

POS-1 and GLD-1 are required to repress the Notch homologue glp-1 translation in Caenorhabditis elegans embryos. Both proteins bind to overlapping elements in the glp-1 3' UTR. Two POS-1 sites are present within this region, but only one is required for repression in vivo. It is proposed that POS-1 restricts access of other RNA-binding proteins to this region.
Abstract
RNA-binding proteins (RBPs) coordinate cell fate specification and differentiation in a variety of systems. RNA regulation is critical during oocyte development and early embryogenesis, in which RBPs control expression from maternal mRNAs encoding key cell fate determinants. The Caenorhabditis elegans Notch homologue glp-1 coordinates germline progenitor cell proliferation and anterior fate specification in embryos. A network of sequence-specific RBPs is required to pattern GLP-1 translation. Here, we map the cis-regulatory elements that guide glp-1 regulation by the CCCH-type tandem zinc finger protein POS-1 and the STAR-domain protein GLD-1. Our results demonstrate that both proteins recognize the glp-1 3′ untranslated region (UTR) through adjacent, overlapping binding sites and that POS-1 binding excludes GLD-1 binding. Both factors are required to repress glp-1 translation in the embryo, suggesting that they function in parallel regulatory pathways. It is intriguing that two equivalent POS-1–binding sites are present in the glp-1 3′ UTR, but only one, which overlaps with a translational derepression element, is functional in vivo. We propose that POS-1 regulates glp-1 mRNA translation by blocking access of other RBPs to a key regulatory sequence.
INTRODUCTION
The physiology of a cell is governed by the identity and extent of genes that it expresses. Gene expression is regulated at the transcriptional, posttranscriptional, and posttranslational levels. Each aspect is important and necessary to ensure appropriate expression for a given cell type. The relative importance of each varies, depending on cell lineage and activity. For example, control of gene expression after transcription is of primary importance during early embryogenesis, when transcription is repressed due to continuous DNA replication (Newport and Kirschner, 1982; Batchelder et al., 1999; Tadros and Lipshitz, 2009), and in cells in which physiology is partially decoupled from the nucleus due to size or morphology, such as neurons (Swanger and Bassell, 2011). In such situations, RNA-binding proteins and small RNA–protein complexes function as critical regulatory factors that control mRNA stability, subcellular localization, and translation efficiency to guide cell function.
To coordinate gene expression, RNA-regulatory factors must select specific target transcripts from the set of all transcripts present in the cell. A variety of high-throughput methods, including cross-linked immunoprecipitation with deep sequencing (Licatalosi et al., 2008; Hafner et al., 2010) and RNA immunoprecipitation coupled with array- or deep sequencing-based detection (Kershner and Kimble, 2010; Wright et al., 2011), reveal that some RNA-regulatory factors interact with hundreds or thousands of mRNA targets. Although the number is large, it is in many cases less than one might expect based solely upon predictions from corresponding in vitro binding studies (Ryder et al., 2004; Bernstein et al., 2005; Wright et al., 2011). In contrast, functional assays routinely show that only a few RNA targets contribute to the phenotype observed upon loss of the RNA-regulatory factor (Lee and Schedl, 2001; Hansen et al., 2004; Kalchhauser et al., 2011), suggesting that most binding events are either unproductive or not important to cellular function. The apparent disparity between in vitro binding, in vivo binding, and functional data reveals that the basis for specific target selection is not well understood.
Posttranscriptional regulation of mRNA is primarily mediated through cis-regulatory elements present in the untranslated regions (UTRs) of the transcript. These regions are not subject to the evolutionary constraint of the genetic code and thus can primarily serve a regulatory role. A survey of germline-expressed genes in the nematode Caenorhabditis elegans demonstrates that 3′ UTRs are sufficient to drive patterned gene expression in the germline (Merritt et al., 2008). Transgenic worms carrying a fluorescent reporter that included only the 3′ UTR of the gene being investigated recapitulated the expression pattern of the endogenous protein in 24 of 30 cases (Merritt et al., 2008). In addition, most transcripts do not contain a unique 5′ UTR, as 5′-end formation in nematodes is mediated primarily by trans-splicing of one of two leader sequences (Allen et al., 2011). Thus the C. elegans germline provides an ideal model system in which the selection of biologically relevant mRNA targets by sequence-specific RNA-binding proteins can be examined.
Studies of translationally regulated transcripts in C. elegans development reveal that multiple RNA-binding proteins contribute to the regulation of a single mRNA (Jadhav et al., 2008; Pagano et al., 2009). One example of this is the transcript encoding the C. elegans Notch receptor homologue glp-1. GLP-1 is a critical regulator of at least two distinct developmental pathways. It is the central regulator of the mitosis-to-meiosis switch in the distal arm of the gonad (Austin and Kimble, 1987), and it is required for specifying endodermal cell fates in the anterior of the four-cell embryo (Mickey et al., 1996). GLP-1 protein is restricted to these locations in the germline and embryo (Figure 1A); however, glp-1 mRNA is present throughout the entire gonad and all cells of the early embryo (Evans et al., 1994). glp-1 translational repression requires at least five different RNA-binding proteins, each repressing translation at different times during development: the STAR-domain protein GLD-1 acts in germ cells entering meiosis (Francis et al., 1995; Kadyk and Kimble, 1998), the PUF family members PUF-5/6 and PUF-7 act during oogenesis (Lublin and Evans, 2007), the KH-domain protein MEX-3 acts after fertilization (Pagano et al., 2009), and both GLD-1 and the CCCH-type tandem zinc finger protein POS-1 are required in the posterior of early embryos (Ogura et al., 2003; Marin and Evans, 2003; Figure 1B). In addition, the RRM-domain protein SPN-4 is required for translational activation of glp-1 in the early embryo (Ogura et al., 2003).
FIGURE 1:

POS-1 and GLD-1 expression is anticorrelated with GLP-1 expression. (A) Schematic of GLD-1 and GLP-1 expression in the germline. GLP-1 expression (green) is restricted to germ cells in mitosis, whereas GLD-1 (red) is expressed in the meiotic syncytium. GLD-1 is expressed diffusely in the cytoplasm, as well as in P-body–like granules. (B) Schematic of GLP-1, POS-1, and GLD-1 expression in embryos. Embryos are oriented with the anterior to the left. glp-1 mRNA (light green) is expressed in all cells of the early embryo, whereas GLP-1 protein (dark green) is not expressed until the four-cell stage and is restricted to the surface of the two anterior blastomeres (ABa and ABp). POS-1 (blue) is expressed in the posterior cytoplasm of early embryos from the one-cell stage. POS-1 localizes to perinuclear P-granules in the germline P-lineage of embryos. GLD-1 (red) expression begins at the four-cell stage, and it is present in the two posterior blastomeres. GLD-1 is found in the cytoplasm, in perinuclear P-granules in the P-lineage, and in granules distributed throughout both cells of the four-cell stage.
Mutational analysis of the glp-1 3′ UTR has identified a 34-nucleotide region that is sufficient to spatially pattern a reporter (Marin and Evans, 2003). This region can be further subdivided into two regulatory elements: the glp-1 repression element (GRE) and the glp-1 derepression element (GDE; Marin and Evans, 2003). Mutations within the GRE result in an expanded reporter expression pattern in the gonad and excess reporter expression in the posterior of early embryos. On the other hand, mutations of the GDE result in either decreased or no reporter expression in either the gonad or embryos (Marin and Evans, 2003). GLD-1 directly associates with the GRE in a sequence-specific manner, suggesting that GLD-1 translationally represses glp-1 through the GRE (Marin and Evans, 2003). Given the proximity of the GDE and the GRE, it has been hypothesized that another RNA-binding protein inhibits GLD-1 association with the GRE by binding to the GDE (Marin and Evans, 2003; Figure 2).
