

Cdc14A phosphatase regulates Wee1 kinase through dephosphorylation of two Cdk phosphorylation sites in its regulatory domain, Ser-123 and -139, both involved in the degradation of Wee1 at the entry into mitosis. In this way, Cdc14A interferes with the negative feedback loop between Wee1 and Cdk1 to regulate the mitotic switch.
Abstract
The activity of Cdk1–cyclin B1 mitotic complexes is regulated by the balance between the counteracting activities of Wee1/Myt1 kinases and Cdc25 phosphatases. These kinases and phosphatases must be strictly regulated to ensure proper mitotic timing. One masterpiece of this regulatory network is Cdk1, which promotes Cdc25 activity and suppresses inhibitory Wee1/Myt1 kinases through direct phosphorylation. The Cdk1-dependent phosphorylation of Wee1 primes phosphorylation by additional kinases such as Plk1, triggering Wee1 degradation at the onset of mitosis. Here we report that Cdc14A plays an important role in the regulation of Wee1 stability. Depletion of Cdc14A results in a significant reduction in Wee1 protein levels. Cdc14A binds to Wee1 at its amino-terminal domain and reverses CDK-mediated Wee1 phosphorylation. In particular, we found that Cdc14A inhibits Wee1 degradation through the dephosphorylation of Ser-123 and Ser-139 residues. Thus the lack of phosphorylation of these two residues prevents the interaction with Plk1 and the consequent efficient Wee1 degradation at the onset of mitosis. These data support the hypothesis that Cdc14A counteracts Cdk1–cyclin B1 activity through Wee1 dephosphorylation.
INTRODUCTION
The regulation of mitotic entry is critical in cell cycle control. In higher eukaryotes, the activation of Cdk1–cyclin B1 complexes, which is subject to strict control, drives the major events of mitosis. During the S and G2 phases, B-type cyclins accumulate, bind to Cdk1, and promote phosphorylation on three regulatory sites of Cdk1: Thr-14, Tyr-15 and Thr-161. The phosphorylation of Cdk1 on Thr-161 is required for maximal kinase activity, whereas phosphorylation on Thr-14 and Tyr-15 inhibits the enzymatic activity of Cdk1. In vertebrate cells, the Wee1 and Myt1 kinases are responsible for these inhibitory modifications, in which Wee1 appears to be dominant (McGowan and Russell, 1995; Mueller et al., 1995a, b; Perry and Kornbluth, 2007). The activation of Cdk1 is achieved by dephosphorylation of these two residues by Cdc25 phosphatases (Nilsson and Hoffmann, 2000). An important mechanism that tips the balance toward Cdc25 phosphatase activity to allow entry into mitosis is the down-regulation of Wee1/Myt1 kinases. Wee1 activity rises during the S and G2 phases of the cell cycle and declines at the G2/M transition, when high Cdk1 activity is required. In human cells, Wee1 is down-regulated primarily through proteasome-dependent degradation after phosphorylation by multiple kinases, among which Cdk1 is present (Mueller et al., 1995a, b; Watanabe et al., 2004, 2005). Cdk1 phosphorylates Wee1 on Ser-123, which primes additional phosphorylation by other kinases, leading to the formation of phosphodegrons responsible for SCF (Skp1/cullin/F-box) ubiquitin-mediated degradation of Wee1 (Watanabe et al., 2005). Two different F-box proteins, β-TrCP and Tome-1 ubiquitin ligases, contribute directly to Wee1 destruction (Ayad et al., 2003; Watanabe et al., 2005; Smith et al., 2007). Other regions of Wee1 are also involved in its degradation. Thus the Wee1 activation domain, through phosphorylation at the Ser-472 residue, has been implicated in an efficient turnover of Wee1 (Owens et al., 2010). Cdk2, bound to cyclin A, could well be the kinase responsible for this phosphorylation (Owens et al., 2010). In other organisms, such as fission yeast and Xenopus, phosphorylation events inhibit the catalytic activity of Wee1 (Mueller et al., 1995a; Parker et al., 1993; Wu and Russell, 1993). In particular, Xenopus Wee1 can be phosphorylated and inhibited by Cdk1 in vitro (Mueller et al., 1995a). However, the phosphorylation of human Wee1 by Cdk1 does not inhibit its kinase activity in vitro (Watanabe et al., 1995). More recently, studies carried out in Xenopus suggested a probably conserved mechanism for Wee1 inactivation in which specific phosphorylation by Cdk1 at the well-conserved N-terminal region, called the Wee-box, induces Wee1 inactivation through binding with the cis/trans prolyl isomerase Pin1 (Okamoto and Sagata, 2007). In addition, other phosphorylation events carried out by different kinases are also involved in human Wee1 inactivation. Thus it was suggested that Akt promotes the G2/M transition by phosphorylation-dependent 14-3-3 binding and the cytoplasmic localization of Wee1 (Katayama et al., 2005). Moreover, Cdk2–cyclin A also contributes to Wee1 nuclear export through binding to a conserved RXL motif and probably phosphorylation on the Thr-239 residue (Li et al., 2010). Although export of Wee1 to the cytoplasm does not seem to be essential for entry into mitosis, it might be needed to prevent early activation of Cdk1–cyclin B1 complexes on centrosomes (Jackman et al., 2003). Therefore multiple mechanisms seem to be involved in the inhibition of human Wee1 at entry into mitosis.
The highly conserved Cdc14 family of dual-specificity phosphatases reverses CDK-dependent phosphorylation events. In Saccharomyces cerevisiae, Cdc14 is essential for the inactivation of mitotic Cdk and mitotic exit by reversing phosphorylation of many Cdk substrates (Stegmeier and Amon, 2004; Queralt and Uhlmann, 2008). In Schizosaccharomyces pombe, the Cdc14 homologue Flp1/Clp1 inhibits mitotic Cdk activity by promoting, at least in part, the down-regulation of Cdc25 protein through its direct dephosphorylation (Esteban et al., 2004, 2008; Wolfe and Gould, 2004). It was also suggested that Flp1 is required for efficient DNA-damage response (Díaz-Cuervo and Bueno, 2008).
Cdc14 homologues also exist in metazoans. In mammals, there are two Cdc14 homologues, Cdc14A and Cdc14B, whose functions and substrates are poorly known. The two homologues are able to rescue Cdc14-deficient yeast cells, suggesting that some functions are conserved (Li et al., 1997; Vázquez-Novelle et al., 2005). However, their participation in the dephosphorylation of mitotic Cdk substrates at the exit of mitosis is not clear. Cdc14A, located at the centrosome during interphase, was implicated in the regulation of the centrosome cycle (Kaiser et al., 2002; Mailand et al., 2002). Moreover, Cdc14A phosphatase prevents premature entry into mitosis by inhibiting Cdc25 activity (Vázquez-Novelle et al., 2010; Sacristán et al., 2011). Cdc14B seems to be involved in many functions during the cell cycle (Mocciaro and Schiebel, 2010) and is able to promote malignant transformation in vitro (Chiesa et al., 2011). It has also been suggested that both Cdc14A and Cdc14B phosphatases play a role in DNA repair (Berdougo et al., 2008; Mocciaro et al., 2010). Recent studies show that Cdc14 phosphatases regulate transcription during the cell cycle through the dephosphorylation of RNA polymerase II. This function is conserved in yeast and mammals (Clemente-Blanco et al., 2011; Guillamot et al., 2011). Here we identify a new Cdc14 substrate: the Wee1 kinase. Depletion of Cdc14A results in the degradation of Wee1 in human cells. This effect correlates with the ability of Cdc14A to reverse Cdk-mediated phosphorylation of Wee1 at Ser-123, a site known to be involved in Wee1 regulation, and Ser-139, a newly identified site phosphorylated by Cdks and also involved in the control of Wee1 stability. Given that Wee1 kinase is a crucial part of the regulatory network controlling Cdk1 activity, our data suggest that Cdc14A interferes with Cdk1 activity through Wee1 regulation.
RESULTS
Down-regulation of Cdc14A results in decreased Wee1 protein levels
The cooperative phosphorylation of Wee1 by Cdk1 and other kinases stimulates its degradation at the onset of mitosis to allow the G2/M transition (Watanabe et al., 2004, 2005). On the basis of the finding that depletion of Cdc14A accelerates entry into mitosis (Vázquez-Novelle et al., 2010) and that Cdc14 phosphatases reverse CDK phosphorylation, we wondered whether Cdc14A might regulate Wee1 kinase directly. To check this possibility, we first analyzed the levels of Wee1 in cells depleted for Cdc14A. U-2-OS cells were transfected with already validated Cdc14A small interfering RNAs (siRNAs; Vázquez-Novelle et al., 2010), and Wee1 was examined at different time points after transfection. As shown in Figure 1A, after 48 h of siRNA Cdc14A treatment the cells showed a reduction in Wee1 protein levels. Consistent with this result, the phosphorylation of Cdk1 at Tyr15 was also reduced at this time point (Figure 1A). Treatment of cells with the proteasome inhibitor MG132 abolished the reduction in Wee1 protein levels (Figure 1B), suggesting that Cdc14A regulates Wee1 degradation. To evaluate siRNA duplex specificity, we performed rescue experiments with a siRNA-resistant Cdc14A construct. In U-2-OS cells infected with empty vector or with retrovirus expressing wild-type (wt) Cdc14A, transfection with siRNA Cdc14A induced the already observed reduction in Wee1 protein level as compared with siRNA control cells (Figure 1C, lanes 5 and 6 and lanes 11 and 12, respectively). However, this effect was significantly reversed by expression of siRNA-resistant Cdc14A (Figure 1C, lanes 17 and 18). Reduction in the levels of Wee1 kinase was also observed in nontransformed human BJ-TERT fibroblasts after transfection with Cdc14A siRNAs (Supplemental Figure S1). We also analyzed the effect of Cdc14A down-regulation on Wee1 protein levels at the G2/M transition. As shown in Figure 1D, the degradation of Wee1 started earlier and was increased in cells depleted for Cdc14A as compared with control cells, suggesting that Cdc14A regulates Wee1 at the G2/M transition.
