

The production, purification, crystallization and crystallographic analysis of H-1 Parvovirus, a gene-therapy vector, are reported.
Keywords: H-1 Parvovirus, viruses, antitumor gene delivery
Abstract
Crystals of H-1 Parvovirus (H-1PV), an antitumor gene-delivery vector, were obtained for DNA-containing capsids and diffracted X-rays to 2.7 Å resolution using synchrotron radiation. The crystals belonged to the monoclinic space group P21, with unit-cell parameters a = 255.4, b = 350.4, c = 271.6 Å, β = 90.34°. The unit cell contained two capsids, with one capsid per crystallographic asymmetric unit. The H-1PV structure has been determined by molecular replacement and is currently being refined.
1. Introduction
H-1 Parvovirus (H-1PV) is a member of the Parvovirus genus of the Parvoviridae, a family of single-stranded (ss) DNA-packaging viruses (Berns & Parrish, 2007 ▶). H-1PV and a number of other rodent parvoviruses, including Minute Virus of Mice (MVM) and LuIII, are being developed as vectors for antitumor gene-delivery applications (reviewed in Blechacz & Russell, 2004 ▶; Rommelaere et al., 2010 ▶). The viruses replicate autonomously in rapidly proliferating and oncogene-transformed cells and exert oncoselective toxicity and oncosuppressive activity in human tumor cell lines, while being nonpathogenic to humans (Rommelaere & Cornelis, 1991 ▶; Blechacz & Russell, 2004 ▶; Herrero Y Calle et al., 2004 ▶; Angelova, Aprahamian, Grekova et al., 2009 ▶; Angelova, Aprahamian, Balboni et al., 2009 ▶). In preclinical studies, H-1PV induced regression of advanced rat and human gliomas in rat models by local, intravenous or intranasal treatment (Geletneky et al., 2010 ▶; Kiprianova et al., 2011 ▶). These observations led to a phase I/IIa clinical trial evaluating the efficacy of H-1PV for the treatment of patients with recurrent glioblastoma multiforme (Geletneky et al., 2012 ▶).
H-1PV virions package a linear ssDNA genome of ∼5 kb into a T = 1 icosahedral capsid with a diameter of ∼260 Å. The genome encodes two nonstructural (NS) proteins, NS1 and NS2, and two overlapping capsid viral proteins (VPs), VP1 and VP2 (Cotmore & Tattersall, 1987 ▶). VP1 (81 kDa) and VP2 (65 kDa) are produced by alternative splicing from the same mRNA. The entire sequence of VP2 is contained within VP1, which has a unique N-terminal region (VP1u) of 142 amino acids. VP3 (63 kDa) is generated by post-translational cleavage of 18 or 21 amino acids from the N-terminus of VP2 after DNA packaging and is thus only present in virions. H-1PV empty (no DNA) capsids are reported to be assembled from ∼10 copies of VP1 and 50 copies of VP2, while full capsids (with DNA) contain ten copies of VP1 and a mixture of VP2 and VP3, with VP3 being the major component.
The three-dimensional structures of several members of the Parvovirus genus have been determined by X-ray crystallography (Tsao et al., 1991 ▶; Agbandje-McKenna et al., 1998 ▶; Agbandje et al., 1993 ▶; Simpson et al., 2002 ▶). In all of these structures, only the overlapping C-terminal polypeptide sequence (∼550 amino acids) common to all three VPs is ordered with T = 1 icosahedral symmetry. The VP structural topology is highly conserved among the parvoviruses and contains a core eight-stranded β-barrel domain and an α-helical region (Chapman & Agbandje-McKenna, 2006 ▶). Structural variations localized to loops between these core regions dictate differences in biological functions such as receptor binding, tissue tropism, pathogenicity and antigenicity (Agbandje-McKenna & Chapman, 2006 ▶; Halder et al., 2012 ▶). Here, we report the production, purification, crystallization and structure determination of H-1PV towards the structural characterization of the capsid regions that dictate packaged genome interactions and its tumor tropism.