FIGURE 2:
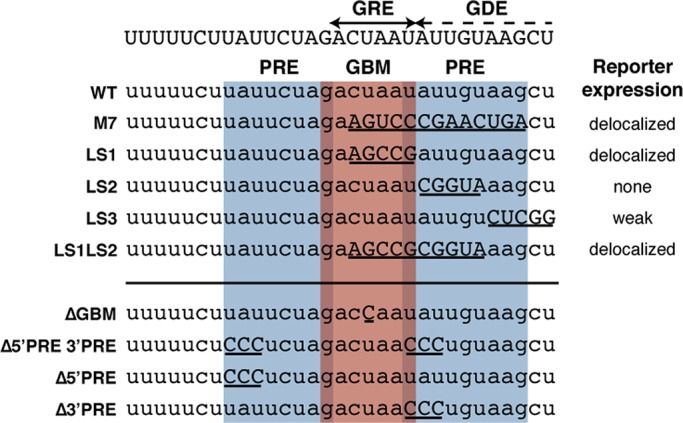
POS-1–and GLD-1–binding sites lie within regulatory elements of the glp-1 3′ UTR. The GRE is adjacent to the GDE (top), and both overlap with predicted POS-1 recognition elements (PRE, blue) and a GLD-1 binding motif (GBM, red). Mutations across this region in the context of a reporter (left, above the line) result in various expression patterns, listed to the right. The mutations used in this study are listed below the line.
We previously mapped a binding site for POS-1 (POS-1 recognition element [PRE]; Farley et al., 2008) within the GDE that partially overlaps with the GLD-1–binding motif (GBM; Wright et al., 2011) in the GRE, suggesting that POS-1 could function as the glp-1 activator through competition with GLD-1 (Figure 2). In contrast, mutational studies suggest that POS-1 acts to repress glp-1 translation, possibly in complex with GLD-1, and a different factor functions as the activator (Marin and Evans, 2003). Of importance, none of the mutations that have been made across the GRE and GDE is predicted to exclusively perturb POS-1 binding (Figure 2), so it remains unclear what role POS-1 plays in regulating glp-1 or whether regulation is direct.
POS-1 and GLD-1 exhibit different expression patterns in the early embryo. POS-1 is first observed in the posterior of the one-cell embryo and is expressed in the posterior blastomeres of the embryo through the four-cell stage. POS-1 is present throughout the cytoplasm of the cells in which it is expressed, as well as in perinuclear granules called P-granules in the germline precursor blastomeres (Tabara et al., 1999). In contrast, GLD-1 is first expressed in the posterior blastomeres of the four-cell stage and it localizes to cytoplasmic granules in the cells in which it is expressed (Jones et al., 1996). In addition to the cytoplasmic granules, GLD-1 also localizes to P-granules (Jones et al., 1996; Figure 1B). Thus POS-1 and GLD-1 may associate with glp-1 mRNA in different subcellular locations.
To determine the individual roles of both POS-1 and GLD-1 in regulating glp-1, we identify mutations that perturb binding of POS-1 or GLD-1 individually and use them to generate fluorescent reporters to measure their relative contribution to the regulation of glp-1 expression in vivo. The results reveal that POS-1 is indeed a repressor of glp-1 translation, but it acts independently of GLD-1, and its activity is highly context dependent.
RESULTS
Identification of a second PRE in the glp-1 3′ UTR
In a previous report (Farley et al., 2008), we used quantitative in vitro binding studies to define the POS-1 consensus recognition element and used the pattern-matching tool PatScan (Dsouza et al., 1997) to identify putative PREs in annotated C. elegans 3′ UTRs based upon this sequence. PatScan identifies binding motifs using a text-string matching algorithm (Dsouza et al., 1997). This analysis revealed a single PRE in the glp-1 3′ UTR; however, we noticed that the search pattern does not accurately reflect the thermodynamic measurements in a special case in which a single-nucleotide deletion compensates for an otherwise deleterious mutation. Specifically, mutation of the adenosine at position 6 of the PRE to a cytosine reduces binding by 0.6 kcal/mol (Farley et al., 2008), whereas reducing the number of intervening nucleotides between positions 6 and 10 improves binding by the same amount (Farley et al., 2008). When we apply the revised pattern to the glp-1 3′ UTR, we observe a second putative PRE that lies immediately upstream from the GBM, such that the last nucleotide of the PRE corresponds to the first nucleotide of the GBM. As such, the GBM is flanked by two PREs, each overlapping the GBM by one nucleotide (Figure 2).
POS-1 and GLD-1 recognize the glp-1 3′ UTR in a sequence-specific manner
To determine whether POS-1 and GLD-1 recognize their predicted binding sites in the glp-1 3′ UTR, we expressed and purified N-terminal maltose-binding protein fusions of the RNA-binding domains of POS-1 (amino acids 1–206) and GLD-1 (amino acids 135–336) for use in quantitative fluorescence electrophoretic mobility shift assays (F-EMSAs). Truncations of both proteins were used because of the difficulty of purifying sufficient quantities of full-length protein to perform in vitro binding experiments. The in vitro specificity of GLD-1(135-336) correlates well with enriched sequence motifs present in mRNAs coimmunoprecipitated with GLD-1 from whole-worm extract (Wright et al., 2011), whereas POS-1(1-206) lacks 58 amino acids with no homology to known domains. These proteins were used in F-EMSA along with a 34-nucleotide RNA fragment of the glp-1 3′ UTR that contains both PREs, the GBM, and flanking sequences (Table 1 and Figure 3). The fraction of bound RNA was plotted as a function of total protein concentration and fit to the Hill equation to determine the apparent dissociation constant (Kd,app) and Hill coefficient (n). By this method, both POS-1(1-206) and GLD-1(135-336) bind to this fragment with high affinity (POS-1(1-206), Kd,app = 19 ± 2 nM and n = 1.3 ± 0.2; GLD-1(135-336), Kd,app = 70 ± 10 nM and n = 1.0 ± 0.2; Table 1 and Figure 3).
TABLE 1:
Dissociation constants of POS-1 and GLD-1 for variants of the glp-1 fragment.
| Identifier | Sequence | POS-1 Kd,app (nM) | GLD-1 Kd,app (nM) |
|---|---|---|---|
| glp-1 WT | UUUUUCUUAUUCUAGACUAAUAUUGUAAGCU | 19 ± 2 | 70 ± 10 |
| ∆GBM | UUUUUCUUAUUCUAGACCAAUAUUGUAAGCU | 21 ± 1 | 2000 |
| ∆5′ PRE | UUUUUCUCCCUCUAGACUAAUAUUGUAAGCU | 55 ± 4 | 33 ± 7 |
| ∆3′ PRE | UUUUUCUUAUUCUAGACUAACCCUGUAAGCU | 53 ± 1 | 25 ± 3 |
| ∆5′ 3′ PRE | UUUUUCUCCCUCUAGACUAACCCUGUAAGCU | 310 ± 20 | 20 ± 3 |
Reported Kd,app values are the mean ± one SD of three independent replicates.
FIGURE 3:

POS-1 and GLD-1 recognize the glp-1 3′ UTR in a sequence-specific manner. Fluorescence electrophoretic mobility shift assays with recombinant POS-1(1-206) (left) and GLD-1(135-329) (right) and fluorescently labeled fragments of the glp-1 3′ UTR. Top, gel shift images. Bottom, quantifications and fits. POS-1(1-206) binds to the wild-type glp-1 fragment with Kd,app = 19 ± 2 nM, mutation of either PRE individually reduces binding ∼2.5-fold (∆5′ PRE, Kd,app = 54 ± 4 nM; ∆3′ PRE, Kd,app = 52 ± 1 nM), and mutation of both reduces binding ∼15-fold (∆5′ 3′ PRE, Kd,app = 310 ± 20 nM). GLD-1(135-329) binds to the wild-type glp-1 fragment with Kd,app = 70 ± 10 nM, and mutation of the GBM almost completely abrogates binding (∆GBM, Kd,app > 2000 nM). Reported Kd,app values are the average ± SD of three independent replicates.
To determine whether binding of each protein is dependent on its respective binding sites, we designed RNA oligonucleotides containing mutations in either the PREs or the GBM (Table 1 and Figure 3). Based on previous studies, the mutations were predicted to reduce the affinity of each protein for this sequence by >10-fold. Mutation of both PREs results in an 15-fold reduction in affinity for POS-1(1-206) (Kd,app = 310 ± 20 nM), whereas mutation of the GBM results in almost complete abrogation of binding of GLD-1(135-336) (Kd,app > 2000 nM). Thus both POS-1(1-206) and GLD-1(135-336) recognize the glp-1 3′ UTR in a binding site–dependent manner (Figure 3B).