FIGURE 1:

Depletion of Cdc14A causes a reduction of Wee1 protein levels. (A) Asynchronously growing U-2-OS cells were transfected with Cdc14A or control siRNAs and grown for 24, 36, or 48 h. Cellular extracts were obtained and analyzed by immunoblot against the indicated proteins. Cdc14A mRNA levels were analyzed at each time point using real-time PCR in triplicate measurements (±SD). The value given for Cdc14A mRNA in control cells was set as 1. The percentage of mitotic cells was measured by fluorescence-activated cell sorting analysis using anti–phospho-histone H3 antibody. (B) U-2-OS cells were treated with Cdc14A (A) or control (C) siRNAs for 48 h. Where indicated, MG132 (20 μM) was added to the culture and maintained during the last 6 h. The levels of Wee1 protein were analyzed by immunoblot in the corresponding cell lysates. (C) U-2-OS cells infected with retroviruses expressing empty vector (control), Cdc14A wild type (wt), or siRNA-resistant Cdc14A (R) (a Cdc14A cDNA with four silent mutations in the sequence corresponding to siRNA sequence) were transfected with control or Cdc14A siRNAs. Cellular extracts were obtained at the indicated times and probed for Wee1 and Cdc14A. Relative Wee1 degradation was quantified by densitometry. Wee1 signal intensity was normalized to the loading control (actin) and expressed as the amount relative to the corresponding 36-h sample, which was set as 1. Note that the anti-Cdc14A antibody is able to recognize ectopically expressed Cdc14A but not the endogenous protein levels. (D) U-2-OS cells were synchronized in G1/S by double thymidine treatment and transfected with control or Cdc14A siRNAs after the first thymidine arrest. Cells were then released into fresh medium containing nocodazole to trap mitotic cells, and samples were taken at different time points as they progressed to mitosis for the immunoblotting and phospho–histone H3 positivity analyses. Asterisk, unspecific cross-reacting band. These blots are representative from at least three different experiments.
Ectopic expression of Cdc14 phosphatases results in the dephosphorylation of Wee1
To investigate Wee1 regulation by Cdc14A, we next analyzed Wee1 in cells with high levels of Cdc14A phosphatase. We used U-2-OS clones conditionally expressing Myc-tagged Cdc14A or its phosphatase-dead (PD) mutant form (Mailand et al., 2002). After the ectopic expression of active Cdc14A, but not of Cdc14A(PD), a striking change in the gel mobility of Wee1, detected as a faster-migrating form, was observed (Figure 2A). The treatment of Wee1, immunoprecipitated from both Cdc14A-overproducing and control cells, with λ phosphatase indicated that the observed change in the Wee1 electrophoretic mobility corresponded to the dephosphorylated forms of the protein (Figure 2B). However, overexpression of Cdc14A did not affect other Cdk1 substrates, such as the centrosomal Nek9 mitotic kinase (Bertran et al., 2011), nor did it cause an indiscriminate effect on all Cdk phosphoepitopes (Supplemental Figure S2), suggesting that the effect on Wee1 is specific. The dephosphorylation and accumulation of Wee1 were also observed when Cdc14A was overexpressed at the G2/M transition (Figure 2C). This effect was correlated, as already reported, with cells accumulating in G2 as consequence of high levels of Cdc14A (Figure 2C; Vázquez-Novelle et al., 2010). The dephosphorylation of Wee1 was also observed in HCT116 or HEK293T cells transiently transfected with active Cdc14A phosphatase (Supplemental Figure S3A). Moreover, the ectopic expression of Cdc14B, but not of PTEN or Cdc25 dual-specificity phosphatases, also resulted in Wee1 modification (Supplemental Figure S3B). Because the depletion of Cdc14B did not have any effect on Wee1 protein levels in normally cycling cells (Supplemental Figure S3C), we conclude that Cdc14A and Cdc14B phosphatases might regulate Wee1 in a different manner. Our work henceforth focuses on the role of the human Cdc14A isoform in Wee1 regulation.
FIGURE 2:
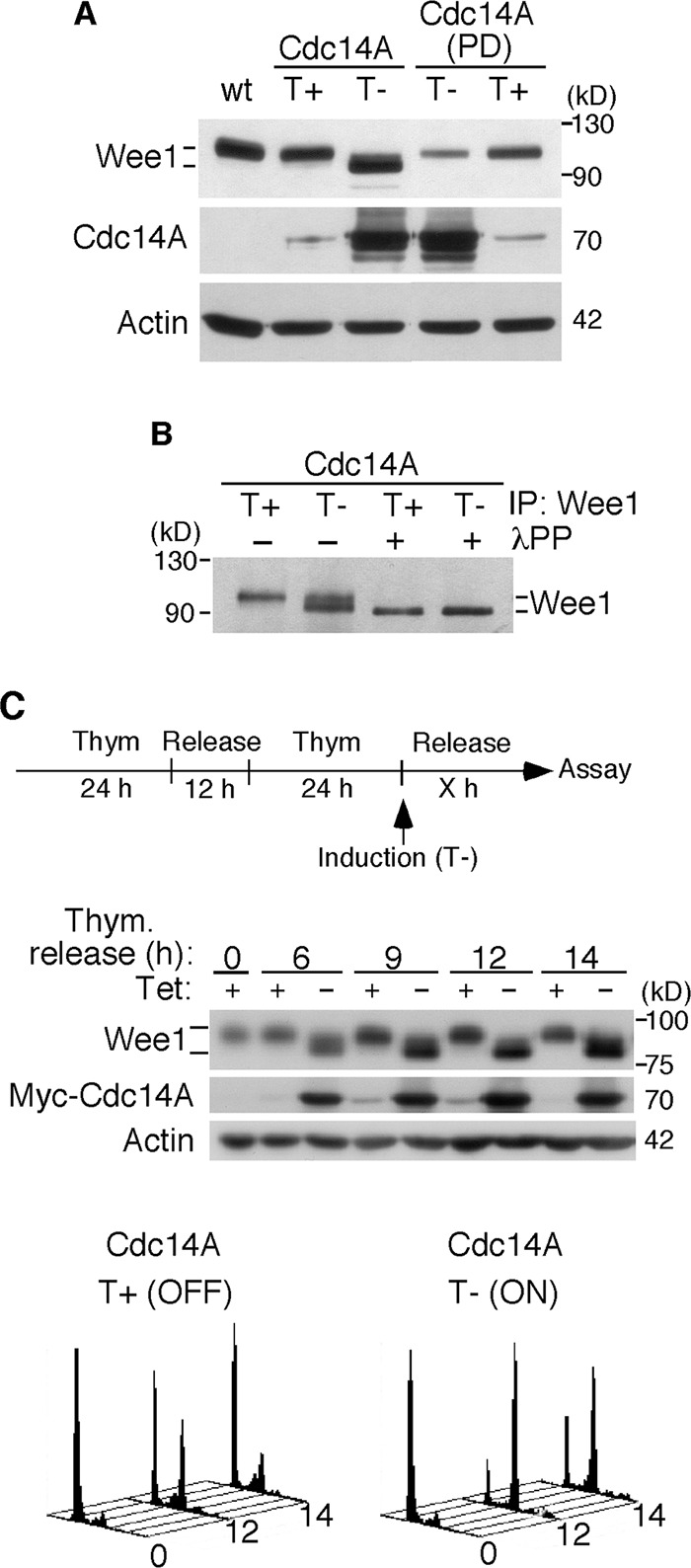
Wee1 is dephosphorylated in cells overexpressing Cdc14A. (A) U-2-OS cell lines conditionally expressing active Cdc14A or its catalytically inactive form, Cdc14A(PD), were left untreated (T+) or induced (T–) to express the transgenes by removal of tetracycline, and cell lysates were processed for immunoblotting with the indicated antibodies. (B) The U-2-OS Cdc14A cell line was left untreated or induced to express wild-type Cdc14A for 24 h, and cell extracts were obtained. Wee1 protein was then immunoprecipitated with anti-Wee1 antibodies and incubated with λ phosphatase (λPP). The dephosphorylation of Wee1 was monitored by immunoblotting. (C) U-2-OS–Myc-Cdc14A cells, synchronized at the G1/S transition by double thymidine treatment, were released and induced to express the transgene (top) or kept noninduced (control conditions). Samples were taken for flow cytometry (bottom) and immunoblot analyses at the indicated times after release. The data shown are representative of three different experiments.
