

Abstract
The protein complex of toll-like receptor 1 and 2 (TLR1/2) is an important regulator of innate immunity, and therefore provides an attractive target for the treatment of various immune disordres. Here we report a novel compound (CU-CPT22) that can compete with the synthetic triacylated lipoprotein (Pam3CSK4) binding to TLR1/2 with high inhibitory activity and specificity. Repression of downstream signaling from TNF-α and IL-1β has also been observed.
Keywords: drug discovery, toll-like receptors, innate immunity, inhibitors
Toll-like receptors (TLRs) are type I transmembrane proteins that recognize pathogen-derived macromolecules, playing a key role in the innate immune system.[1–3] These pathogen-derived macromolecules are broadly shared by pathogens but distinguishable from host molecules, collectively referred to as pathogen-associated molecular patterns (PAMPs).[1,4] In human, 10 TLRs respond to a variety of PAMPs, including lipopolysaccharide (TLR4), lipopeptides (TLR2 associated with TLR1 or TLR6), bacterial flagellin (TLR5), viral dsRNA (TLR3), viral or bacterial ssRNA (TLRs 7 and 8), and CpG-rich unmethylated DNA (TLR9), among others.[5–7]
TLR dimerization leads to the activation of nuclear factor-κB (NF-κB) and interferon-regulatory factors (IRFs), and these transcription factors in turn induce the production of proinflammatory cytokines and type I interferons (IFNs), respectively.[1,8] Finally, the key output from TLR activation are inflammatory cytokines such as TNF and IL-1β, which have proven to be directly relevant to inflammatory diseases.[9] TLR2, signaling as a heterodimer with either TLR1 or TLR6, recognizes a wide range of ligands, many of which are from Gram-positive bacteria.[10] The molecular recognition by TLR2 was largely explained when the crystal structure of the TLR1/TLR2 heterodimer in complex with its specific lipoprotein ligand, Pam3CSK4, was solved.[11] In this structure (Supplementary Figure S1a), the extracellular domains of TLR1 and TLR2 form an “M”-shaped heterodimer, with the two N-termini extending outward in opposite directions. The lipid chains of Pam3CSK4 bridge the two TLRs, contributing to the formation of the heterodimer. Two of the three lipid chains of Pam3CSK4 interact with a hydrophobic pocket in TLR2, and the amide-bound lipid chain lies in a hydrophobic channel within TLR1. The ligand-bound complex of TLR1 and TLR2 is stabilized by protein-protein contacts near the ligand-binding pocket.[9,11]
Previous reports have demonstrated that the cytokine response to human cytomegalovirus (CMV), lymphocytic choriomeningitis virus (LCMV), and herpes simplex virus 1 (HSV-1) is regulated by TLR1/2.[12,13] TLR1/2 antagonists have been suggested to have beneficial effects in both chronic and acute inflammatory diseases ranging from acne[12] to sepsis,[13] and can also attenuate pulmonary metastases of tumor.[14] However, a significant bottleneck in the field can be attributed to the lack of efficient and specific probes for the TLR1/2 signaling pathway. Even though there is limited success of TLR2 regulators reported in literature,[15] low molecular weight inhibitors with high potency and specificity against TLR1/2 have not been reported as to the best of our knowledge.
Novel inhibitors of TLR1/2 were obtained by cell-based screening of the 1,363-compound NCI-2 Diversity small molecule library. Screening was performed in a 96-well plate format using our previously established high-throughput nitric oxide (NO) assay in RAW 264.7 macrophage cells (For detailed screening method, see Supplementary Figure S2).[16,17] Synthetic triacylated lipoprotein Pam3CSK4 was employed to selectively activate TLR1/2 signaling, resulting in the expression of inducible nitric oxide synthase (iNOS) and the production of NO in RAW 264.7 macrophage cells.[16] We monitored the NO level as an indicator of Pam3CSK4-induced TLR1/2 activation to determine the potency of inhibitors.