The PREs are equivalent and independent
Given that only one of the PREs in the glp-1 fragment perfectly matches the previously published consensus, we wanted to determine whether POS-1(1-206) recognized each binding site. To test the contribution of each site individually, we designed oligonucleotides bearing mutations in only one of the PREs and performed EMSA experiments with POS-1(1-206). Mutation of either PRE resulted in a 2.5-fold reduction in binding compared with the wild-type sequence (Kd,app WT = 19 ± 2 nM, Kd,app ∆5′ PRE = 54 ± 4 nM, and Kd,app ∆3′ PRE = 52 ± 1 nM), demonstrating that each site is recognized by POS-1(1-206) with equivalent affinity (Figure 3B).
To determine whether the two equivalent PREs in the glp-1 3′ UTR are independent, we analyzed the relationship between the macroscopic and microscopic dissociation constants. An RNA with two equivalent, independent binding sites for a protein for which only one site is occupied at any given time should have a macroscopic dissociation constant that is twofold tighter than the microscopic dissociation constants observed for either in isolation due to statistical effects. We observe a 2.5-fold decrease in affinity between the glp-1 fragments with one intact PRE versus the wild-type sequence, suggesting that the two PREs are both equivalent and independent. This hypothesis is further supported by the unchanged Hill coefficients of the individual-site mutants versus the wild-type sequence (nWT = 1.3 ± 0.2, n∆5′ PRE = 1.7 ± 0.5, and n∆3′ PRE = 1.5 ± 0.3), as cooperative binding to the wild-type sequence is expected to increase the Hill coefficient.
POS-1(1-206) antagonizes GLD-1(135-336) binding to the glp-1 3′ UTR
Given that each of two independent POS-1–binding sites overlaps with the GBM, we hypothesized that POS-1(1-206) binding may inhibit GLD-1(135-336) binding to the glp-1 3′ UTR. To test this hypothesis, we performed in vitro competition gel shift experiments with POS-1(1-206) and GLD-1(135-336). In these experiments, a range of concentrations of the competitor protein was titrated into a fixed, trace concentration of fluorescently labeled RNA and a fixed subsaturating concentration of the other protein. The differently bound species of RNA were resolved from one another by electrophoresis on a native polyacrylamide gel.
When POS-1(1-206) was titrated into samples containing 400 nM GLD-1(135-336) and labeled RNA, we observed a decrease in the amount of GLD-1(135-336)–RNA complex and corresponding formation of faster-mobility POS-1(1-206)–RNA complex (Figure 4, A and B). At high POS-1(1-206) concentration (>300 nM), nonspecific POS-1(1-206) binding obscured visibility of residual GLD-1(135-336) complex (Figure 4, A and B). In contrast, when GLD-1(135-336) is titrated into samples containing 100 nM POS-1(1-206), no GLD-1(135-336) complex is observed, even when GLD-1(135-336) is present at a concentration that is 10-fold greater than POS-1(1-206) (Figure 4, A and B). No evidence of a slower-mobility species is apparent, suggesting that efficient ternary complex formation does not happen. Because the RNA contains two POS-1–binding sites that overlap with the single GLD-1–binding site and because the apparent affinity of POS-1(1-206) is 3.5-fold tighter than the apparent affinity of GLD-1(135-336), this is the expected result if the proteins compete for binding to the RNA fragment. Similar results were obtained when the experiment was repeated with a shorter variant of POS-1 containing only the RNA-binding domain (POS-1-RBD, amino acids 80–180; Figure 4, C and D). This construct binds to the glp-1 fragment with similar affinity to that of the longer construct (Kd,app,POS-1-RBD = 30 ± 17 nM; Supplemental Figure S1) but provides greater resolution of the POS-1(80-180)– and GLD-1(135-336)–bound complexes. POS-1(80-180) efficiently competed with GLD-1(135-336) for binding to the glp-1 fragment, whereas GLD-1(135-336) complex formation was strongly inhibited by POS-1(80-180). Taken together, the data show that POS-1 and GLD-1 do not simultaneously bind the glp-1 3′ UTR fragment and suggest that POS-1 could inhibit GLD-1 binding to the glp-1 3′ UTR in vivo.
FIGURE 4:

POS-1 antagonizes GLD-1 to the glp-1 fragment. (A) Gels of competition experiments with POS-1(1-206) and GLD-1(135-329). Top, POS-1(1-206) is titrated into a fixed concentration of GLD-1(135-329). Bottom, GLD-1(135-329) is titrated into a fixed concentration of POS-1(1-206). The mobilities of POS-1–bound RNA, GLD-1–bound RNA, and free RNA are labeled to the left of the gels. (B) Quantifications of POS-1(1-206) competition experiments. Left, POS-1(1-206) is titrated into a fixed concentration of GLD-1(135-329). Right, GLD-1(135-329) is titrated into a fixed concentration of POS-1(1-206). In both plots, fraction of total RNA bound by each protein is plotted against the concentration of the titrated protein. Filled circles, POS-1–bound RNA. Open circles, GLD-1–bound RNA. (C, D). Competition experiments with POS-1(80-180) and GLD-1(135-329). The gels in C are labeled as in A, and the plots in D are labeled as in B.
Mutations in the glp-1 3′ UTR are specific for either POS-1 or GLD-1
To individually determine the regulatory contribution of direct binding of POS-1 or GLD-1 to the glp-1 3′ UTR, we designed mutants that would exclusively affect either POS-1 or GLD-1 binding. To determine whether the mutations designed to inhibit binding of either POS-1 or GLD-1 to the glp-1 3′ UTR do not interfere with the binding of the other protein, we used quantitative EMSA to measure the affinity of POS-1(1-206) for the ∆GBM version of the glp-1 fragment and the affinity of GLD-1(135-336) for the ∆5′, 3′ PRE sequence. Mutation of the GBM does not change the apparent affinity of POS-1(1-206) (Kd,app WT = 19 ± 2 nM and Kd,app ∆GBM = 21 ± 1 nM), indicating that single-nucleotide GBM mutation does not affect POS-1(1-206) binding. Mutation of both PREs results in a threefold increase in GLD-1(135-336) affinity (Kd,app WT = 70 ± 10 nM and Kd,app ∆PRES = 20 ± 4 nM; Supplemental Figure S2), which is statistically significant (p = 0.008), suggesting that mutations in the POS-1–binding sites weakly increase the affinity of GLD-1(135-336) for the glp-1 3′ UTR.
Both POS-1 and GLD-1 binding are required for repression of glp-1 translation in embryos
To determine whether direct binding of POS-1 to the glp-1 3′ UTR antagonizes GLD-1 and thus derepresses glp-1 translation in early embryos, we generated green fluorescent protein (GFP) reporters carrying the wild-type glp-1 3′ UTR or the mutant variations characterized in vitro (Table 2). To ensure that we were observing only the posttranscriptional regulation of glp-1, our reporters used the mex-5 promoter, a germline promoter with a similar expression pattern to the glp-1 promoter. The open reading frame of each reporter encodes a protein fusion of GFP and C. elegans histone 2B (H2B), which concentrates the fluorescence signal in the nucleus and facilitates cell identification. Because the half-life of both GFP and H2B is long relative to oogenesis (Frand et al., 2005), the open reading frame also contains the mouse ornithine decarboxylase PEST domain, which destabilizes the protein (Frand et al., 2005). To enable direct comparison of the reporter expression patterns resulting from different transgenic constructs, the transgenes were integrated site specifically into chromosome II using Mos1-mediated single-copy insertion of transgenes (MosSCI; Frøkjaer-Jensen et al., 2008; Figure 5A).