Cdc14A interacts with and dephosphorylates Wee1
To check whether Wee1 kinase is a Cdc14A substrate, we first analyzed the molecular interactions between Cdc14A and Wee1 in the cell. Because the antibodies available do not detect endogenous Cdc14A, we used the Myc-Cdc14A fusion proteins expressed in HEK293T cells. As shown in Figure 3A, the overexpressed Myc-Cdc14A coimmunoprecipitated with the endogenous Wee1. No interaction was observed, however, between Wee1 and the dual Cdc25A phosphatase under the same overexpression conditions (Figure 3B), suggesting a specific interaction between Cdc14A and Wee1 proteins. Wee1 kinase contains 16 potential phosphorylation sites for CDKs, which, except for one of them (Ser-472), lie within the amino-terminal regulatory domain (Figure 3C). To test whether Cdc14A interacted with Wee1 through this domain, we coexpressed hemagglutinin (HA)-tagged, full-length Wee1, N-terminal (amino acids [aa] 1–257) or C-terminal (aa 258–646) halves of Wee1 and Myc-Cdc14A proteins in HEK293T cells. As shown in Figure 3, D and E, only the full-length or amino-terminal domain of Wee1 bound to Myc-Cdc14A, suggesting that the CDK-dependent phosphorylation state of Wee1 could mediate binding with the Cdc14A phosphatase.
FIGURE 3:

Wee1 interacts with Cdc14A through its amino regulatory region. (A, B) HEK293T cells were transfected with Myc-Cdc14A, Myc-Cdc14A(PD), or HA-Cdc25A. After 24 h of transfection, endogenous Wee1 was immunoprecipitated with anti-Wee1 antibody and analyzed by immunoblotting to detect the presence of tagged Cdc14A (A) or Cdc25A (B), respectively, and Wee1. Cellular extracts were also probed for the indicated antigens. Asterisk, unspecific cross-reacting band. (C) Distribution of CDK phosphorylation consensus sites (bars) on Wee1. Ser-123 and -139 correspond to the full consensus sequence Ser/Thr-Pro-X-Arg/Lys, where X represents any amino acid. (D) HEK293T cells were cotransfected with Myc-Cdc14A and full-length (FL) HA-Wee1. After 40 h of transfection, cellular extracts were obtained, and the presence of Cdc14A and Wee1 was analyzed by immunoblotting in anti-Myc-Cdc14A immunoprecipitates. (E) HEK293T cells were cotransfected with Myc-Cdc14A and HA-Wee1 N-terminal (ND; aa 1–257) or C-terminal (CD; aa 258–646) domains of Wee1. After 40 h of transfection, cellular extracts were immunoprecipitated with anti-Myc antibodies and analyzed by immunoblotting to detect the presence of the two proteins. The data shown are representative of at least two independent experiments.
Next, to see whether Wee1 was a substrate of Cdc14A phosphatase, we first performed in vitro phosphatase assays using Wee1 immunoprecipitates obtained from Cdc14A U-2-OS noninduced cells (control conditions) and recombinant GST-Cdc14A or GST-Cdc14A(PD) proteins purified from Escherichia coli. As shown in Figure 4A, incubation of Wee1 with wild-type Cdc14A, but not with the inactive Cdc14A(PD) form, resulted in some degree of Wee1 dephosphorylation, indicating that Cdc14A was able to dephosphorylate Wee1. As mentioned, Wee1 is phosphorylated by several kinases, among which mitotic Cdk1 is present. Because Cdc14A preferentially reverses CDK-dependent phosphorylations, we then tested whether Cdc14A was able to remove Cdk1-cyclin B1–dependent Wee1 phosphoresidues. To prevent the autophosphorylation of Wee1, we generated a kinase-dead (KD) derivative by replacing lysine by arginine-328 of the kinase domain. In vitro autophosphorylation assays confirmed that the Wee1(KD) mutant was inactive (Figure 4B, lane 1). Recombinant glutathione S-transferase GST-Wee1(KD) was first phosphorylated by Cdk1–cyclin B1 complexes and then incubated with GST-Cdc14A or the inactive form GST-Cdc14A(PD). As shown in Figure 4B, Cdc14A was able to decrease the phosphorylation signal of Wee1. Finally, to further test the ability of Cdc14A to reverse CDK-mediated serine phosphorylations (Bremmer et al., 2011), we immunoprecipitated protein extracts obtained from HEK293T cells coexpressing HA-tagged Wee1 and Myc-tagged Cdc14A or Cdc14A(PD) with anti-HA and immunoblotted them with specific anti–phospho-Ser (CDK substrates) antibody. As shown in Figure 4C, the ectopic expression of Cdc14A efficiently reversed the Cdk-mediated serine phosphorylations of Wee1. Taken together, all these results suggest that Cdc14A interacts with and dephosphorylates Cdk-dependent phosphorylation sites of Wee1.
FIGURE 4:
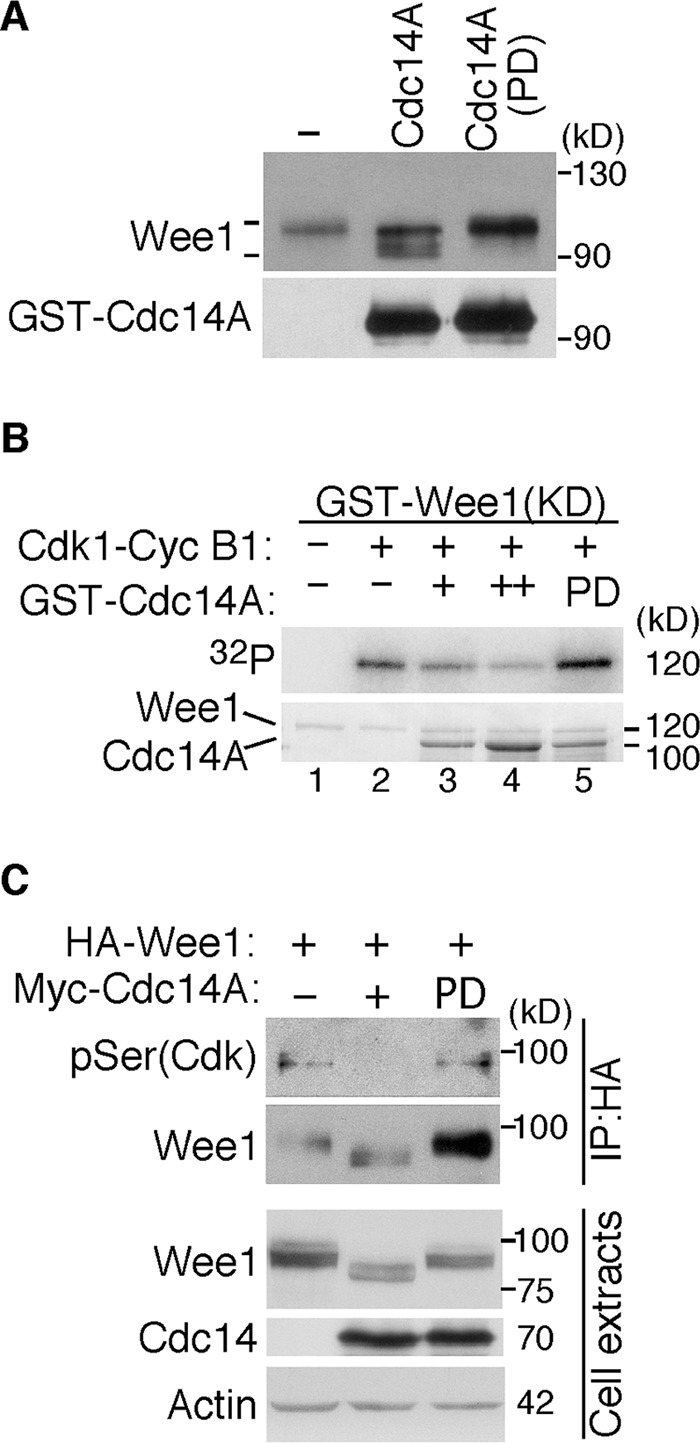
Cdc14A dephosphorylates Wee1. (A) Wee1 protein was obtained by immunoprecipitation with anti-Wee1 antibodies from the noninduced U-2-OS–Myc-Cdc14A cellular extracts (500 μg) and incubated with 100 ng of GST-Cdc14A or its inactive form, GST-Cdc14A(PD). Dephosphorylation of Wee1, detected by its different SDS–PAGE gel mobility, and the presence of the indicated Cdc14A forms detected by immunoblotting. (B) GST-Wee1(KD), GST-Cdc14A, and GST-Cdc14A(PD) fusion proteins were expressed and purified from E. coli. Inactive Wee1 kinase was first incubated with recombinant Cdk1–cyclin B1 in the presence of [γ-32P]ATP. Then it was washed extensively and treated with active or inactive GST-Cdc14A phosphatases. Reactions were resolved by SDS–PAGE and analyzed by Coomassie staining (bottom) and autoradiography (top). (C) HEK293T cells were cotransfected with HA-tagged Wee1 and Myc-tagged Cdc14A or Myc-tagged Cdc14A(PD). After 40 h, protein extracts were obtained, and Wee1 was immunoprecipitated with anti-HA antibodies and subjected to immunoblotting with phospho-serine (Cdk substrates) and anti–total Wee1 antibodies. Cell lysates were also analyzed by immunoblotting with the indicated antibodies. The data shown are representative of three different experiments.