2. Materials and methods
2.1. Production and purification
The NB324K cells (simian virus 40-transformed human newborn kidney fibroblast cells) used for virus production were maintained in Eagle’s minimal essential medium with 5% fetal calf serum, glutamine and antibiotics as recommended by the manufacturer (Gibco-BRL). Cell monolayers at 50% confluency (cell density of 4 × 106 per 10 cm plate) were infected with wild-type H-1PV at 310 K for 1 h with a multiplicity of infection of 0.1 PFU per cell, which was followed by occasional rocking of the plates. The cells were incubated for an additional 5–7 d until a cytopathic effect was observed (∼80% cell lysis). Cells were harvested by scraping and pelleted by low-speed centrifugation at 500g and 277 K for 15 min. The cell pellet was resuspended in TE buffer (50 mM Tris–HCl, 0.5 mM EDTA pH 8.7) and stored at 253 K. The virus capsids were released from the frozen cells by three cycles of rapid freeze–thawing. The cellular debris was removed by centrifugation at 12 100g and 277 K for 15 min. The supernatant was treated with nuclease and purified by three rounds of CsCl density (1.32–1.46 g cm−3) gradients using equilibrium centrifugation at 209 490g and 277 K for 24 h. Visible blue virus-capsid bands corresponding to empty capsids (1.32 g cm−3) and full capsids (1.41–1.46 g cm−3) were extracted from the density tubes by side puncture and dialyzed into Tris–HCl buffer (10 mM Tris–HCl pH 7.5, 150 mM NaCl, 8 mM CaCl2.2H2O). The capsid concentrations were assessed by hemagglutination of sheep erythrocytes as well as optical density measurements (assuming extinction coefficients of 1.0 and 7.0 for calculations in mg ml−1 for empty and full capsids, respectively) and adjusted to 10 mg ml−1 using Amicon Ultra-0.5 centrifugal filter units (100 kDa molecular-weight cutoff, Millipore) by centrifugation at 2300g at 277 K. The purity and integrity of the viral capsids were monitored using SDS–PAGE (15% resolving) with Coomassie Blue staining and negative-stain electron microscopy (EM), respectively.
2.2. Electron microscopy
For the EM visualization, 5 µl purified virus sample at an estimated concentration of 2.0 mg ml−1 was spotted onto a 400-mesh carbon-coated copper grid (Ted Pella Inc., Redding, California, USA) for 1 min before blotting with filter paper (Whatman No. 5). The grid was washed twice by addition of 5 µl filtered water followed by blotting with filter paper after 15 s. The sample was then negatively stained with 5 µl NanoW (Nanoprobes) for 1 min, blotted dry and viewed with a Hitachi 3000 EM.
2.3. Crystallization
Crystallization conditions for the H-1PV full and empty capsids were screened based on conditions previously reported for other autonomous parvoviruses (Tsao et al., 1992 ▶; Agbandje et al., 1993 ▶; Hernando et al., 2000 ▶; Kontou et al., 2005 ▶; Llamas-Saiz et al., 1997 ▶) using the hanging-drop vapor-diffusion method (McPherson, 1982 ▶) with VDX 24-well plates and siliconized cover slips (Hampton Research, Laguna Niguel, California, USA). The reservoir solution consisted of 1–3%(w/v) polyethylene glycol 8000 (PEG 8000), 150 mM NaCl, 8 mM CaCl2.2H2O as precipitant in 10 mM Tris–HCl pH 7.5 (reservoir solution). The drops were prepared by mixing 2 µl virus solution (at 10 mg ml−1) in Tris–HCl buffer with 2 µl reservoir solution and were equilibrated against 1 ml reservoir solution at RT. The data collection, processing and structure determination of H-1PV from only the full capsid crystals are described below.