TABLE 2:
Transgenic worm strains used in this study.
| glp-1 3′ UTR variant | Strain identifier | Genotype |
|---|---|---|
| WT | WRM5 | sprSi5[Pmex-5::MODC PEST:GFP:H2B::glp-1 3′UTR cb-unc-119(+)] II, unc-119(ed3) III |
| ∆GBM | WRM6 | sprSi6[Pmex-5::MODC PEST:GFP:H2B::glp-1 3′UTR(∆GBM) cb-unc-119(+)] II, unc-119(ed3) III |
| ∆5′ PRE | WRM7 | sprSi7[Pmex-5::MODC PEST:GFP:H2B::glp-1 3′UTR(∆5′ PRE) cb-unc-119(+)] II, unc-119(ed3) III |
| ∆3′ PRE | WRM8 | sprSi8[Pmex-5::MODC PEST:GFP:H2B::glp-1 3′UTR(∆3′ PRE) cb-unc-119(+)] II, unc-119(ed3) III |
| ∆5′ 3′ PRE | WRM9 | sprSi9[Pmex-5::MODC PEST:GFP:H2B::glp-1 3′UTR(∆5′ 3′ PRE) cb-unc-119(+)] II, unc-119(ed3) III |
FIGURE 5:
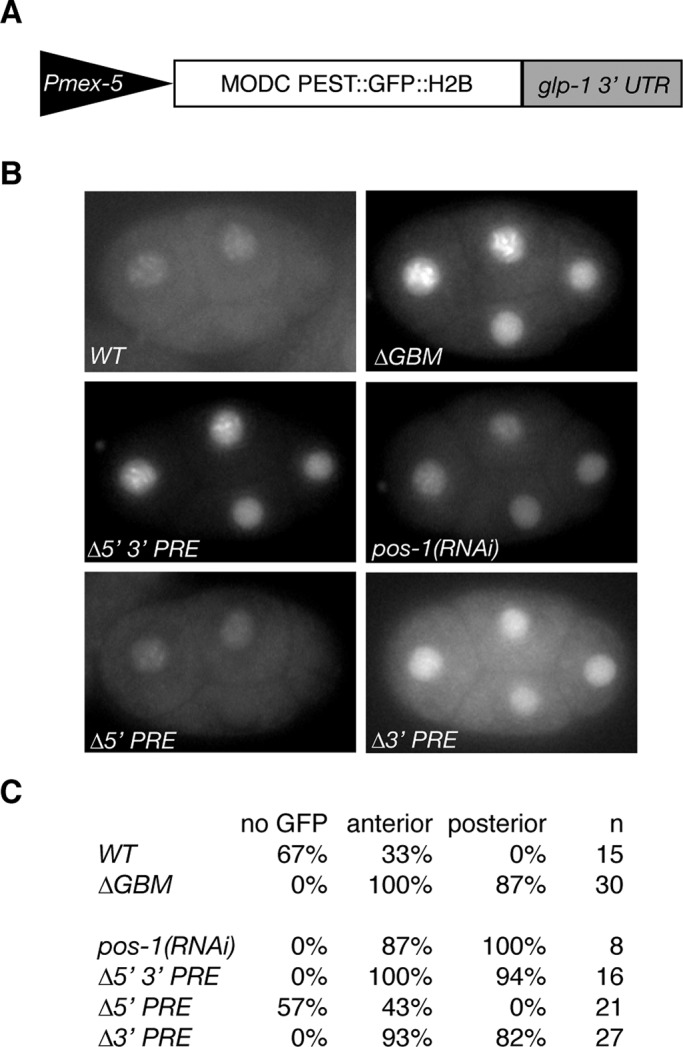
POS-1 and GLD-1 binding are independently required to repress a glp-1 3′ UTR reporter in embryos. (A) Schematic of reporter constructs used in this study. (B) Representative images of four-cell embryos with the listed reporter or experimental condition. (C) Table of results. No GFP, the percentage of embryos with no detectable GFP; anterior, the percentage of embryos expressing GFP in the two anterior blastomeres; posterior, the percentage of embryos expressing GFP in the two posterior blastomeres; n is the number of embryos observed.
To determine the effects on reporter translation in embryos of disrupting POS-1 or GLD-1 binding to the glp-1 3′ UTR, we dissected adult worms and observed live four-cell-stage embryos. Endogenous GLP-1 is expressed only in the two anterior blastomeres at the four-cell stage (Evans et al., 1994). If direct binding of GLD-1 is required for translational repression, mutating the GBM should result in reporter expression in the posterior as well as the anterior of the four-cell-stage embryo. If POS-1 antagonizes GLD-1 binding and thus leads to derepression, neither anterior nor posterior expression of the reporter should be observed when the PREs are mutated. Thirty-three percent of embryos carrying the WT reporter express GFP only in the anterior blastomeres of the four-cell stage, whereas 67% express no detectable GFP (n = 15) at this stage (Figure 5, B and C). The substantial fraction of transgene-bearing, four-cell-stage embryos that lack detectable GFP fluorescence is likely due to the time required for maturation of the GFP chromophore (Reid and Flynn, 1997), which is long relative to the duration of the four-cell stage. The anterior expression pattern matches that of endogenous GLP-1 protein, indicating that the glp-1 3′ UTR is sufficient for appropriate patterning, and the reporter mimics the expression pattern of endogenous GLP-1, as previously reported (Evans et al., 1994). On treatment with pos-1(RNAi), 100% of embryos express GFP in all cells of the four-cell-stage embryo (n = 8; Figure 5, B and C), suggesting that POS-1 protein plays an inhibitory role in the translational regulation of glp-1. POS-1 may have indirect effects on the translation of glp-1, so to investigate the requirement for POS-1 binding, we examined a reporter with both PREs mutated. Mutation of both PREs results in a similar expression pattern as that for embryos carrying the wild-type reporter treated with pos-1(RNAi) (94% express in posterior, n = 16; Figure 5, B and C), suggesting that the PREs are required for translational repression rather than derepression of glp-1 and that POS-1 directly regulates glp-1. Because the two PREs are equivalent and independent, we hypothesized that the two binding sites are redundant. To test this hypothesis and determine the individual contribution of each PRE, we generated transgenic strains bearing mutations in either the 5′ or 3′ PRE. Mutating the 5′ PRE results in embryos exhibiting wild-type GFP expression (anterior, 43%; no expression, 57%; n = 21; Figure 5, B and C), whereas mutating the 3′ PRE results in ubiquitously expressed GFP at the four-cell stage (82% express in posterior, n = 27; Figure 5, B and C). This suggests that despite the equivalent affinities of the PREs in vitro, only the 3′ PRE is required for POS-1–mediated translational repression of glp-1.
Given that POS-1 binding is essential for translational repression of glp-1 and that GLD-1 does not bind to the glp-1 3′ UTR in the presence of POS-1 in vitro, we wanted to revisit the role of the GBM in embryos compared with the germline, where GLD-1 is present but POS-1 is not. To test this hypothesis, we generated a transgenic line carrying a mutation in the GBM and observed four-cell-stage embryos. Embryos carrying this transgene express GFP in all cells of the early embryo (87% express in posterior, n = 30; Figure 5, B and C), suggesting that the GBM is also required for translational repression of glp-1 in the embryo. This matches the previously published results for both the endogenous GLP-1 expression pattern in gld-1(RNAi) embryos (Marin and Evans, 2003) and for reporters carrying mutations in the GRE (Marin and Evans, 2003), confirming that GLD-1 acts through the GRE to repress glp-1 translation in the embryo.
The 3′ PRE mutations disrupt the GDE in the germline
The 3′ PRE mutation lies entirely within the GDE, which is required for translational activation of glp-1 but results in delocalized expression in the embryo. We wanted to determine the effect of the PRE mutations on germline expression of the glp-1 reporter. The gonads of live worms were observed by confocal fluorescence microscopy (Figure 6A). To compare the GFP expression level between strains, we imaged confocal sections through the syncytial region of the gonad of multiple worms for each strain. For each image, the mean pixel intensity across one row of nuclei within the distal arm of the gonad was determined using the image quantification software ImageJ (Schneider et al., 2012). To normalize for differences in the lengths of gonad arms, the measured pixel intensities for each image were divided into 30 segments, and intensities within segments were averaged together. Single-worm segmented intensity measurements for one transgenic strain were averaged to give a composite average intensity for each strain (Figure 6B).
FIGURE 6:

Mutation of the 3′ PRE decreases glp-1 reporter expression independently of POS-1. (A) Representative confocal fluorescence images of the listed reporter strains or experimental condition. White asterisks mark the distal end of the gonad, and white arrows mark decreased reporter expression in the meiotic region of ∆5′ 3′ PRE and ∆3′ PRE reporter strains. (B) Quantification of confocal pixel intensities. Reporter expression for each strain was quantified using the image quantification software ImageJ as described in Materials and Methods. Each of the untreated transgenic strains was normalized to the average pixel intensity of the first 10% of the gonad of worms expressing the glp-1 WT reporter, and each of the RNAi experiments was normalized to the average pixel intensity of the first 10% of mock-treated animals. Plotted data represent the mean intensity per bin ± one SEM. Traces represent the quantification of images taken of the strain listed in the legends at the upper right of each plot.