Cdc14A dephosphorylates Wee1 at Ser-123 and Ser-139 residues
To understand the mechanism by which Cdc14A regulates Wee1 turnover, we first investigated the phosphorylation sites reversed by Cdc14A phosphatase. It was shown that Cdk1 phosphorylation at Ser-123 is involved in Wee1 degradation at the G2/M transition (Watanabe et al., 2004, 2005). In light of this, we postulated that this residue might be the ideal candidate as target for Cdc14A. Moreover, an as-yet-uncharacterized residue close to Ser-123 and conserved in mammals, Ser-139, also corresponds to the full canonical site for CDK phosphorylation, Ser/Thr-Pro-X-Arg/Lys. These observations prompted us to focus on these two residues as putative Cdc14A targets. In vitro phosphatase assays performed with the corresponding nonphosphorylatable mutant (double S123/139A, 2A mutant) afforded the first evidence concerning the possibility that these two residues might be Cdc14A dephosphorylation sites. Thus Cdc14A removed some Cdk1 phosphorylation in the wild-type amino-terminal domain of Wee1 (Figure 5A, lane 2). However, when both Ser-123 and Ser-139 were modified (2A), although Wee1 was still phosphorylated by Cdk1, treatment with active Cdc14A did not decrease the phosphorylation signal to any significant extent (Figure 5A, lane 5), indicating that Cdc14A dephosphorylates Wee1 at Ser-123 and Ser-139 residues. These data suggested that phosphorylation of Ser-139 could also be involved in the control of Wee1 turnover and that Cdc14A could regulate Wee1 through these two residues.
FIGURE 5:

Cdc14A dephosphorylates Wee1 at Ser-123 and Ser-139. (A) The Ser-123 and Ser-139 residues were mutated to the nonphosphorylatable Ala by site-directed mutagenesis (2A mutant) in the recombinant GST–N-terminal domain of Wee1 (GST-Wee1(ND)). Wild-type and Wee1 2A mutant were expressed and purified from E. coli. GST-Wee1 fusion proteins were first phosphorylated with Cdk1–cyclin B1 complexes in the presence of [γ-32P]ATP and then incubated with GST-Cdc14A phosphatase or its inactive form, GST-Cdc14A(PD). Proteins were resolved by SDS–PAGE gels, and phosphorylation was detected by autoradiography. The same membrane was then reprobed with anti-Wee1 and anti-Cdc14A antibodies to check the amount of Wee1 and Cdc14A proteins loaded. (B, C) HEK293T cells were transfected with HA-tagged Wee1, and after 12 h of transfection, cells were treated with nocodazole (12 h) to accumulate cells in mitosis. Cell lysates were prepared, and Wee1 was immunopurified with anti-HA antibodies and treated with active Cdc14A, the inactive phosphatase form Cdc14A(PD), or λPP, where indicated. Reactions were resolved by SDS–PAGE gels, and phosphorylation at Ser-123 or Ser-139 was analyzed with anti–pSer-123(Wee1) or anti–pSer-139(Wee1) antibodies, respectively. The amount of total Wee1 was analyzed on the same filter by sequentially immunoprobing with anti-Wee1 antibodies. Immunoblotting with anti-Wee1 antibody also demonstrated a shift in mobility after treatment with active Cdc14A or λPP. The presence of Cdc14A was determined with anti-Cdc14A antibodies. Two mutants of Wee1 in which the Ser-123 or -139 was changed to alanine, S123A and S139A, respectively, were immunoprecipitated, as indicated for wild-type HA-Wee1 protein, and used as anti–pSer-123 or anti–pSer-139 Wee1 negative controls. NT, nontreated immunoprecipitates. (D) U-2-OS cells were treated with Cdc14A (A) or control (C) siRNAs for 48 h. Where indicated, MG132 (20 μM) was added to the culture and maintained during the last 6 h. Cellular extracts were prepared and analyzed by immunoblotting with the indicated antibodies. The data shown are representative of at least three independent experiments.
To confirm in vivo phosphorylation of Wee1 at Ser-139, we generated an affinity-purified phosphospecific antibody directed against sequences surrounding Wee1-phosphorylated Ser-139 (pSer-139-Wee1). To facilitate the immunopurification of Wee1, we used HEK293T cells expressing HA-tagged Wee1. Wee1 was immunoprecipitated from these cells and subjected to immunoanalysis with pSer-139-Wee1 antibody. As shown in Figure 5B, HA-tagged Wee1 immunopurified from enriched mitotic HEK293T cells was phosphorylated at Ser-139 and dephosphorylated by active Cdc14A. Moreover, Cdc14A also dephosphorylated pSer-123-Wee1 in vitro, as detected by commercially available antibodies (Figure 5C). Then, to confirm in vivo Cdc14A dephosphorylation of Wee1, we examined whether depletion of Cdc14A would cause hyperphosphorylation of Wee1 at Ser-123 and Ser-139. Because hyperphosphorylation of Wee1 drives it to its degradation, making technically difficult the detection of these phosphoserines before degradation, we tested phosphorylation of Wee1 in U-2-OS cells depleted for Cdc14A and treated with the proteasome inhibitor MG132. As shown in Figure 5D, Wee1 kinase from cells treated with Cdc14A siRNAs and MG132 had increased levels of both Ser-123 and Ser-139 phosphorylation as compared with control cells.
Wee1 Ser-139 is a newly identified Cdk phosphoresidue with a role in the regulation of its turnover
The phosphorylation of Wee1 at Ser-139 was also checked by studying how the change of this residue to the nonphosphorylatable amino acid alanine (S139A mutant) affected the total Wee1 phosphorylation state in the cell. In parallel, we also tested Wee1 phosphorylation when preventing phosphorylation of the already characterized Ser-123 residue (S123A mutant). We engineered U-2-OS cells that stably expressed retrovirally transduced, HA-tagged Wee1 wild type or the different alanine mutants at a moderate level. As shown in Figure 6A, both the S123A and S139A mutants already displayed a faster electrophoretic mobility shift than wild-type Wee1 during interphase. Moreover, we observed that the S139A mutant failed to undergo mitotic hyperphosphorylation to an even greater extent than the S123A mutant (Figure 6A). Accordingly, when the two residues were mutated, the gel mobility of Wee1 was even faster (Figure 6A). These results show that the phosphorylation of Ser-139 has a clear effect on the total Wee1 phosphorylation state, indicating that Ser-139 is phosphorylated in vivo. Moreover, and as expected, the treatment with the specific CDK inhibitor purvalanol abolished this phosphorylation, indicating that Ser-139 is a CDK phosphorylation site (Figure 6B). We next analyzed the phosphorylation of Ser-139 at different phases during the cell cycle. Cells were synchronized at mitosis, G1, or G1/S phases and processed for the immunoprecipitation of HA-Wee1 and the detection of Ser-139 phosphorylation. We found that although some level of Ser-139 phosphorylation was already detected at G1/S, it was higher in mitosis (Figure 6C). As expected, at the G1 phase, when Wee1 was still low in abundance and appeared hypophosphorylated, no phosphorylation of Ser-139 was detected (Figure 6C). These results indicate that Wee1 is phosphorylated on Ser-139 as of early S phase, probably mediated by Cdk2, and that Cdk1 might be responsible for this modification at the G2/M transition.
FIGURE 6:

Human Wee1 is phosphorylated by Cdk1–cyclin B1 at Ser-139. (A) U-2-OS cells stably expressing retrovirally transduced, HA-tagged Wee1 wild type (wt) or the different phosphorylation Wee1 mutants at a moderate level were treated with nocodazole for 12 h to obtain mitotic cells. Rounded mitotic cells were collected, and the cells that remained attached were also harvested and considered as interphase cells. Cellular extracts were obtained and analyzed by immunoblotting with the indicated antibodies. (B) HEK293T cells transiently expressing HA-tagged Wee1 were treated with the vehicle dimethyl sulfoxide or the CDK inhibitor purvalanol A for 6 h. Wee1 was immunoprecipitated with anti-HA antibody and analyzed with both specific p-Ser-139(Wee1) and anti-Wee1 antibodies. Cell lysates were also analyzed by immunoblotting with the indicated antibodies. (C) U-2-OS cells stably expressing retrovirally transduced, HA-tagged Wee1 were synchronized at the indicated phases of the cell cycle. Cells were arrested in mitosis by nocodazole treatment (12 h). Mitotic cells were collected (mitosis) or released from the arrest and harvested after 4 h of growth (G1). Cells that remained attached to the plates after nocodazole treatment were harvested and considered as interphase cells. For G1/S-synchronized cells, the cultures were subjected to a double thymidine treatment. Synchronization was checked by fluorescence-activated cell sorting (right) and immunoblot analyses. HA-Wee1 was immunoprecipitated with anti-HA antibodies and subjected to immunoblot with anti–pSer-139(Wee1) and anti-Wee1 antibodies. These results are representative of three independent experiments.
To understand the biological significance of Ser-139 phosphorylation, we first tested whether the change of Ser-139 to the nonphosphorylatable residue alanine affected the function of Wee1. To achieve this, we assayed its effect on Cdk1 Tyr15 phosphorylation in transfected cells. As shown in Supplemental Figure S4, wild-type Wee1 increased Cdk1 Tyr15 phosphorylation as compared with empty vector or the kinase-dead Wee1 form. Moreover, the S139A mutant was as potent as wild-type Wee1, and the mutants S123A and 2A also showed similar kinase activities. Although we cannot exclude that the endogenous Wee1 protein cooperates with the ectopically expressed Wee1 versions, these results suggest that S139A does not affect the overall structure or function of Wee1 and that phosphorylation of this residue does not regulate Wee1 kinase activity.