2.4. Data collection and reduction
Crystals were soaked for 30 s in cryoprotectant solution consisting of the reservoir solution plus 10% PEG 8000 and 30% glycerol and were flash-cooled to 100 K in a liquid-nitrogen stream prior to X-ray diffraction data collection. Diffraction data were collected on the F1 beamline (λ = 0.9186 Å) of the Cornell High Energy Synchrotron Source (CHESS) using an ADSC Quantum 270 CCD detector at crystal-to-detector distances of 230 and 300 mm, with an oscillation angle of 0.3° and a exposure time of 30 s per image. The measured diffraction intensities were indexed and integrated with the HKL-2000 suite of programs and were scaled and merged with SCALEPACK (Otwinowski & Minor, 1997 ▶). The intensities were converted to structure-factor amplitudes using the TRUNCATE program in CCP4 (Winn et al., 2011 ▶) for the structure-determination process.
2.5. Molecular replacement: determination of particle orientation and position
The orientation of the H-1PV full capsids in the crystal unit cell was determined with a self-rotation function (Rossmann & Blow, 1962 ▶) using the General Lock Rotation Function (GLRF) program (Tong & Rossmann, 1997 ▶). The calculations used the top 10% (largest amplitudes) of the experimentally observed data between 10.0 and 5.0 Å resolution to represent the second Patterson. The radius of integration was set to 120 Å with κ = 72°, 120° and 180° to search for fivefold, threefold and twofold symmetry elements, respectively. This procedure was followed by molecular replacement using the available MVM prototype strain (MVMp) structure. A Cα model of the MVMp VP2 crystal structure (PDB entry 1z14; Kontou et al., 2005 ▶) was generated by the MOLEMAN program (Kleywegt & Jones, 1997 ▶), expanded to 60 subunits (one empty capsid) by icosahedral matrix multiplication using the Oligomer Generator subroutine available at the VIPERdb website (Carrillo-Tripp et al., 2009 ▶) and used as the phasing model. Structure factors were calculated in the 10.0–5.0 Å resolution range using the SFALL program in CCP4 (Winn et al., 2011 ▶) and used for cross-rotation and translation function searches using the AMoRe program (Navaza, 2001 ▶). The solution with the highest structure-factor correlation coefficient (CC) and the lowest R factor {CC = 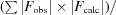
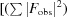 ×
× 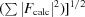 and R factor =
and R factor = 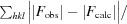
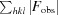 × 100, where F
obs are the observed structure factors and F
calc are structure factors calculated from the model} was used in the translation-function search for determining the correct solution, and the top solution from the translation function was further refined using a rigid-body option, FITING, in AMoRe. The MVMp Cα capsid was rotated and translated into the unit cell according to the final molecular-replacement solution. The solutions were confirmed by a self-rotation function search with structure factors calculated from the oriented and positioned model in the 10.0–5.0 Å resolution range using the SFALL program in CCP4 (Winn et al., 2011 ▶), as described above for the experimental data sets, prior to structure refinement.
× 100, where F
obs are the observed structure factors and F
calc are structure factors calculated from the model} was used in the translation-function search for determining the correct solution, and the top solution from the translation function was further refined using a rigid-body option, FITING, in AMoRe. The MVMp Cα capsid was rotated and translated into the unit cell according to the final molecular-replacement solution. The solutions were confirmed by a self-rotation function search with structure factors calculated from the oriented and positioned model in the 10.0–5.0 Å resolution range using the SFALL program in CCP4 (Winn et al., 2011 ▶), as described above for the experimental data sets, prior to structure refinement.
To calculate initial phases for model refinement and electron-density map calculation, an MVMp VP2 polyalanine model was generated from the VP2 crystal structure (PDB entry 1z14; Kontou et al., 2005 ▶) using the MOLEMAN program (Kleywegt & Jones, 1997 ▶) and superimposed individually onto the 60 subunits of the oriented and positioned MVMp Cα capsid model. The phases calculated from this model were improved using simulated-annealing, energy-minimization, conventional positional and individual temperature-factor (B-factor) refinement in the CNS program (Brünger et al., 1998 ▶). This procedure was followed by real-space electron-density averaging using a VP2 molecular mask. The refinement and averaging procedures were conducted while applying strict 60-fold noncrystallographic symmetry (NCS) in the CNS program (Brünger et al., 1998 ▶). Reflections with I/σ(I) > 0 were used in refinement. 5% of the total data set was partitioned for monitoring of the refinement process with an R
free calculation (
× 100, calculated with a 5% randomly selected fraction of the reflection data not included in refinement; Brünger, 1992 ▶). The refinement and averaging cycles were followed by interactive model building into the averaged σ-weighted 2F
obs − F
calc electron-density map by substitution, insertion and deletion of amino acids relative to the starting MVMp polyalanine model using the Coot program (Emsley et al., 2010 ▶). The model building was followed by a second round of refinement conducted as described above.