Worms carrying the wild-type (WT) reporter have maximum GFP expression in the distal end of the germline, with diminishing expression in the meiotic region (Figure 6B; glp-1 WT, n = 17). This closely matches the expression of endogenous GLP-1 in the germline (Crittenden et al., 1994). Mutation of the 5′ PRE has no effect on expression of GFP, consistent with the effects observed in embryos (Figure 6B; glp-1 ∆5′ PRE, n = 18). In contrast, mutation of the 3′ PRE results in slightly reduced GFP expression in the mitotic region of the germline (Figure 6B; glp-1 ∆3′ PRE, n = 16) relative to the wild-type glp-1 reporter. Mutation of both the 5′ and 3′ PREs results in a further reduction in reporter expression throughout the distal arm of the gonad (Figure 6B; glp-1 ∆5′ 3′ PRE, n = 18). This apparent decrease is not dependent on POS-1, as pos-1(RNAi) has no apparent effect on expression of the wild-type reporter in the germline relative to worms fed with an empty feeding vector (Figure 6B; glp-1 WT; mock, n = 12, vs. glp-1 WT; pos-1(RNAi), n = 8). Taken together, these results suggests that the 5′ and 3′ PRE mutations also disrupt a regulatory element that participates in translational activation of glp-1 mRNA in the distal arm of the germline and that POS-1 does not regulate glp-1 mRNA in the germline. Thus another RNA-binding protein may recognize this element and promote glp-1 translation. This additional factor is unlikely to be GLD-1, as the GBM is involved in translational repression of glp-1 in the syncytial region. A reporter carrying a mutation in the GBM displays slightly increased expression of GFP in the syncytial region of the germline (Figure 6B; glp-1 ∆GBM, n = 19).
SPN-4 does not directly activate glp-1 translation in the germline
The RRM-containing protein SPN-4 is a potential candidate for the activating factor that operates through the GDE in early embryos. SPN-4 is expressed throughout all cells of the embryo during the four-cell stage (Ogura et al., 2003), and GLP-1 expression is undetectable in embryos lacking SPN-4 (Ogura et al., 2003). In addition, SPN-4 and POS-1 compete for binding to the nos-2 3′ UTR in vitro (Jadhav et al., 2008). To determine whether SPN-4 activates expression of the glp-1 3′ UTR reporter, we observed the pattern of GFP expression in four-cell embryos as a function of SPN-4 knockdown. Forty-eight percent of untreated four-cell embryos carrying the glp-1 WT reporter described earlier exhibit GFP expression in the anterior cells (n = 28; Figure 7A), whereas only 11% of spn-4(RNAi) four-cell embryos had detectable GFP in the anterior cells (n = 38). This suggests that SPN-4 is indeed required to activate expression from the glp-1 3′ UTR reporter in the early embryo. However, this activation is not mediated through the GDE, as SPN-4 knockdown has no effect on GFP expression in four-cell embryos carrying a reporter bearing the ∆3′ PRE mutation (∆3′ PRE: 100% of embryos have anterior and posterior GFP expression, n = 30; ∆3′ PRE spn-4(RNAi): 93% of embryos have anterior GFP expression and 97% have posterior GFP expression, n = 30; Figure 7A). Furthermore, purified SPN-4–RBD does not bind to either the glp-1 fragment (Figure 7B) or a longer RNA containing the entire glp-1 GDE (unpublished data) in vitro. Thus SPN-4 is not likely to compete with POS-1 for binding to the GDE. Consistent with this interpretation, SPN-4 was previously observed to interact with a distant element of the glp-1 3′-UTR by yeast three-hybrid assay (Ogura et al., 2003).
FIGURE 7:

SPN-4 activates glp-1 translation indirectly. (A) Representative images of four-cell embryos carrying the listed reporter construct (left) and treated as described (top). Bottom, table of results. Anterior and posterior, the percentage of four-cell embryos with detectable GFP fluorescence in either the anterior or posterior cells, respective. None, the percentage of embryos with no detectable GFP fluorescence. (B) Representative gel shift data for SPN-4(50-135). The protein concentration used in each lane is labeled above the gel.
The cytoplasmic poly(A) polymerase GLD-2 and its specificity factors GLD-3 and RNP-8 are not required for translational activation of glp-1 mRNA
To identify proteins that are responsible for the translational activation of glp-1 mRNA in the germline, we took a candidate approach and screened the expression pattern of the glp-1 WT reporter in the context of RNA interference (RNAi) knockdown by feeding. One potential candidate is the cytoplasmic poly(A) polymerase GLD-2, which mediates the translational activation of multiple transcripts in the distal arm of the germline by extending their shortened poly(A) tails and thus permitting translation (Wang et al., 2002; Hansen et al., 2004; Eckmann et al., 2004; Suh et al., 2006). GLD-2 requires the association of a specificity factor to target it to mRNAs. Two such specificity factors have been identified: GLD-3 and RNP-8 (Wang et al., 2002; Eckmann et al., 2004; Kim et al., 2010). The GLD-2/GLD-3 complex promotes meiotic entry and spermatogenesis (Wang et al., 2002; Eckmann et al., 2002, 2004), whereas the GLD-2/RNP-8 complex promotes oogenesis (Kim et al., 2009, 2010). Given the catalytic activity and biological roles of these complexes, they may play a role in the translational activation of glp-1.
To test the requirement for each of these proteins in the regulation of glp-1 translation, we reduced the level of their expression in worms carrying the glp-1 WT reporter by feeding worms bacteria expressing double-stranded RNA targeted against the gene of interest. If the proteins are required for translational activation of glp-1 mRNA, the level of reporter expression observed in the germline should decrease relative to worms treated with an empty feeding vector. We do not observe a decrease in reporter expression (Supplemental Figure S3), suggesting that translational activation of glp-1 mRNA in the germline is not dependent on GLD-2–mediated cytoplasmic polyadenylation.
Taken together, consistent with previous work, the results lead to the conclusion that translational control of glp-1 requires both repressive and activating elements. GLD-1 binds to the GRE and is required for repression. POS-1 also binds to the GRE in vitro, but binding to the GRE has no effect on glp-1 expression in worms. POS-1(1-206) also binds with equivalent affinity to the GDE in vitro, but binding is required for repression, not activation. We suggest that POS-1 represses translation by competing with an unidentified factor that binds to the GDE to promote translation. This would explain why two POS-1–binding sites with equivalent affinities, separated by only five nucleotides, contribute disparately to glp-1 regulation, and why the PRE is necessary but not sufficient to confer regulation in worms.
DISCUSSION
In this report, we describe the translational regulation of the C. elegans Notch homologue glp-1 by the CCCH-type tandem zinc finger protein POS-1 and the STAR-domain protein GLD-1. The glp-1 3′ UTR contains a cluster of regulatory elements comprising two PREs, each of which overlaps with a GBM. POS-1 and GLD-1 each bind to their respective sites in vitro, but POS-1 recognizes its sites with a higher affinity than GLD-1. In the presence of POS-1, GLD-1 binding to its site is inhibited. However, both the 3′ PRE and the GBM are required for translational repression of glp-1 3′ UTR reporter in the four-cell embryo, suggesting that both POS-1 and GLD-1 translationally repress glp-1 mRNA through this cluster of binding sites. In addition, the 3′ PRE but not the 5′ PRE is required for regulation of glp-1. Mutation of both PREs or the 3′ PRE alone results in decreased reporter expression in the distal germline, where POS-1 is not expressed. This suggests that another factor is required for translational activation of glp-1 mRNA, and it recognizes the PREs. Here, we present evidence for a cluster of functional overlapping binding sites within the glp-1 3′ UTR.
Translational repression by POS-1 and GLD-1
Our data suggest that POS-1 and GLD-1 directly and independently repress glp-1 translation in the early embryo. Although binding sites for both proteins within the glp-1 3′ UTR are required for translational repression of a glp-1 3′ UTR reporter in vivo, the in vitro competition results support the hypothesis that binding of POS-1 and GLD-1 is mutually exclusive. GLD-1 forms homodimers in the absence of RNA, and these dimers bind to RNA directly (Ryder et al., 2004). Moreover, GLD-1 contacts sequence outside its seven-nucleotide motif (Ryder et al., 2004; Wright et al., 2011), suggesting that tight binding of proteins to immediately flanking sequences would antagonize GLD-1 binding. Thus, although POS-1 and GLD-1 cooperate to ensure that glp-1 translation remains repressed, our data show no evidence for positive cooperativity in their ability to recognize the glp-1 3′-UTR.