Because human Wee1 is negatively regulated at the G2/M transition by phosphorylation-dependent degradation (Watanabe et al., 2004, 2005), we analyzed the possible involvement of Ser-139 in Wee1 stability. First, we checked the turnover of Ser-139 Wee1 mutants in parallel with wild-type and S123A versions, the latter as an already described stabilizing mutation (Watanabe et al., 2005), by using the inhibitor of protein synthesis cycloheximide. As shown in Figure 7A, both the S123A and S139A mutants were more stable than wild-type Wee1. Moreover, the turnover of 2A mutant was significantly slower than that of wild type, although this double mutation did not fully block Wee1 degradation. Furthermore, the change of Ser-139 to Asp (S139D), an amino acid that mimics constitutive phosphorylation, resulted in a more labile protein as compared with wild-type Wee1 (Figure 7A). These data suggest that the phosphorylation of Ser-139 is required for proper Wee1 turnover and that additional phosphorylation sites (other than Ser-123 and Ser-139) are involved.
FIGURE 7:

(A) Phosphorylation of Wee1 at Ser-139 is required for normal Wee1 turnover. HEK293T cells were transfected with HA-tagged, wild-type Wee1 (wt) or the different HA-tagged Wee1 mutants (S123A, S139A, S139D, and S123/139A or 2A). At 24 h posttransfection, cells were treated with cycloheximide to inhibit protein synthesis and then harvested at the indicated time points to check HA-Wee1 protein levels by immunoblotting. HA-Wee1 bands were quantified and normalized with respect to actin. HA-Wee1 protein levels from 0 h were considered as 1. The data are representative of three independent experiments. (B) Cdc14A dephosphorylates Wee1 at Ser-123 and Ser-139 to regulate Wee1 stability. U-2-OS cells stably expressing retrovirally transduced, HA-tagged Wee1 wt or the double 2A mutant were treated with control or Cdc14A siRNAs for 48 or 72 h. Cellular extracts were obtained and analyzed by immunoblotting against the indicated proteins. These blots are representative of three independent experiments. (C) Recombinant HA-Wee1 proteins (wt, S123A, S139A, or 2A mutants) were expressed in HEK293T cells, immunoprecipitated with anti-HA antibodies, and analyzed by immunoblotting with the indicated antibodies. This experiment was performed twice.
Cdc14A regulates Wee1 stability through dephosphorylation on Ser-123 and Ser-139
To finally corroborate that the mechanism by which Cdc14A regulates Wee1 stability was indeed the dephosphorylation of Ser-123 and Ser-139 residues, we tested the effect of Cdc14A deficiency in cells expressing the Wee1 2A mutant, whose turnover cannot be regulated by these two residues. U-2-OS cells stably expressing retrovirally transduced, HA-tagged Wee1 wild-type or 2A mutant at a moderate level were transfected with control or Cdc14A siRNAs and analyzed by immunoblot at 48 and 72 h posttransfection. As observed in Figure 7B, the levels of the Wee1 2A mutant were not significantly reduced after treatment with Cdc14A siRNAs. Taken together, these data suggest that Cdc14A phosphatase regulates Wee1 stability through Ser-123 and Ser-139 dephosphorylation.
Because it was shown that at the G2/M transition Wee1 degradation is mediated by Cdk1-primed Plk1 phosphorylation (Watanabe et al., 2005), we then investigated whether the phosphorylation of Ser-139 might be involved in the Wee1–Plk1 interaction. As expected, Plk1 was efficiently coimmunoprecipitated with wild-type Wee1 (Figure 7C). However, this molecular interaction was strongly reduced with both S123A (as already reported by Watanabe et al., 2004) and S139A mutants and was not detected with the 2A mutant (Figure 7C). These results suggested that both Ser-123 and Ser-139 residues must be phosphorylated to allow efficient interaction between Wee1 and Plk1 kinases.
DISCUSSION
In vertebrate cells, a role for Cdc14 phosphatases in the control of Cdk1 activity has been reported. This function is carried out, at least in part, through the regulation of Cdc25 phosphatase activity (Krasinska et al., 2007; Vázquez-Novelle et al., 2010; Sacristán et al., 2011; Tumurbaatar et al., 2011). The activation of Cdk1–cyclin B1 complexes is controlled by the opposing activities of Wee1/Myt1 kinases and Cdc25 phosphatases, which regulate the Cdk1 Thr-14 and Tyr-15 phosphorylation state. When active, these kinases and phosphatases are in turn inhibited or positively regulated, respectively, by direct Cdk1 phosphorylation, thereby further amplifying Cdk1 activation. Human Cdc14A interferes with the positive Cdc25-mediated feedback loop that controls the activation of Cdk1–cyclin B1 to promote entry into mitosis (Vázquez-Novelle et al., 2010).
In mammalian cells, the phosphorylation of Wee1 by Cdk1 promotes its degradation at the G2/M transition (Watanabe et al., 2004). By using siRNA-mediated treatment, here we show that Cdc14A plays a role in the regulation of Wee1 protein levels. Cdc14A-depleted cells displayed reduced levels of Wee1 when growing asynchronously and also when the cells were synchronized at late G2 phase. The decrease in the level of Wee1 protein was a result not of altering Wee1 transcription, but instead of stabilizing the protein. We also observed that the ectopic expression of Cdc14A during G2 resulted in the accumulation of dephosphorylated Wee1. These results suggest that Cdc14A modulates entry into mitosis by counteracting phosphorylation-dependent Wee1 degradation.
The Cdk-dependent phosphorylation of Wee1 at Ser-123 allows binding with β-TrCP E3 ubiquitin ligase (Watanabe et al., 2004, 2005). Moreover, phospho–Ser-123 promotes additional Wee1 phosphorylation at Ser-121 and Ser-53 by CK2 and Plk1 kinases, respectively, which creates another β-TrCP binding site responsible for the ubiquitination and proteasome-mediated degradation of Wee1 at the G2/M transition (Watanabe et al., 2004, 2005). We show here that Cdc14A dephosphorylates Wee1 at Ser-123, suggesting that Cdc14A deficiency may enhance phosphorylation-dependent Wee1 degradation. We further show that the instability of Wee1 in Cdc14A-depleted cells also reflects an additional site-specific dephosphorylation, at Ser-139, which fulfills the strict requirement for a CDK consensus site (Nigg, 1995). The phosphorylation of Wee1 at Ser-139 was already detectable at the G1/S transition and was higher in mitosis, when Cdk1–cyclin B1 complexes become active. Our results clearly suggest that Cdc14A regulates Wee1 stability through the dephosphorylation of both the Ser-123 and Ser-139 residues.
The newly identified Cdk-mediated Wee1 phosphorylation site, Ser-139, appears to be involved in its degradation at entry into mitosis. Thus mutation of this residue to alanine confers marked stability to the protein, and the protein turnover of the corresponding mutant mimicking a constitutive phosphorylation is slightly accelerated when compared with the wild-type protein. Moreover, although the single mutation of the previously characterized Ser-123 interferes with the interaction of Wee1 with Plk1 kinase (Watanabe et al., 2005, and the present work), this interaction was also altered in a S139A mutant, with the concomitant mutation of Ser-123 and Ser-139 completely disrupting the Wee1–Plk1 interaction. This key observation suggests that Cdk1–cyclin B1 might phosphorylate these two nearby residues to prime Plk1 phosphorylation and the consequent protein degradation and that the lack of modification in any of them reduces binding efficiency with Plk1 kinase. The Ser-139 is not, however, within a known consensus motif for a Plk1-docking site. It could be that phosphorylation of those two proximal residues (Ser-123, Ser-139) favors a particular protein conformation, which, in turn, facilitates pSer-123–mediated Plk1 interaction and the consequent recognition by the ubiquitin ligase SCF (Skp1/cullin/F-box)-β-TrCP. As mentioned, although the phosphorylation of Ser-139 was higher in mitosis, it was also observed during S and G2 phases. Thus Cdk2 might also be the kinase responsible for this phosphorylation, which together with phosphorylation of Ser-123 (Watanabe et al., 2005) may contribute to the basal rate of Wee1 turnover during interphase.
We also show that the double 2A Wee1 mutant, although more stable than wild-type Wee1, is still degraded in mitosis, suggesting that additional mechanisms are involved in the regulation of human Wee1 stability at the G2/M transition. Different regions of Wee1, among which the activation domain is included, are also involved in the control of its degradation (Owens et al., 2010). Moreover, the phosphorylation of Ser-472, a potential CDK site in this domain, also seems to affect the rate of Wee1 turnover (Owens et al., 2010). Taken together, these data indicate that Wee1 degradation is mediated by a precise and complex pattern of phosphorylations in which different kinases are involved. The Wee1 hyperphosphorylated state is reached at the onset of mitosis, making the protein labile at this point of the cell cycle. In addition, it was suggested that a hypophosphorylated form of Wee1 could also render the protein more unstable (Watanabe et al., 1995). Wee1 kinase is also regulated by changes in its subcellular localization in human cells. Thus the phosphorylation of Wee1 at Ser642 by Akt accelerates the cytoplasmic localization of Wee1 at the G2/M transition (Katayama et al., 2005). The nuclear export of Wee1 is also mediated by Cdk2–cyclin A phosphorylation at Thr-239 (Li et al., 2010). The Wee1 nuclear export does not seem to be essential, however, for entry into mitosis (Li et al., 2010). Moreover, it is possible that different posttranslational modifications, other than reversible phosphorylation events, also regulate Wee1 stability in human cells.