3. Results and discussion
3.1. H-1PV production, purification and crystallization
Wild-type H-1PV full and empty capsids produced in NB324K cells and purified using a CsCl density gradient were observed to be assembled from VP1–3 and VP1–2, respectively, as expected (Fig. 1 ▶ a). The higher abundance of VP3 compared with VP2 in the H-1PV full capsids is consistent with cleavage of >50% of the VP2 to generate VP3 following genome packaging in the sample crystallized. The appearance of the H-1PV full and empty capsids in the negative-stain electron micrographs, stain excluded and stain penetrated (Figs. 1 ▶ b and 1 ▶ c), respectively, is consistent with the presence and the absence of packaged genome, respectively. Crystals with a rod-shaped habit (Fig. 1 ▶ d) were obtained from both the full and empty capsids in ∼3–4 weeks at room temperature from the Tris–HCl buffer with 3% PEG 8000. The approximate crystal dimensions were 0.15 × 0.01 × 0.005 mm (Fig. 1 ▶ d).
Figure 1.

Purification and crystallization of H-1PV full and empty capsids. (a) A 15% SDS–PAGE gel of the purified H-1PV capsids showing the relative ratios and positions of VP1, VP2 and VP3 (molecular weights of 81, 65 and 63 kDa, respectively). The positions of low-molecular-weight standards (labeled in kDa; Bio-Rad, Hercules, California, USA) are indicated on the right-hand side. (b, c) Negatively stained electron micrographs of H-1PV full capsids viewed at 100 000× magnification (b) and H-1PV empty capsids viewed at 60 000× magnification (c). (d) Optical photograph of H-1PV full capsid crystals in a rod-shaped habit.
3.2. Data collection and processing
A total of 392 usable images were collected from two crystals of H-1PV full capsids. The crystals diffracted X-rays to beyond 2.7 Å resolution (Fig. 2 ▶). Indexing of the two data sets showed that the crystals belonged to the primitive monoclinic crystal system, with a β angle that was very close to 90° (Table 1 ▶). The unique b axis was determined during indexing by assuming twofold symmetry along either the a, the b or the c axis and comparing the R
sym [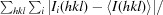
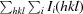 × 100, where I
i(hkl) is the intensity of an individual reflection with indices hkl and 〈I(hkl)〉 is the average intensity of all symmetry-equivalent measurements of that reflection; the summation is over all intensities]. Assumption of a twofold along a or c gave R
sym values of ∼30% or ∼50%, respectively, compared with an R
sym of 13.2% for a twofold along the b axis. A similar strategy was used to identify the unique b axis for the Hong Kong 97 (HK97) bacteriophage (Wikoff et al., 1999 ▶). As also reported for HK97, the proximity of the β angle to 90° resulted in an orientation ambiguity in indexing, in which each crystal could be indexed in two possible orientations because the positions of the reflections are consistent with mmm symmetry. The correct orientation(s) was determined by scaling the data sets independently and then together. While the data sets scaled well individually (R
sym of ∼13%), when scaled together the correct orientation had an R
merge (the same as R
sym but for merged data sets) of 13.2%, while the incorrect orientation had an R
merge of ∼60%. Inspection of the 0k0 class of reflections (for k = 2n) implied systematic absences for the odd reflections, indicating the presence of a 21 screw axis; thus, the crystals belonged to space group P21. The data-collection and processing statistics are summarized in Table 1 ▶.