The apparent independence of POS-1 and GLD-1 regulation of glp-1 may be a consequence of the differing spatial, temporal, and subcellular localization patterns of each protein. Because POS-1 is expressed earlier than GLD-1 (Tabara et al., 1999; Jones et al., 1996), glp-1 is likely regulated only by POS-1 during the one- and two-cell stages. Once GLD-1 translation begins, it may become the primary negative regulator of glp-1. Because POS-1 antagonizes binding of GLD-1 to the glp-1 3′ UTR, the handoff to GLD-1 mediated repression would likely require inactivation or turnover of POS-1.
Alternatively, the differences in subcellular localization between POS-1 and GLD-1 may be indicative of different locations of glp-1 repression in the early embryo. In the germline, GLD-1 is present in cytoplasmic granules that also contain the DDX6-like RNA helicase CGH-1 (Noble et al., 2008). Both GLD-1 and CGH-1 have been implicated in the translational repression and stabilization of a number of maternal mRNAs in the germline that are destined for translation after fertilization (Scheckel et al., 2012). This suggests that these granular structures play a role in the storage of maternal mRNAs. Indeed, the glp-1 3′ UTR is sufficient for localization of a reporter mRNA to these granules in the meiotic region of the germline (Noble et al., 2008), and removal of the GLD-1–binding site results in decreased subcellular localization of reporter mRNA in the germline (Noble et al., 2008). GLD-1 also localizes to granular structures in the cytoplasm of early embryos (Jones et al., 1996), but the role of embryonic cytoplasmic granules is unknown. It is possible that glp-1 mRNA localizes to GLD-1–containing granules in the embryo in a GLD-1–dependent manner. If this is the case, two distinct pools of glp-1 mRNA could exist within the posterior of the four-cell embryo: a cytoplasmic pool that requires POS-1 for translational repression and a granular pool that requires GLD-1 for translational repression. Thus each protein would play a role in the translational regulation of glp-1 without forming a ternary complex.
POS-1–mediated translational repression
Our data demonstrate that the 5′ and 3′ PREs within the glp-1 3′ UTR are not functionally equivalent. In four-cell embryos, mutation of the 3′ PRE results in aberrant translation of a reporter in the anterior blastomeres, whereas mutation of the 5′ PRE has no effect on the expression of a reporter. In the germline, mutation of the 3′ PRE results in decreased reporter expression relative to a reporter bearing the wild-type glp-1 3′ UTR, whereas mutation of the 5′ PRE has no apparent effect. The discrepancy between the function of these binding sites is likely explained by the overlap of the 3′ PRE (but not the 5′ PRE) with a previously identified activating regulatory element called the glp-1 derepression element (Marin and Evans, 2003). Mutations within this element cause either no or reduced expression of a reporter without having a detectable difference on the expression level of the reporter mRNA (Marin and Evans, 2003). Thus the GDE is required for translational activation, and mutations that target the 3′ PRE are likely to perturb the function of this element as well. This also explains why mutations of the 3′ PRE result in decreased reporter expression in a region of the gonad where POS-1 is not expressed, as this mutation likely targets a regulatory element that is recognized by another protein in the germline.
The overlap between regulatory elements suggests a competition model for POS-1–mediated translational regulation of mRNAs. In this speculative model, POS-1 regulates its targets not by binding to mRNA and recruiting regulatory machinery, but instead by interfering with the binding of other RNA-binding proteins that do recruit regulatory machinery. Thus the regulatory role of a PRE in vivo is likely heavily dependent on its sequence context, as functional PREs should overlap with or be near regulatory elements recognized by other proteins. As a result, many PREs are likely to be bound by POS-1, but binding of POS-1 may not result in a change in the rate of translation or the stability of the associated mRNA. In support of this model is the observation that the addition of multiple PREs is not sufficient to confer patterned expression on an otherwise unpatterned 3′ UTR (Farley et al., 2008). More than 40% of C. elegans transcripts are predicted to contain at least one PRE (Farley et al., 2008), and so finding the functional binding sites likely presents multiple challenges in bioinformatics and systems biology. Thus identifying functional PREs in vivo will likely require the integration of multiple data sets describing the binding sites for other RNA-binding proteins in vivo. By investigating the specificity of additional factors, we may identify additional binding sites that overlap with PREs and may be occupied by POS-1 in vivo.
MATERIALS AND METHODS
Cloning and purification of POS-1, GLD-1, and SPN-4
DNA encoding amino acids 1–206 of POS-1 cloned into the protein expression vector pMAL-c2x (New England BioLabs, Ipswich, MA) was graciously provided by Tom Evans (University of Colorado Anshutz Medical Campus, Aurora, CO). The plasmid was transformed into Escherichia coli strain BL21 (DE3). Protein expression was induced with 1 mM isopropyl-β-d-thiogalactoside (IPTG) and 100 μM Zn(OAc)2. The cells were lysed using a microfluidizer, and the lysate was purified using an amylose column (New England BioLabs), followed by a Source Q column (GE Healthcare Life Sciences, Piscataway, NJ) and a HiPrep 16/60 Sephacryl S-200 column (GE Healthcare Life Sciences). After the final column, the protein was dialyzed into 25 mM Tris, pH 8.0, 25 mM NaCl, 2 mM dithiothreitol (DTT), and 100 μM Zn(OAc)2, concentrated to ∼30 μM, and used for experiments.
DNA encoding amino acids 50–135 of SPN-4 was cloned into the protein expression vector pHMTc (Ryder et al., 2004), transformed into E. coli strain BL21 (DE3), and induced as described for POS-1 1–206 but omitting the zinc acetate. Cells were lysed using a microfluidizer, and the lysate was purified using an amylose column (New England BioLabs), followed by a HiTrap SP HP column (GE Healthcare Life Sciences) and a SourceQ column. Following the last column, the protein was dialyzed into 25 mM Tris, pH 8.0, 25 mM NaCl, and 2 mM DTT, concentrated to ∼20 μM, and used for experiments.
POS-1–RBD was expressed and purified from pHMTc-POS-1(80-180) as described previously (Farley et al., 2008). GLD-1 was expressed and purified from pHMTc-GLD1(135-336) as described (Ryder et al., 2004).
Fluorescence labeling of RNAs
RNA oligonucleotides were synthesized by Integrated DNA Technologies (Coralville, IA) and fluorescently labeled on their 3′ ends via periodate oxidation, followed by reaction with fluorescein-5-thiosemicarbazide (Sigma-Aldrich, St. Louis, MO). Unreacted label was purified away via G-25 spin column. A detailed protocol is available (Pagano et al., 2011).
Fluorescence electrophoretic mobility shift assays
Fluorescence electrophoretic mobility shift assays were essentially performed and analyzed as described in Pagano et al. (2011). Briefly, 2 nM fluorescently labeled RNA in equilibration buffer (50 mM Tris, pH 8.0, 100 mM NaCl, 5 μM Zn(OAc)2, 0.01% IGEPAL CA-630, and 0.01 mg/ml tRNA) was mixed with various concentrations of either POS-1 or GLD-1 and equilibrated at room temperature for 3 h. The protein-bound and free RNA was then resolved on a 1× Tris-borate buffer (TB) native 5% polyacrylamide slab gel run at 120 V for approximately 1 h at 4 ºC.
Competition assays were performed in essentially the same manner, except that a fixed concentration of POS-1 or GLD-1 was added to the labeled RNA in equilibration buffer to achieve ∼70% bound RNA in the absence of competitor protein. Then various amounts of competitor protein were added to each reaction and incubated for at least 3 h. The reactions were then loaded onto a 1× TB native 5% polyacrylamide slab gel run at 120 V for 3 h at 4ºC to resolve POS-1– and GLD-1–bound complexes. Gels were quantified using Image Gauge (Fujifilm, Tokyo, Japan), and the fraction of protein-bound RNA was determined by quantifying the ratio of the background-corrected pixel intensity of the protein-bound RNA relative to the sum of the background-corrected pixel intensities of each RNA species. Two independent replicates of each competition experiment involving POS-1 were performed, and five independent replicates were performed with POS-1–RBD.