Our results suggest that human Cdc14A constitutes a key element in the complex network for Wee1 regulation at the entry into mitosis. By promoting Wee1 stability, Cdc14A participates in maintaining the interphase state when cells are not ready for nuclear division (see model in Figure 8). Thus siRNA-mediated down-regulation of Cdc14A accelerates entry into mitosis (Vázquez-Novelle et al., 2010). However, this phenotype has not been observed in Cdc14-knockout cell lines, which do not show any obvious defect in cell cycle progression (Mocciaro et al., 2010). Among previously discussed possibilities trying to explain discrepancies between knockout and siRNA depletion data (Mocciaro and Schiebel, 2010), we favor the idea that the permanent lack of Cdc14A could be compensated in the long term by other phosphatases in knockout cells. This is based on the fact that knockout cell lines are generated by selection. However, the nature of siRNA-mediated protein depletion likely avoids compensation because of the short window of time for compensatory phosphatases to act, thus revealing some phenotypes undetectable in knockout cell lines.
FIGURE 8:
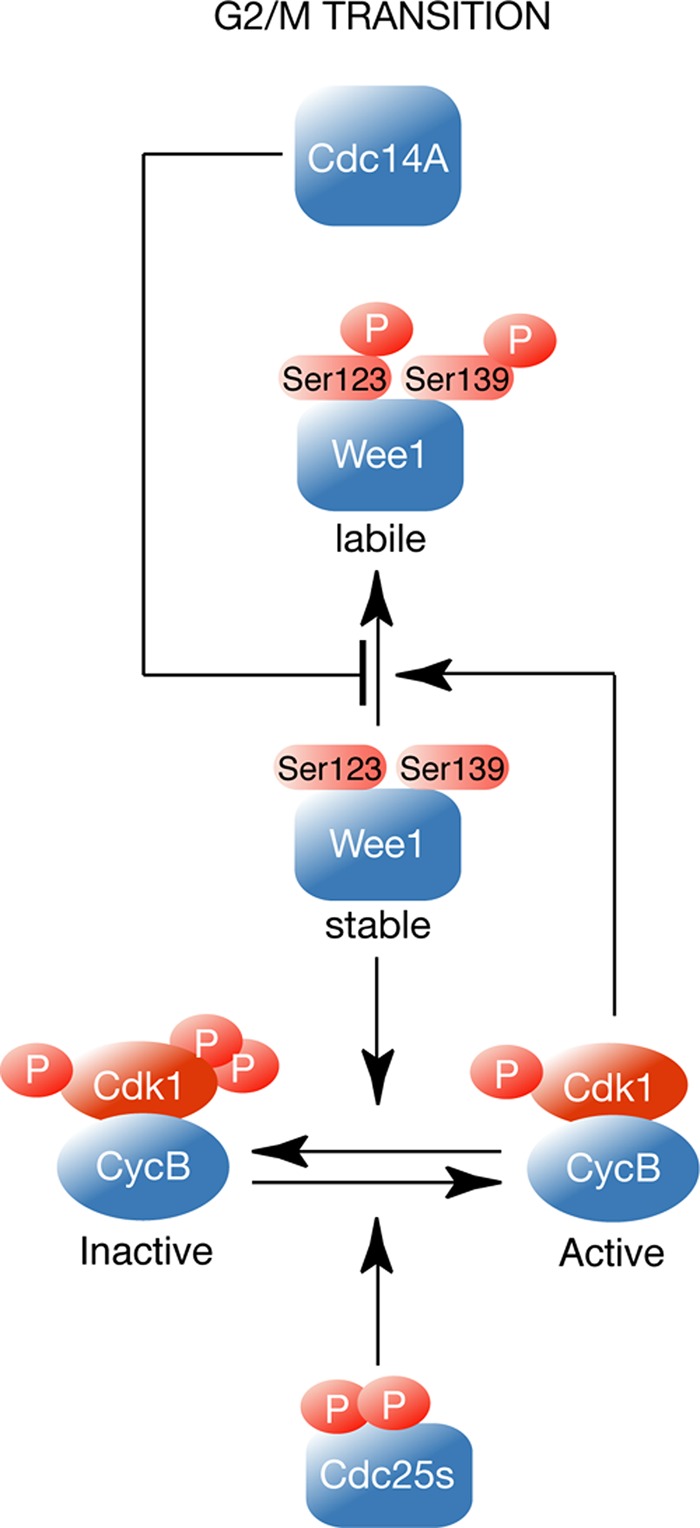
Model of Cdc14A function in the regulation of human Wee1. At the G2/M transition Cdc14A reverses Cdk1-mediated Wee1 phosphorylation at serines 123 and 139 to control Wee1 degradation.
Wee1 is predominantly nuclear, but it has also been found to associate with centrosomes. Cdc14A localizes to centrosomes during interphase, and it spreads throughout the cell during mitosis (Kaiser et al., 2002; Mailand et al., 2002). Thus Cdc14A-mediated Wee1 regulation could take place at the centrosomes during interphase. Alternatively, Cdc14A could act when released from the centrosome at the onset of mitosis. Cdc14B is able to dephosphorylate Wee1; however, in clear contrast with the observed function for Cdc14A, Wee1 protein levels remain unaffected in asynchronous Cdc14B-depleted cells. Given that Cdc14B is maintained in the nucleolus during G2, one possibility is that regulation of Wee1 is specific for the centrosomal Cdc14A isoform and that it is at the centrosome where Cdc14A regulates the Wee1 kinase in a normal cell cycle. It has been shown that after G2 DNA damage, no change in subcellular localization is observed for Cdc14A; however, Cdc14B isoform moves from the nucleolus to the nucleoplasm (Bassermann et al., 2008). In fission yeast, replication stress also induces changes in the subcellular localization of Flp1/Cdc14 from the nucleolus to the nucleus (Díaz-Cuervo and Bueno, 2008). These findings support the possibility that Cdc14B isoform could be involved in the regulation of Wee1 upon genotoxic stress. This hypothesis deserves further investigation.
The regulation of Wee1 has been addressed in numerous studies, from which it has become evident that multiple phosphorylation events, carried out by different kinases, play an important role. Our results indicate that Wee1 is a substrate of Cdc14 phosphatases in human cells and that by reversing specific Cdk1 phosphorylation, the Cdc14A isoform induces Wee1 stability, which in turn directly inhibits Cdk1 activity. Given that Cdc14A also acts on Cdc25 phosphatases (Krasinska et al., 2007; Vázquez-Novelle et al., 2010), taken together, our data suggest that Cdc14A modulates Cdk1 activity by directly controlling the two major CDK regulators, Wee1 and Cdc25, responsible for the negative and positive feedback loops, respectively, established with Cdk1. It also has been suggested that PP2A phosphatase regulates Cdc25 and Wee1 phosphorylation, thus controlling Cdk1 activity during interphase (Mochida et al., 2009; Wicky et al., 2011). Therefore Wee1 could be regulated by different phosphatases, as it is targeted by different kinases responsible for its inhibitory hyperphosphorylation state in mitosis. An attractive hypothesis is that these phosphatases could act on different or the same Wee1 phosphoresidues to integrate diverse signaling inputs; they could act at different subcellular locations or under different cellular conditions. All of these regulatory steps would finally contribute to refine the control of entry into mitosis.
MATERIALS AND METHODS
Cell culture, synchronization, and drug treatments
Derivatives of the U-2-OS cell line conditionally expressing Cdc14A alleles were from the laboratory of J. Lukas (Mailand et al., 2002). Cells were maintained and induced to express the transgenes as described previously (Vázquez-Novelle et al., 2010). HCT116-Cdc14Bflox/flox cells were from P. Jallepalli (Berdougo et al., 2008). U-2-OS and HEK293T cells were cultured in DMEM supplemented with 2 mM glutamine, 100 U/ml penicillin G, 0.1 mg/ml streptomycin, and 10% fetal bovine serum at 37ºC/5% CO2 and used for infection and transient transfection experiments, respectively. HEK293T cells were transfected with the calcium phosphate/HEBS (HEPES-buffered saline solution) method. U-2-OS cells were synchronized in G1/S phase by double thymidine treatment. Briefly, thymidine, 2.5 mM, was added for 24 h, followed by a release period of 12 h and a second treatment with thymidine for a further 24 h. Then the cells were released to fresh culture medium after extensive washing with phosphate-buffered saline (PBS). Where indicated, nocodazole (50 ng/ml) was added to avoid exit from mitosis. Mitotic cells were obtained by shaking off the rounded cells after 12 h of treatment with nocodazole (50 ng/ml). The proteasome inhibitor MG132 (20 μM; Calbiochem, La Jolla, CA) was added to the culture medium for 6 h. For estimation of the protein half-life, the cultures were treated with cycloheximide (25 μg/ml) for the indicated times.
Plasmids, mutagenesis, and virus production
Human Wee1 cDNA was cloned into the pCEFL-HA mammalian expression vector using BamHI and EcoRI restriction enzyme sites. Two truncated constructions corresponding to the amino domain (aa 1–257) and carboxyl domain (aa 258–646) were generated by PCR using the primers 5′-CGCGGATCCATGAGCTTCCTGAGCCGACAG-3′, 5′-CCGGAATTCTCAGTATATAGTAAGGCTG-3′, 5′-CCGGAATTCGGATCCTATTGGAATGATTCCTGTGGTG-3′, and 5′-CCGGAATTCTCACGTTCTCTTTCTACGACGACAC-3′. Kinase-dead (K328R), Ser-123A, Ser-139A, or Ser-123/139A (2A) mutants were generated using site-directed mutagenesis. For bacterial expression in E. coli and the subsequent purification of recombinant GST proteins, wild-type and mutant Wee1 constructs were subcloned into a pGEX-4T-GST vector. GST-Cdc14A or GST-Cdc14A(PD) was cloned into pGEX-KG-GST. For viral infection, wild-type and mutant Wee1 cDNAs were subcloned into pBabe-puro retroviral expression vector. Retroviruses were produced in HEK293T cells and used to infect U-2-OS cells. Infected cells were selected by adding puromycin (0.5 μg/ml) to the culture medium. pBabe-puro-EGFP plasmid was used to control infection efficiency.