× 100, where I
i(hkl) is the intensity of an individual reflection with indices hkl and 〈I(hkl)〉 is the average intensity of all symmetry-equivalent measurements of that reflection; the summation is over all intensities]. Assumption of a twofold along a or c gave R
sym values of ∼30% or ∼50%, respectively, compared with an R
sym of 13.2% for a twofold along the b axis. A similar strategy was used to identify the unique b axis for the Hong Kong 97 (HK97) bacteriophage (Wikoff et al., 1999 ▶). As also reported for HK97, the proximity of the β angle to 90° resulted in an orientation ambiguity in indexing, in which each crystal could be indexed in two possible orientations because the positions of the reflections are consistent with mmm symmetry. The correct orientation(s) was determined by scaling the data sets independently and then together. While the data sets scaled well individually (R
sym of ∼13%), when scaled together the correct orientation had an R
merge (the same as R
sym but for merged data sets) of 13.2%, while the incorrect orientation had an R
merge of ∼60%. Inspection of the 0k0 class of reflections (for k = 2n) implied systematic absences for the odd reflections, indicating the presence of a 21 screw axis; thus, the crystals belonged to space group P21. The data-collection and processing statistics are summarized in Table 1 ▶.
Figure 2.

X-ray diffraction image for a crystal of H-1PV full capsids. The image is a typical 0.3° oscillation photograph. The concentric rings indicate the 10.0, 5.0 and 3.0 Å resolution shells. (b) The inset shows a close-up of the boxed region in the upper right-hand corner.
Table 1. Data-collection, processing and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Wavelength (Å) | 0.9186 |
| No. of images | 392 |
| Space group | P21 |
| Unit-cell parameters (Å, °) | a = 255.4, b = 350.4, c = 271.6, β = 90.34 |
| Resolution (Å) | 50–2.7 (2.8–2.7) |
| Total No. of reflections | 16673125 |
| No. of unique reflections | 1210268 (123000) |
| Crystal mosaicity (°) | 0.3–0.5 |
| Completeness (%) | 93.1 (94.7) |
| R merge † (%) | 13.2 (37.8) |
| Multiplicity | 2.6 (2.4) |
| 〈I/σ(I)〉 | 6.9 (2.4) |
| V M (Å3 Da−1) | 2.1 |
| R factor‡/R free § (%) | 23.9/23.9 |
R
merge =
× 100, where Ii(hkl) is the intensity of an individual reflection with indices hkl and 〈I(hkl)〉 is the average intensity of all symmetry-equivalent measurements of that reflection; the summation is over all intensities.
R factor =
× 100, where F
obs and F
calc are the observed and calculated structure-factor amplitudes, respectively.
R free is calculated in the same way as the R factor, except that it uses 5% of the reflection data partitioned from the refinement process.
Packing considerations based on the unit-cell parameters and the P21 space group suggested the presence of two capsids per unit cell related by the 21 screw axis and one capsid in the crystallographic asymmetric unit. Based on the unit-cell volume and the molecular weight of full capsids (∼5.7 × 106 Da), and assuming the presence of one capsid per asymmetric unit, the Matthews coefficient (V M) was calculated to be 2.1 Å3 Da−1, with a solvent content of 48%.
3.3. Molecular replacement: particle orientation and position
The orientations of the two capsids in the crystal unit cell were determined with a self-rotation function that searched for the fivefold, threefold and twofold icosahedral symmetry axes of the virus capsid with κ = 72° (Fig. 3 ▶ a), κ = 120° (Fig. 3 ▶ b) and κ = 180° (Fig. 3 ▶ c), respectively. The κ = 72° search showed that the two capsids in the unit cell have different orientations but are related by a twofold rotation along the b axis (Fig. 3 ▶ a). The κ = 180° search showed the presence of a crystallographic peak at ϕ = 90°, ψ = 90° corresponding to the twofold axis along b, thus establishing the orientation of the icosahedral symmetry axes relative to this crystallographic axis, and confirmed that the two capsids in the unit cell are related by the crystallographic twofold axis (b unique). The κ = 72° and κ = 120° peaks that do not belong to either of the two capsids in the unit cell (5f1 and 5f2 in Fig. 3 ▶ a and 3f1–4 in Fig. 3 ▶ b) were interpreted as Klug peaks resulting from the superimposition of the self-Pattersons of crystallographically related capsids at a common origin (Johnson et al., 1975 ▶). These peaks were not packing peaks resulting from pseudo-symmetry at the ‘low’ resolution (10.0−5.0 Å) used to compute the self-rotation functions as they were also observed for ‘high’ resolution data between 4.0 and 3.0 Å (data not shown), for which such packing peaks would be expected to disappear.