Cloning of reporter constructs
The glp-1 3′ UTR was amplified via PCR using Elongase (Invitrogen, Carlsbad, CA) from worm genomic DNA using primers that added the attB2R and attB3 sites to the 5′ and 3′ ends of the product, respectively. The PCR product was then cloned into pDONRP2RP3 using BP Clonase II (Invitrogen). Site-specific mutations in the glp-1 3′ UTR were introduced via QuikChange mutagenesis using Pfu Turbo. Each of the resulting variants of the glp-1 3′ UTR was then used in a multisite gateway reaction with plasmids bearing the mex-5 promoter (pCM1.111) and MODC PEST::GFP::H2B ORF (pBMF2.7) to generate constructs for integration. The gateway reaction was catalyzed with LR Clonase II Plus (Invitrogen), and the promoter::ORF::3′ UTR fusions were cloned into the MosSCI integration vector pCFJ150.
Generation and verification of transgenic strains
Single-copy integrated transgenic worms strains were generated by MosSCI (Frøkjaer-Jensen et al., 2008). Plasmids bearing the transgene to be integrated were microinjected into the gonads of young adult worms of strain EG4322 along with pharyngeal- and body wall–expressed mCherry markers and a constitutive germline-expressed Mos1 transposase. Prior to injection, worms were maintained at 15ºC on NGM agar plates seeded with Comamonas (DA1877). Worms were propagated for two generations and screened for successful integration by checking for wild-type movement without expression of the mCherry extrachromosomal array markers. Putative integrants were confirmed by PCR using a transgene-specific primer and a worm genome–specific primer. These PCR products were then sequenced to validate the mutations in the reporter's 3′ UTR.
Imaging and quantification of fluorescent reporter strains
Before imaging, worms were maintained at 25ºC for at least 24 h to promote GFP folding. Embryos were obtained by dissecting adult worms and then mounted on 2% agarose pads, and whole worms were paralyzed with 0.4 mM levamisole and mounted on 2% agarose pads. Both differential interference contrast and GFP images were collected using a 40× Plan Neofluar oil immersion objective on a Zeiss Axioskop 2 plus microscope (Carl Zeiss, Jena, Germany).
Confocal microscopy was performed on a Leica DMIRE2 microscope outfitted with a Leica TCS SP2 scanner (Leica, Wetzlar, Germany) using a 40× oil immersion/1.25 numerical aperture objective. A single confocal section through the syncytial region of the distal gonad arm was imaged for each worm. Samples were excited with a 488-nm laser at 100% intensity, and the photomultiplier tube collected emitted light between 500 and 600 nm with a gain of 610 HV. Each scan line is the average of eight reads.
Confocal images were quantified using the segmented line tool in ImageJ (National Institutes of Health, Bethesda, MD; Schneider et al., 2012), using a method adapted from Wright et al. (2011). A 7-pixel-wide line was drawn through a row of nuclei beginning at the distal end of the gonad and ending at the bend in the distal arm, and the average pixel intensity across this line was segmented into 30 equally sized bins. Segmentation and averaging were performed using Perl scripts that are available upon request. The measured average pixel intensity was determined for multiple worms, and values were averaged together to give a composite pixel intensity for each strain.
RNAi knockdown
Embryos were harvested from adult worms by treatment with 0.5 N NaOH and 2% Clorox bleach, washed twice with water, and then transferred to NGM plates supplemented with 1 mM IPTG and 100 μg/ml ampicillin and seeded with HT115(DE3) bacteria transformed with a construct expressing double-strand RNA targeting the gene of interest. Before seeding, bacteria transformed with spn-4–expressing, gld-2–expressing (Kim et al., 2010), gld-3–expressing (Eckmann et al., 2004), or rnp-8–expressing (Kim et al., 2010) constructs were induced at ∼0.4 OD600 with 1 mM IPTG. Constructs targeting pos-1, gld-2, gld-3, and rnp-8 were obtained from the Ahringer feeding library (Kamath et al., 2003). The construct targeting spn-4 was generated by cloning the PCR product amplified from spn-4 cDNA with the following primers into the NcoI site of the C. elegans feeding vector L4440: forward, 5′-GAGCCATGGTGCAAAACACACAGATATTTACTAAC-3′; reverse, 5′-GAGCCATGGACTGGCTTGACGATTCTTTTGG-3′; Kamath and Ahringer, 2003). Worms were maintained at 25ºC for 2 d, and the P0 generation was imaged as described earlier. Greater than 95% embryonic lethality was observed in the F1 generation of pos-1– or spn-4–treated worms.
Supplementary Material
Acknowledgments
We thank Rob Stefani for technical assistance with the competition gel shifts, Craig Mello for guidance and microinjection training, Nick Rhind for graciously sharing equipment, and Ruth Zearfoss and Ebru Kaymak for critical comments and discussion. We also thank Tom Evans for providing the POS-1 expression plasmid. This work was supported by National Institutes of Health Grant GM081422 to S.P.R.
Abbreviations used:
- EMSA
electrophoretic mobility shift assay
- GBM
GLD-1 binding motif
- GDE
glp-1 derepression element
- GFP
green fluorescent protein
- GRE
glp-1 repression element
- H2B
histone 2B
- Kd,app
apparent equilibrium dissociation constant
- KH
hnRNP K homology
- MosSCI
Mos1-mediated single-copy insertion of transgenes
- PRE
POS-1 recognition element
- RBD
RNA-binding domain
- RBP
RNA-binding protein
- RRM
RNA recognition motif
- STAR
signal transduction activator of RNA metabolism
- UTR
untranslated region
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-03-0216) on October 3, 2012.
REFERENCES
- Allen MA, Hillier LW, Waterston RH, Blumenthal T. A global analysis of C. elegans trans-splicing. Genome Res. 2011;21:255–264. doi: 10.1101/gr.113811.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin J, Kimble J. glp-1 is required in the germ line for regulation of the decision between mitosis and meiosis in C. elegans. Cell. 1987;51:589–599. doi: 10.1016/0092-8674(87)90128-0. [DOI] [PubMed] [Google Scholar]
- Batchelder C, Dunn MA, Choy B, Suh Y, Cassie C, Shim EY, Shin TH, Mello C, Seydoux G, Blackwell TK. Transcriptional repression by the Caenorhabditis elegans germ-line protein PIE-1. Genes Dev. 1999;13:202–212. doi: 10.1101/gad.13.2.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein D, Hook B, Hajarnavis A, Opperman L, Wickens M. Binding specificity and mRNA targets of a C. elegans PUF protein, FBF-1. RNA. 2005;11:447–458. doi: 10.1261/rna.7255805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crittenden SL, Troemel ER, Evans TC, Kimble J. GLP-1 is localized to the mitotic region of the C. elegans germ line. Development. 1994;120:2901–2911. doi: 10.1242/dev.120.10.2901. [DOI] [PubMed] [Google Scholar]
- Dsouza M, Larsen N, Overbeek R. Searching for patterns in genomic data. Trends Genet. 1997;13:497–498. doi: 10.1016/s0168-9525(97)01347-4. [DOI] [PubMed] [Google Scholar]
- Eckmann CR, Crittenden SL, Suh N, Kimble J. GLD-3 and control of the mitosis/meiosis decision in the germline of Caenorhabditis elegans. Genetics. 2004;168:147–160. doi: 10.1534/genetics.104.029264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckmann CR, Kraemer B, Wickens M, Kimble J. GLD-3, a bicaudal-C homolog that inhibits FBF to control germline sex determination in C. elegans. Dev Cell. 2002;3:697–710. doi: 10.1016/s1534-5807(02)00322-2. [DOI] [PubMed] [Google Scholar]
- Evans TC, Crittenden SL, Kodoyianni V, Kimble J. Translational control of maternal glp-1 mRNA establishes an asymmetry in the C. elegans embryo. Cell. 1994;77:183–194. doi: 10.1016/0092-8674(94)90311-5. [DOI] [PubMed] [Google Scholar]
