolution (Å) | 50–1.92 |
| No. of observed reflections | 26278 |
| No. of unique reflections | 6642 |
| Completeness (%) | 99.7 (99.8) |
| Multiplicity | 4.0 |
| Mean I/σ(I) | 13.77 (3.7) |
| R merge † (%) | 9.3 (35.5) |
R
merge = 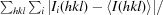
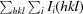 .
.
Structure solution was performed by molecular replacement using the structure of human cell-death activator CIDE-A (PDB entry 2eel; RIKEN Structural Genomics/Proteomics Initiative, unpublished work) as a search model, and refinement is in progress.
Acknowledgments
We are grateful to Professors Maojun Yang and Peng Li of Tsinghua University and Shentao Li of Capital Medical University for providing the plasmid, technical assistance with data collection and processing, critical discussion and valuable comments. We are also grateful to the staff of the SSRF BL17U beamline for assistance in data collection. This work was supported by the Ministry of Science and Technology (grant No. 2011CB910501) and the National Natural Science Foundation (NSFC grant No. 31030020). We declare that the experiments performed in this study comply with the current laws and ethical standards of our country. The authors declare that they have no conflicts of interest.
References
- Danesch, U., Hoeck, W. & Ringold, G. M. (1992). J. Biol. Chem. 267, 7185–7193. [PubMed]
- Gesta, S., Tseng, Y.-H. & Kahn, C. R. (2007). Cell, 131, 242–256. [DOI] [PubMed]
- Gong, J., Sun, Z. & Li, P. (2009). Curr. Opin. Lipidol. 20, 121–126. [DOI] [PubMed]
- Gong, J., Sun, Z., Wu, L., Xu, W., Schieber, N., Xu, D., Shui, G., Yang, H., Parton, R. G. & Li, P. (2011). J. Cell Biol. 195, 953–963. [DOI] [PMC free article] [PubMed]
- Inohara, N., Koseki, T., Chen, S., Wu, X. & Núñez, G. (1998). EMBO J. 17, 2526–2533. [DOI] [PMC free article] [PubMed]
- Jancarik, J., Scott, W. G., Milligan, D. L., Koshland, D. E. & Kim, S.-H. (1991). J. Mol. Biol. 221, 31–34. [DOI] [PubMed]
- Li, F., Gu, Y., Dong, W., Li, H., Zhang, L., Li, N., Li, W., Zhang, L., Song, Y., Jiang, L., Ye, J. & Li, Q. (2010). FEBS J. 277, 4173–4183. [DOI] [PubMed]
- Li, J. Z., Ye, J., Xue, B., Qi, J., Zhang, J., Zhou, Z., Li, Q., Wen, Z. & Li, P. (2007). Diabetes, 56, 2523–2532. [DOI] [PubMed]
- Liang, L., Zhao, M., Xu, Z., Yokoyama, K. K. & Li, T. (2003). Biochem. J. 370, 195–203. [DOI] [PMC free article] [PubMed]
- Liu, K., Zhou, S., Kim, J.-Y., Tillison, K., Majors, D., Rearick, D., Lee, J. H., Fernandez-Boyanapalli, R. F., Barricklow, K., Houston, M. S. & Smas, C. M. (2009). Am. J. Physiol. Endocrinol. Metab. 297, E1395–E1413. [DOI] [PubMed]
- Matsusue, K., Kusakabe, T., Noguchi, T., Takiguchi, S., Suzuki, T., Yamano, S. & Gonzalez, F. J. (2008). Cell Metab. 7, 302–311. [DOI] [PMC free article] [PubMed]
- Miura, S., Gan, J.-W., Brzostowski, J., Parisi, M. J., Schultz, C. J., Londos, C., Oliver, B. & Kimmel, A. R. (2002). J. Biol. Chem. 277, 32253–32257. [DOI] [PubMed]
- Murphy, D. J. & Vance, J. (1999). Trends Biochem. Sci. 24, 109–115. [DOI] [PubMed]
- Nishino, N. et al. (2008). J. Clin. Invest. 118, 2808–2821. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Puri, V., Konda, S., Ranjit, S., Aouadi, M., Chawla, A., Chouinard, M., Chakladar, A. & Czech, M. P. (2007). J. Biol. Chem. 282, 34213–34218. [DOI] [PubMed]
- Rajala, M. W. & Scherer, P. E. (2003). Endocrinology, 144, 3765–3773. [DOI] [PubMed]
- Tauchi-Sato, K., Ozeki, S., Houjou, T., Taguchi, R. & Fujimoto, T. (2002). J. Biol. Chem. 277, 44507–44512. [DOI] [PubMed]
- Toh, S. Y., Gong, J., Du, G., Li, J. Z., Yang, S., Ye, J., Yao, H., Zhang, Y., Xue, B., Li, Q., Yang, H., Wen, Z. & Li, P. (2008). PLoS One, 3, e2890. [DOI] [PMC free article] [PubMed]
- Wolins, N. E., Brasaemle, D. L. & Bickel, P. E. (2006). FEBS Lett. 580, 5484–5491. [DOI] [PubMed]
- Wu, C., Zhang, Y., Sun, Z. & Li, P. (2008). BMC Evol. Biol. 8, 159. [DOI] [PMC free article] [PubMed]
- Zhang, L., Xiang, H., Gao, J., Hu, J., Miao, S., Wang, L., Deng, X. & Li, S. (2010). Protein Expr. Purif. 69, 204–208. [DOI] [PubMed]
- Zhou, Z., Yon Toh, S., Chen, Z., Guo, K., Ng, C. P., Ponniah, S., Lin, S.-C., Hong, W. & Li, P. (2003). Nature Genet. 35, 49–56. [DOI] [PubMed]
- Zweytick, D., Athenstaedt, K. & Daum, G. (2000). Biochim. Biophys. Acta, 1469, 101–120. [DOI] [PubMed]