Figure 3.

Stereographic projections of the self-rotation function search for the H-1PV full capsid X-ray diffraction data. (a) κ = 72°, (b) κ = 120° and (c) κ = 180°, searching for fivefold, threefold and twofold icosahedral symmetry elements, respectively. The peaks representing fivefold positions are delineated by the dashed pentagons in (a). The peaks belonging to each of the two viral capsids in the unit cell are circled in red and blue, respectively. The peaks interpreted as Klug peaks (5f1, 5f2 and 3f1–4) are circled in green and labeled in (a) and (b). The a*, b* and c* axes are labeled.
Molecular-replacement procedures using an MVMp Cα capsid model, in which the highest peak obtained for the cross-rotation function was used for the translation-function calculation in both primitive monoclinic space groups P2 and P21, resulted in a solution with the highest CC and lowest R factor in P21 (35.4% and 46.5%, respectively, compared with CC = 21.9% and R factor = 50.6% in P2). This observation confirmed the space-group assignment from the data indexing and scaling. Rigid-body refinement (FITING in AMoRe) improved the CC and R factor to 40.3% and 44.9%, respectively (Navaza, 2001 ▶). The cross-rotation function Eulerian angles were α = 22.31°, β = 70.69°, γ = 28.97° and the fractional coordinate position from the translation search was 0.2510, 0.0000, 0.2489. A self-rotation function for the oriented and translated MVMp Cα capsid model in the P21 unit cell confirmed the molecular-replacement solution with respect to the experimental data (not shown). In addition, a self-rotation function calculated separately for the two capsids in space group P1 confirmed the contribution of peaks from each capsid in the unit cell and also confirmed that the extra peaks interpreted as Klug peaks (Figs. 3 ▶ a and 3 ▶ b) did not belong to either capsid alone but were a result of the packing arrangement of the two capsids in this unit cell.
A single cycle of refinement and real-space electron-density averaging in the CNS program (Brünger et al., 1998 ▶) to improve the initial phases calculated to 2.7 Å resolution from an oriented and positioned MVMp VP2 polyalanine capsid model resulted in an interpretable structure. H-1PV residues 38–593 (the last C-terminal residue; VP2 numbering) could be built into the averaged σ-weighted 2F obs − F calc electron-density map (not shown). The average NCS correlation coefficient was 0.86. The R factor and R free values from a second round of refinement (as described above) following the first round of model building were 23.9% and 23.9%, respectively (Table 1 ▶). The similarity of the R factor and R free for virus structures is a consequence of the high noncrystallographic icosahedral symmetry of the capsid, which causes strong correlations between the reflections in the working data set and those partitioned for R free calculation.
Interestingly, the averaged F obs − F calc difference electron-density map with the experimental data and the current VP2 capsid model structure factors revealed positive density inside the capsid that is likely to be icosahedrally ordered DNA nucleotides based on previous reports for other autonomous parvoviruses (Xie & Chapman, 1996 ▶; Govindasamy et al., 2003 ▶; Kontou et al., 2005 ▶). This possibility will be verified following further model building and structure refinement, which is in progress. This interior density was not present in a preliminary structure determined for H-1PV empty capsids (data not shown) using the same protocols as described above for the H-1PV full capsid structure. A detailed comparison of the H-1PV full and empty capsids, once refinement is complete, will provide information on the interior capsid regions involved in packaged genomic DNA interactions for autonomous parvoviruses. In addition, a comparison of this structure with other available parvovirus structures such as MVMp (Kontou et al., 2005 ▶), Canine Parvovirus (Tsao et al., 1991 ▶) and Porcine Parvovirus (Simpson et al., 2002 ▶) could provide insight into capsid regions that play a role in the tumor tropism for the rodent parvoviruses. This work will ultimately provide information on capsid regions that can be manipulated for improved genome packaging and targeted cell/tissue tropism, which will positively impact gene-delivery efficacy.