- Farley BM, Pagano JM, Ryder SP. RNA target specificity of the embryonic cell fate determinant POS-1. RNA. 2008;14:2685–2697. doi: 10.1261/rna.1256708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis R, Barton MK, Kimble J, Schedl T. gld-1, a tumor suppressor gene required for oocyte development in Caenorhabditis elegans. Genetics. 1995;139:579–606. doi: 10.1093/genetics/139.2.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frand AR, Russel S, Ruvkun G. Functional genomic analysis of C. elegans molting. PLoS Biol. 2005;3:e312. doi: 10.1371/journal.pbio.0030312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frøkjaer-Jensen C, Davis MW, Hopkins CE, Newman BJ, Thummel JM, Olesen S-P, Grunnet M, Jorgensen EM. Single-copy insertion of transgenes in Caenorhabditis elegans. Nat Genet. 2008;40:1375–1383. doi: 10.1038/ng.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen D, Wilson-Berry L, Dang T, Schedl T. Control of the proliferation versus meiotic development decision in the C. elegans germline through regulation of GLD-1 protein accumulation. Development. 2004;131:93–104. doi: 10.1242/dev.00916. [DOI] [PubMed] [Google Scholar]
- Jadhav S, Rana M, Subramaniam K. Multiple maternal proteins coordinate to restrict the translation of C. elegans nanos-2 to primordial germ cells. Development. 2008;135:1803–1812. doi: 10.1242/dev.013656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones AR, Francis R, Schedl T. GLD-1, a cytoplasmic protein essential for oocyte differentiation, shows stage- and sex-specific expression during Caenorhabditis elegans germline development. Dev Biol. 1996;180:165–183. doi: 10.1006/dbio.1996.0293. [DOI] [PubMed] [Google Scholar]
- Kadyk LC, Kimble J. Genetic regulation of entry into meiosis in Caenorhabditis elegans. Development. 1998;125:1803–1813. doi: 10.1242/dev.125.10.1803. [DOI] [PubMed] [Google Scholar]
- Kalchhauser I, Farley BM, Pauli S, Ryder SP, Ciosk R. FBF represses the Cip/Kip cell-cycle inhibitor CKI-2 to promote self-renewal of germline stem cells in C. elegans. EMBO J. 2011;30:3823–3829. doi: 10.1038/emboj.2011.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath RS, Ahringer J. Genome-wide RNAi screening in Caenorhabditis elegans. Methods. 2003;30:313–321. doi: 10.1016/s1046-2023(03)00050-1. [DOI] [PubMed] [Google Scholar]
- Kamath RS, et al. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 2003;421:231–237. doi: 10.1038/nature01278. [DOI] [PubMed] [Google Scholar]
- Kershner AM, Kimble J. Genome-wide analysis of mRNA targets for Caenorhabditis elegans FBF, a conserved stem cell regulator. Proc Natl Acad Sci USA. 2010;107:3936–3941. doi: 10.1073/pnas.1000495107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KW, Nykamp K, Suh N, Bachorik JL, Wang L, Kimble J. Antagonism between GLD-2 binding partners controls gamete sex. Dev Cell. 2009;16:723–733. doi: 10.1016/j.devcel.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KW, Wilson TL, Kimble J. GLD-2/RNP-8 cytoplasmic poly(A) polymerase is a broad-spectrum regulator of the oogenesis program. Proc Natl Acad Sci USA. 2010;107:17445–17450. doi: 10.1073/pnas.1012611107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MH, Schedl T. Identification of in vivo mRNA targets of GLD-1, a maxi-KH motif containing protein required for C. elegans germ cell development. Genes Dev. 2001;15:2408–2420. doi: 10.1101/gad.915901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licatalosi DD, et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. 2008;456:464–469. doi: 10.1038/nature07488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lublin AL, Evans TC. The RNA-binding proteins PUF-5, PUF-6, and PUF-7 reveal multiple systems for maternal mRNA regulation during C. elegans oogenesis. Dev Biol. 2007;303:635–649. doi: 10.1016/j.ydbio.2006.12.004. [DOI] [PubMed] [Google Scholar]
- Marin VA, Evans TC. Translational repression of a C. elegans Notch mRNA by the STAR/KH domain protein GLD-1. Development. 2003;130:2623–2632. doi: 10.1242/dev.00486. [DOI] [PubMed] [Google Scholar]
- Merritt C, Rasoloson D, Ko D, Seydoux G. 3′ UTRs are the primary regulators of gene expression in the C. elegans germline. Curr Biol. 2008;18:1476–1482. doi: 10.1016/j.cub.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mickey KM, Mello CC, Montgomery MK, Fire A, Priess JR. An inductive interaction in 4-cell stage C. elegans embryos involves APX-1 expression in the signalling cell. Development. 1996;122:1791–1798. doi: 10.1242/dev.122.6.1791. [DOI] [PubMed] [Google Scholar]
- Newport J, Kirschner M. A major developmental transition in early Xenopus embryos: II. Control of the onset of transcription. Cell. 1982;30:687–696. doi: 10.1016/0092-8674(82)90273-2. [DOI] [PubMed] [Google Scholar]
- Noble SL, Allen BL, Goh LK, Nordick K, Evans TC. Maternal mRNAs are regulated by diverse P body-related mRNP granules during early Caenorhabditis elegans development. J Cell Biol. 2008;182:559–572. doi: 10.1083/jcb.200802128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura K-I, Kishimoto N, Mitani S, Gengyo-Ando K, Kohara Y. Translational control of maternal glp-1 mRNA by POS-1 and its interacting protein SPN-4 in Caenorhabditis elegans. Development. 2003;130:2495–2503. doi: 10.1242/dev.00469. [DOI] [PubMed] [Google Scholar]
- Pagano JM, Clingman CC, Ryder SP. Quantitative approaches to monitor protein-nucleic acid interactions using fluorescent probes. RNA. 2011;17:14–20. doi: 10.1261/rna.2428111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano JM, Farley BM, Essien KI, Ryder SP. RNA recognition by the embryonic cell fate determinant and germline totipotency factor MEX-3. Proc Natl Acad Sci USA. 2009;106:20252–20257. doi: 10.1073/pnas.0907916106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid BG, Flynn GC. Chromophore formation in green fluorescent protein. Biochemistry. 1997;36:6786–6791. doi: 10.1021/bi970281w. [DOI] [PubMed] [Google Scholar]
- Ryder SP, Frater LA, Abramovitz DL, Goodwin EB, Williamson JR. RNA target specificity of the STAR/GSG domain post-transcriptional regulatory protein GLD-1. Nat Struct Mol Biol. 2004;11:20–28. doi: 10.1038/nsmb706. [DOI] [PubMed] [Google Scholar]
- Scheckel C, Gaidatzis D, Wright JE, Ciosk R. Genome-wide analysis of GLD-1-mediated mRNA regulation suggests a role in mRNA storage. PLoS Genet. 2012;8:e1002742. doi: 10.1371/journal.pgen.1002742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh N, Jedamzik B, Eckmann CR, Wickens M, Kimble J. The GLD-2 poly(A) polymerase activates gld-1 mRNA in the Caenorhabditis elegans germ line. Proc Natl Acad Sci USA. 2006;103:15108–15112. doi: 10.1073/pnas.0607050103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanger SA, Bassell GJ. Making and breaking synapses through local mRNA regulation. Curr Opin Genet Dev. 2011;21:414–421. doi: 10.1016/j.gde.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabara H, Hill RJ, Mello CC, Priess JR, Kohara Y. pos-1 encodes a cytoplasmic zinc-finger protein essential for germline specification in C. elegans. Development. 1999;126:1–11. doi: 10.1242/dev.126.1.1. [DOI] [PubMed] [Google Scholar]
- Tadros W, Lipshitz HD. The maternal-to-zygotic transition: a play in two acts. Development. 2009;136:3033–3042. doi: 10.1242/dev.033183. [DOI] [PubMed] [Google Scholar]
- Wang L, Eckmann CR, Kadyk LC, Wickens M, Kimble J. A regulatory cytoplasmic poly(A) polymerase in Caenorhabditis elegans. Nature. 2002;419:312–316. doi: 10.1038/nature01039. [DOI] [PubMed] [Google Scholar]
- Wright JE, Gaidatzis D, Senften M, Farley BM, Westhof E, Ryder SP, Ciosk R. A quantitative RNA code for mRNA target selection by the germline fate determinant GLD-1. EMBO J. 2011;30:533–545. doi: 10.1038/emboj.2010.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.