Flow cytometry
For cell cycle analysis, cells were fixed in ice-cold 70% ethanol and stained with PBS containing 8 μg/ml propidium iodide and 20 μg/ml RNaseA for 1 h at 37ºC. For phospho–histone H3 positivity, cells were harvested and fixed in ice-cold 70% ethanol. Cells were washed with PBS–0.15% Triton X-100 and subsequently stained with anti–phospho–histone H3 and fluorescein isothiocyanate–conjugated anti-rabbit antibodies. Then they were incubated with 8 mg/ml propidium iodide and 20 mg/ml RNase A solution for 1 h at 37ºC. Stained cells were analyzed using a FACSCalibur device (BD Biosciences, San Diego, CA).
RNA interference
Cdc14A siRNA (5′-GGGACAUUGAUAGCCUGUUAUGUAA-3′) and its corresponding siRNA control (low GC) were from Invitrogen (Carlsbad, CA). The final concentration of the corresponding siRNA duplex in the culture medium was 60–100 nM. Transfection of U-2-OS cells was performed using Lipofectamine RNAiMAX Reagent (Invitrogen) according to the manufacturer's instructions. Four extra silent mutations were introduced into Cdc14A cDNA in the area corresponding to siRNA duplex by PCR mutagenesis. The PCR oligos used are 5′-AGAACAGGGACCCTGATCGCCTGCTATGTAATG-3′ and 5′-CATTACATAGCAGGCGATCAGGGTCCCTGTTCT-3′. The resulting Cdc14A mutant (Cdc14A [R]) was subcloned into pBabe-puro retroviral expression vector. Retrovirus were produced in HEK293T cells and used to infect U-2-OS cells. After 24 h of infection cells were grown in medium containing 0.5 mg/ml of puromycin for 72 h and then transfected with the corresponding siRNAs.
mRNA analyses
RNA was extracted from U-2-OS cells using the RNeasy Kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. cDNA synthesis was performed from 1 μg of RNA using SuperScript First–Strand Kit (Invitrogen). Cdc14A primers were 5′-GTTCCTGAACATCTGTGA-3′ and 5′-GCATGTGTAAACCTGTAG-3′. The 18S rRNA primers, used as internal control, were 5′-CGCCGCTAGAGGTGAAATTC-3′ and 5′-CTTTCGCTCTGGTCCGTCTT-3′. Quantitative PCR analysis was performed according to standard procedures using the iQ5 Multicolor Real-Time PCR Detection System (Bio-Rad, Hercules, CA).
Immunochemical techniques
For Western blotting, cells were harvested and lysed in buffer containing 0.5% NP40, 20 mM Tris-Cl, pH 8.0, 150 mM NaCl, 1 mM dithiothreitol (DTT), 10 mM β-glycerophosphate, 5 mM NaF, and 1 mM Na3VO4 and supplemented with complete protease inhibitor mixture (Roche, Indianapolis, IN). The primary antibodies used in this study were Myc tag (9E10; Sigma-Aldrich, St. Louis, MO), HA tag (12CA5; Boehringer Mannheim, Mannheim, Germany), Wee1 (sc-9037; Santa Cruz Biotechnology, Santa Cruz, CA), phospho–Wee1-Ser-123 (PAB0632; Abnova, Taipei City, Taiwan), Cdk1 (sc-54; Santa Cruz Biotechnology), phospho–Tyr-15-Cdk1 (9111; Cell Signaling, Beverly, MA), β-actin (AC-15; Sigma-Aldrich), phospho–(Ser) CDKs (2324; Cell Signaling), Cdc14A (DCS-291; NeoMarkers, Fremont, CA), cyclin B1 (sc-752; Santa Cruz Biotechnology), Plk1 (33-1700; Zymed, San Francisco, CA), phospho–histone-H3(S10) (06-570; Millipore, Billerica, MA), and Nek9 (kindly provided by J. Roig, Institute for Research in Biomedicine, Barcelona, Spain). Rabbit polyclonal antibody specific for Wee1 phosphorylated at Ser-139 was raised against the Wee1 LGSSFS(p)PVRCG synthetic phosphopeptide. Serum from immunized rabbit was affinity purified on a specific phosphorylated peptide column. For the immunoprecipitation studies, cell lysates (0.5–1 mg of protein) were incubated with the corresponding antibodies (2 μg/mg extract) and protein G Dynabeads (Invitrogen) for 3 h at 4ºC. Immunoprecipitates were collected by centrifugation, washed five times with lysis buffer, and subjected to SDS–PAGE electrophoresis and immunoblot analysis.
Kinase and phosphatase assays
Recombinant GST-Wee1(KD) was phosphorylated by Cdk1–cyclin B1 (New England BioLabs, Ipswich, MA) in the presence of 0.10 μCi of [γ-32P]ATP for 30 min. Samples were then incubated for 30 min at 30°C with GST-Cdc14A or GST-Cdc14A(PD) (100 ng) in phosphatase buffer (20 mM Tris, pH 8.3, 150 mM NaCl, 2 mM EDTA, 0.1% Triton X-100, 5 mM DTT) or with λ phosphatase (New England BioLabs) according to the manufacturer's instructions. Reactions were stopped by the addition of loading buffer and boiled for 5 min at 95ºC. Proteins were resolved by SDS–PAGE and visualized by immunoblotting, Coomassie staining, or autoradiography. Wee1 protein was immunoprecipitated with specific antibodies (anti-Wee1 or anti-HA) and was incubated for 40 min at 30ºC with GST-Cdc14A, GST-Cdc14A(PD), or λ phosphatase. Reactions were stopped by the addition of loading buffer and boiled for 5 min at 95ºC. Dephosphorylation was detected by immunoblotting with anti–total Wee1 antibodies or phosphospecific antibodies.
Supplementary Material
Acknowledgments
We thank E. Díaz and A. Pandiella (Instituto de Biología Molecular y Celular del Cáncer, Salamanca, Spain) for their generous help in generating specific pSer-139(Wee1) antibody; S. Andrés for technical assistance; and the B05–B10 laboratories at the Instituto de Biología Molecular y Celular del Cáncer for helpful discussions. This work was funded by grants from the Spanish Ministry of Science and Innovation (MICINN; BFU2008-04293 and BFU2009-06938). S.O. was supported by a FPU fellowship from the Spanish Ministry of Education, and P.A. was supported by a JAE-Predoc fellowship from the Consejo Superior de Investigaciones Científicas.
Abbreviations used:
- CDK
cyclin-dependent kinase
- GST
glutathione S-transferase
- HA
hemagglutinin
- KD
kinase dead
- λPP
lambda protein phosphatase
- PD
phosphatase dead
- SCF
Skp1/cullin/F-box complex
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-04-0260) on October 10, 2012.