Acknowledgments
The authors would like to thank the staff at the F1 beamline at CHESS, Cornell University. We especially thank Kathy Dedrick for assistance in obtaining beam time and David Schuller, Irina Kriksunov, Marian Szebenyi, Chae Un Kim, Bill Miller, Mike Cook and Scott Smith for help in data collection. CHESS is supported by the National Science Foundation and the National Institutes of Health/National Institute of General Medical Sciences under NSF award DMR-0936384 and the MacCHESS facility through award GM-103485 from the National Institute of General Medical Sciences, National Institutes of Health. This project was funded by NSF project MCB-0718948 (RM and MA-M) and UF COM Research Funds (MA-M).
References
- Agbandje, M., McKenna, R., Rossmann, M. G., Strassheim, M. L. & Parrish, C. R. (1993). Proteins, 16, 155–171. [DOI] [PubMed]
- Agbandje-McKenna, M. & Chapman, M. S. (2006). Parvoviruses, edited by J. R. Kerr, S. F. Cotmore, M. E. Bloom, R. M. Linden & C. R. Parrish, pp. 125–139. London: Hodder Arnold.
- Agbandje-McKenna, M., Llamas-Saiz, A. L., Wang, F., Tattersall, P. & Rossmann, M. G. (1998). Structure, 6, 1369–1381. [DOI] [PubMed]
- Angelova, A. L., Aprahamian, M., Balboni, G., Delecluse, H.-J., Feederle, R., Kiprianova, I., Grekova, S. P., Galabov, A. S., Witzens-Harig, M., Ho, A. D., Rommelaere, J. & Raykov, Z. (2009). Mol. Ther. 17, 1164–1172. [DOI] [PMC free article] [PubMed]
- Angelova, A. L., Aprahamian, M., Grekova, S. P., Hajri, A., Leuchs, B., Giese, N. A., Dinsart, C., Herrmann, A., Balboni, G., Rommelaere, J. & Raykov, Z. (2009). Clin. Cancer Res. 15, 511–519. [DOI] [PubMed]
- Berns, K. & Parrish, C. R. (2007). Fields Virology, 5th ed., edited by D. M. Knipe & P. M. Howley, pp. 2437–2478. Philadelphia: Lippincott Williams & Wilkins.
- Blechacz, B. & Russell, S. J. (2004). Expert Rev. Mol. Med. 6, 1–24. [DOI] [PubMed]
- Brünger, A. T. (1992). Nature (London), 355, 472–475. [DOI] [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Carrillo-Tripp, M., Shepherd, C. M., Borelli, I. A., Venkataraman, S., Lander, G., Natarajan, P., Johnson, J. E., Brooks, C. L. III & Reddy, V. S. (2009). Nucleic Acids Res. 37, D436–442. [DOI] [PMC free article] [PubMed]
- Chapman, M. S. & Agbandje-McKenna, M. (2006). Parvoviruses, edited by J. R. Kerr, S. F. Cotmore, M. E. Bloom, R. M. Linden & C. R. Parrish, pp. 107–123. London: Hodder Arnold.
- Cotmore, S. F. & Tattersall, P. (1987). Adv. Virus Res. 33, 91–174. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Geletneky, K., Huesing, J., Rommelaere, J., Schlehofer, J. R., Leuchs, B., Dahm, M., Krebs, O., von Knebel Doeberitz, M., Huber, B. & Hajda, J. (2012). BMC Cancer, 12, 99. [DOI] [PMC free article] [PubMed]
- Geletneky, K., Kiprianova, I., Ayache, A., Koch, R., Herrero Y Calle, M., Deleu, L., Sommer, C., Thomas, N., Rommelaere, J. & Schlehofer, J. R. (2010). Neuro Oncol. 12, 804–814. [DOI] [PMC free article] [PubMed]
- Govindasamy, L., Hueffer, K., Parrish, C. R. & Agbandje-McKenna, M. (2003). J. Virol. 77, 12211–12221. [DOI] [PMC free article] [PubMed]
- Halder, S., Ng, R. & Agbandje-McKenna, M. (2012). Future Virol. 7, 253–278.
- Hernando, E., Llamas-Saiz, A. L., Foces-Foces, C., McKenna, R., Portman, I., Agbandje-McKenna, M. & Almendral, J. M. (2000). Virology, 267, 299–309. [DOI] [PubMed]
- Herrero Y Calle, M., Cornelis, J. J., Herold-Mende, C., Rommelaere, J., Schlehofer, J. R. & Geletneky, K. (2004). Int. J. Cancer, 109, 76–84. [DOI] [PubMed]
- Johnson, J. E., Argos, P. & Rossmann, M. G. (1975). Acta Cryst. B31, 2577–2583.
- Kiprianova, I., Thomas, N., Ayache, A., Fischer, M., Leuchs, B., Klein, M., Rommelaere, J. & Schlehofer, J. R. (2011). Clin. Cancer Res. 17, 5333–5342. [DOI] [PubMed]
- Kleywegt, G. J. & Jones, T. A. (1997). Methods Enzymol. 277, 208–230. [DOI] [PubMed]
- Kontou, M., Govindasamy, L., Nam, H.-J., Bryant, N., Llamas-Saiz, A. L., Foces-Foces, C., Hernando, E., Rubio, M.-P., McKenna, R., Almendral, J. M. & Agbandje-McKenna, M. (2005). J. Virol. 79, 10931–10943. [DOI] [PMC free article] [PubMed]
- Llamas-Saiz, A. L., Agbandje-McKenna, M., Wikoff, W. R., Bratton, J., Tattersall, P. & Rossmann, M. G. (1997). Acta Cryst. D53, 93–102. [DOI] [PubMed]
- McPherson, A. (1982). Preparation and Analysis of Protein Crystals, 1st ed. New York: Wiley.
- Navaza, J. (2001). Acta Cryst. D57, 1367–1372. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Rommelaere, J. & Cornelis, J. J. (1991). J. Virol. Methods, 33, 233–251. [DOI] [PubMed]
- Rommelaere, J., Geletneky, K., Angelova, A. L., Daeffler, L., Dinsart, C., Kiprianova, I., Schlehofer, J. R. & Raykov, Z. (2010). Cytokine Growth Factor Rev. 21, 185–195. [DOI] [PubMed]
- Rossmann, M. G. & Blow, D. M. (1962). Acta Cryst. 15, 24–31.
- Simpson, A. A., Hébert, B., Sullivan, G. M., Parrish, C. R., Zádori, Z., Tijssen, P. & Rossmann, M. G. (2002). J. Mol. Biol. 315, 1189–1198. [DOI] [PubMed]
- Tong, L. & Rossmann, M. G. (1997). Methods Enzymol. 276, 594–611. [PubMed]
- Tsao, J., Chapman, M. S., Agbandje, M., Keller, W., Smith, K., Wu, H., Luo, M., Smith, T. J., Rossmann, M. G., Compans, R. W. & Parrish, C. R. (1991). Science, 251, 1456–1464. [DOI] [PubMed]
- Tsao, J., Chapman, M. S., Wu, H., Agbandje, M., Keller, W. & Rossmann, M. G. (1992). Acta Cryst. B48, 75–88. [DOI] [PubMed]
- Wikoff, W. R., Duda, R. L., Hendrix, R. W. & Johnson, J. E. (1999). Acta Cryst. D55, 763–771. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Xie, Q. & Chapman, M. S. (1996). J. Mol. Biol. 264, 497–520. [DOI] [PubMed]