REFERENCES
- Ayad NG, Rankin S, Murakami M, Jebanathirajah J, Gygi S, Kirschner MW. Tome-1, a trigger of mitotic entry, is degraded during G1 via the APC. Cell. 2003;113:101–113. doi: 10.1016/s0092-8674(03)00232-0. [DOI] [PubMed] [Google Scholar]
- Bassermann F, Frescas D, Guardavaccaro D, Busino L, Peschiaroli A, Pagano M. The Cdc14B-Cdh1-Plk1 axis controls the G2 DNA-damage-response checkpoint. Cell. 2008;134:256–267. doi: 10.1016/j.cell.2008.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdougo E, Nachury MV, Jackson PK, Jallepalli PV. The nucleolar phosphatase Cdc14B is dispensable for chromosome segregation and mitotic exit in human cells. Cell Cycle. 2008;7:1184–1190. doi: 10.4161/cc.7.9.5792. [DOI] [PubMed] [Google Scholar]
- Bertran MT, Sdelci S, Regue L, Avruch J, Caelles C, Roig J. Nek9 is a Plk1-activated kinase that controls early centrosome separation through Nek6/7 and Eg5. EMBO J. 2011;30:2634–2647. doi: 10.1038/emboj.2011.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremmer SC, Hall H, Martinez JS, Eissler CL, Hinrichsen TH, Rossie S, Parker LL, Hall MC, Charbonneau H. Cdc14 phosphatases preferentially dephosphorylate a subset of cyclin-dependent kinase (Cdk) sites containing phosphoserine. J Biol Chem. 2011;287:1662–1669. doi: 10.1074/jbc.M111.281105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiesa M, Guillamot M, Bueno MJ, Malumbres M. The Cdc14B phosphatase displays oncogenic activity mediated by the Ras-Mek signaling pathway. Cell Cycle. 2011;10:1607–1617. doi: 10.4161/cc.10.10.15566. [DOI] [PubMed] [Google Scholar]
- Clemente-Blanco A, et al. Cdc14 phosphatase promotes segregation of telomeres through repression of RNA polymerase II transcription. Nat Cell Biol. 2011;13:1450–1456. doi: 10.1038/ncb2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz-Cuervo H, Bueno A. Cds1 controls the release of Cdc14-like phosphatase Flp1 from the nucleolus to drive full activation of the checkpoint response to replication stress in fission yeast. Mol Biol Cell. 2008;19:2488–2499. doi: 10.1091/mbc.E07-08-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteban V, Blanco M, Cueille N, Simanis V, Moreno S, Bueno A. A role for the Cdc14-family phosphatase Flp1p at the end of the cell cycle in controlling the rapid degradation of the mitotic inducer Cdc25p in fission yeast. J Cell Sci. 2004;117:2461–2468. doi: 10.1242/jcs.01107. [DOI] [PubMed] [Google Scholar]
- Esteban V, Sacristán M, Andrés S, Bueno A. The Flp1/Clp1 phosphatase cooperates with HECT-type Pub1/2 protein-ubiquitin ligases in Schizosaccharomyces pombe. Cell Cycle. 2008;7:1269–1276. doi: 10.4161/cc.7.9.5947. [DOI] [PubMed] [Google Scholar]
- Guillamot M, Manchado E, Chiesa M, Gómez-López G, Pisano DG, Sacristán MP, Malumbres M. Cdc14b regulates mammalian RNA polymerase II and represses cell cycle transcription. Sci Rep. 2011;1:189. doi: 10.1038/srep00189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman M, Lindon C, Nigg EA, Pines J. Active cyclin B1-Cdk1 first appears on centrosomes in prophase. Nat Cell Biol. 2003;5:143–148. doi: 10.1038/ncb918. [DOI] [PubMed] [Google Scholar]
- Kaiser BK, Zimmerman ZA, Charbonneau H, Jackson PK. Disruption of centrosome structure, chromosome segregation, and cytokinesis by misexpression of human Cdc14A phosphatase. Mol Biol Cell. 2002;13:2289–2300. doi: 10.1091/mbc.01-11-0535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama K, Fujita N, Tsuruo T. Akt/protein kinase B-dependent phosphorylation and inactivation of WEE1Hu promote cell cycle progression at G2/M transition. Mol Cell Biol. 2005;25:5725–5737. doi: 10.1128/MCB.25.13.5725-5737.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasinska L, de Bettignies G, Fisher D, Abrieu A, Fesquet D, Morin N. Regulation of multiple cell cycle events by Cdc14 homologues in vertebrates. Exp Cell Res. 2007;313:1225–1239. doi: 10.1016/j.yexcr.2006.12.022. [DOI] [PubMed] [Google Scholar]
- Li C, Andrake M, Dunbrack R, Enders GH. A bifunctional regulatory element in human somatic Wee1 mediates cyclin A/Cdk2 binding and Crm1-dependent nuclear export. Mol Cell Biol. 2010;30:116–130. doi: 10.1128/MCB.01876-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Ernsting BR, Wishart MJ, Lohse DL, Dixon JE. A family of putative tumor suppressors is structurally and functionally conserved in humans and yeast. J Biol Chem. 1997;272:29403–29406. doi: 10.1074/jbc.272.47.29403. [DOI] [PubMed] [Google Scholar]
- Mailand N, Lukas C, Kaiser BK, Jackson PK, Bartek J, Lukas J. Deregulated human Cdc14A phosphatase disrupts centrosome separation and chromosome segregation. Nat Cell Biol. 2002;4:317–322. doi: 10.1038/ncb777. [DOI] [PubMed] [Google Scholar]
- McGowan CH, Russell P. Cell cycle regulation of human WEE1. EMBO J. 1995;14:2166–2175. doi: 10.1002/j.1460-2075.1995.tb07210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mocciaro A, Berdougo E, Zeng K, Black E, Vagnarelli P, Earnshaw W, Gillespie D, Jallepalli P, Schiebel E. Vertebrate cells genetically deficient for Cdc14A or Cdc14B retain DNA damage checkpoint proficiency but are impaired in DNA repair. J Cell Biol. 2010;189:631–639. doi: 10.1083/jcb.200910057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mocciaro A, Schiebel E. Cdc14: a highly conserved family of phosphatases with non-conserved functions? J Cell Sci. 2010;123:2867–2876. doi: 10.1242/jcs.074815. [DOI] [PubMed] [Google Scholar]
- Mochida S, Ikeo S, Gannon J, Hunt T. Regulated activity of PP2A-B55 delta is crucial for controlling entry into and exit from mitosis in Xenopus egg extracts. EMBO J. 2009;28:2777–2785. doi: 10.1038/emboj.2009.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller PR, Coleman TR, Dunphy WG. Cell cycle regulation of a Xenopus Wee1-like kinase. Mol Biol Cell. 1995a;6:119–134. doi: 10.1091/mbc.6.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller PR, Coleman TR, Kumagai A, Dunphy WG. Myt1: a membrane-associated inhibitory kinase that phosphorylates Cdc2 on both threonine-14 and tyrosine-15. Science. 1995b;270:86–90. doi: 10.1126/science.270.5233.86. [DOI] [PubMed] [Google Scholar]
- Nigg EA. Cyclin-dependent protein kinases: key regulators of the eukaryotic cell cycle. Bioessays. 1995;17:471–480. doi: 10.1002/bies.950170603. [DOI] [PubMed] [Google Scholar]
- Nilsson I, Hoffmann I. Cell cycle regulation by the Cdc25 phosphatase family. Prog Cell Cycle Res. 2000;4:107–114. doi: 10.1007/978-1-4615-4253-7_10. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Sagata N. Mechanism for inactivation of the mitotic inhibitory kinase Wee1 at M phase. Proc Natl Acad Sci USA. 2007;104:3753–3758. doi: 10.1073/pnas.0607357104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens L, Simanski S, Squire C, Smith A, Cartzendafner J, Cavett V, Caldwell Busby J, Sato T, Ayad NG. Activation domain-dependent degradation of somatic Wee1 kinase. J Biol Chem. 2010;285:6761–6769. doi: 10.1074/jbc.M109.093237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker LL, Walter SA, Young PG, Piwnica-Worms H. Phosphorylation and inactivation of the mitotic inhibitor Wee1 by the nim1/cdr1 kinase. Nature. 1993;363:736–738. doi: 10.1038/363736a0. [DOI] [PubMed] [Google Scholar]
- Perry JA, Kornbluth S. Cdc25 and Wee1: analogous opposites? Cell Div. 2007;2:12. doi: 10.1186/1747-1028-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queralt E, Uhlmann F. Cdk-counteracting phosphatases unlock mitotic exit. Curr Opin Cell Biol. 2008;20:661–668. doi: 10.1016/j.ceb.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacristán MP, Ovejero S, Bueno A. Human Cdc14A becomes a cell cycle gene in controlling Cdk1 activity at the G/M transition. Cell Cycle. 2011;10:387–391. doi: 10.4161/cc.10.3.14643. [DOI] [PubMed] [Google Scholar]
- Smith A, Simanski S, Fallahi M, Ayad NG. Redundant ubiquitin ligase activities regulate wee1 degradation and mitotic entry. Cell Cycle. 2007;6:2795–2799. doi: 10.4161/cc.6.22.4919. [DOI] [PubMed] [Google Scholar]
- Stegmeier F, Amon A. Closing mitosis: the functions of the Cdc14 phosphatase and its regulation. Annu Rev Genet. 2004;38:203–232. doi: 10.1146/annurev.genet.38.072902.093051. [DOI] [PubMed] [Google Scholar]
- Tumurbaatar I, Cizmecioglu O, Hoffmann I, Grummt I, Voit R. Human Cdc14B promotes progression through mitosis by dephosphorylating Cdc25 and regulating Cdk1/cyclin B activity. PLoS One. 2011;6:e14711. doi: 10.1371/journal.pone.0014711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vázquez-Novelle MD, Esteban V, Bueno A, Sacristán MP. Functional homology among human and fission yeast Cdc14 phosphatases. J Biol Chem. 2005;280:29144–29150. doi: 10.1074/jbc.M413328200. [DOI] [PubMed] [Google Scholar]
- Vázquez-Novelle MD, Mailand N, Ovejero S, Bueno A, Sacristán MP. Human Cdc14A phosphatase modulates the G2/M transition through Cdc25A and Cdc25B. J Biol Chem. 2010;285:40544–40553. doi: 10.1074/jbc.M110.133009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N, Arai H, Iwasaki J, Shiina M, Ogata K, Hunter T, Osada H. Cyclin-dependent kinase (CDK) phosphorylation destabilizes somatic Wee1 via multiple pathways. Proc Natl Acad Sci USA. 2005;102:11663–11668. doi: 10.1073/pnas.0500410102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N, Arai H, Nishihara Y, Taniguchi M, Watanabe N, Hunter T, Osada H. M-phase kinases induce phospho-dependent ubiquitination of somatic Wee1 by SCFbeta-TrCP. Proc Natl Acad Sci USA. 2004;101:4419–4424. doi: 10.1073/pnas.0307700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N, Broome M, Hunter T. Regulation of the human WEE1Hu CDK tyrosine 15-kinase during the cell cycle. EMBO J. 1995;14:1878–1891. doi: 10.1002/j.1460-2075.1995.tb07180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicky S, Tjandra H, Schieltz D, Yates J 3rd, Kellogg DR. The Zds proteins control entry into mitosis and target protein phosphatase 2A to the Cdc25 phosphatase. Mol Biol Cell. 2011;22:20–32. doi: 10.1091/mbc.E10-06-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe BA, Gould KL. Fission yeast Clp1p phosphatase affects G(2)/M transition and mitotic exit through Cdc25p inactivation. EMBO J. 2004;23:919–929. doi: 10.1038/sj.emboj.7600103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Russell P. Nim1 kinase promotes mitosis by inactivating Wee1 tyrosine kinase. Nature. 1993;363:738–741. doi: 10.1038/363738a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.