We identified nine initial hits (Supplementary Scheme 1) that inhibited TLR1/2 activation by at least 70% at 3.0 µM with no significant cytotoxicity (Supplementary Figure S3). The most potent compound was NCI35676 (Table 1, a natural product from nutgalls and oak barks named purpurogallin) with an IC50 of 2.45 ± 0.25 µM (Supplementary Figure S4). NCI35676 has been reported to possess anti-oxidant[18] and anti-cancer[19,20] properties, as well as modulation of inflammatory response activity.[21] Nonetheless, no previous work has yet been reported on the molecular target of purpurogallin or its derivatives. Further, TLR specificity evaluation was performed, indicating that out of nine initial hits, only NCI35676 specifically inhibited TLR1/2 signaling, but not other homologous TLRs (Supplementary Figure S5).
Table 1.
Structure-activity relationship analysis of the benzotropolone analogs in inhibition of NO production in RAW 264.7 cells.
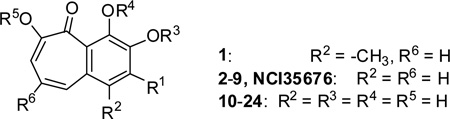 | ||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R3 | R4 | R5 | IC50 (µM)[a] | |
| NCI35676 | OH | H | H | H | 2.45± 0.25 | |
| 1 | H | H | H | H | 3.13± 0.11 | |
| 2 | H | H | H | H | 2.25± 0.31 | |
| 3 | F | H | H | H | 22.7± 1.4 | |
| 4 | OCH3 | H | H | H | 4.83± 0.25 | |
| 5 | OCH3 | CH3 | H | CH3 | 11.7± 1.1 | |
| 6 | OCH3 | CH3 | CH3 | CH3 | 39.9± 0.9 | |
| 7 | OCOCH3 | H | H | H | 4.83± 0.25 | |
| 8 | OCOCH3 | COCH3 | H | COCH3 | 1.42± 0.21 | |
| 9 | OCOCH3 | COCH3 | COCH3 | COCH3 | 2.35± 0.41 | |
| R1 | R6 | |||||
| 10 | OCH3 | COOH | 21.5± 0.4 | |||
| 11 | OCH3 | COOCH3 | 3.11± 0.75 | |||
| 12 | H | COOH | 16.5± 0.8 | |||
| 13 | H | COOCH3 | 9.01± 0.50 | |||
| 14 | OCH3 | COOCH2CH3 | 2.83± 0.44 | |||
| 15 | OCH3 | COOCH(CH3)2 | 2.47± 0.71 | |||
| 16 | OCH3 | COO(CH2)3CH3 | 2.83± 0.44 | |||
| 17 | OCH3 | COO(CH2)7CH3 | 0.72± 0.14 | |||
| 18 | OCH3 | COO(CH2)9CH3 | 1.01± 0.10 | |||
| 19 | OCH3 | COO(CH2)13CH3 | 3.24± 0.13 | |||
| 20 | OCH3 | CONH(CH2)3CH3 | 1.26± 0.31 | |||
| 21 | OCH3 | CONH(CH2)5CH3 | 1.36± 0.21 | |||
| CU-CPT22 | OCH3 | COO(CH2)5CH3 | 0.58± 0.09 | |||
| 23 | OCH3 | CH2OH | 4.11± 0.74 | |||
| 24 | OCH3 | CONH(o-toluene) | 1.36± 0.21 | |||
| 25 | 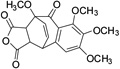 |
74.6± 2.9 | ||||
| 26 | 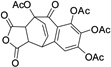 |
14.8± 0.5 | ||||
IC50 and corresponding SD values are determined from at least three independent repeats.
Base on the promising preliminary results, we attempted to optimize NCI35676 in order to improve its inhibitory potency and selectivity. We designed a series of NCI35676 analogs to explore the structure-activity relationship (SAR) around the benzotropolone core scaffold. A one-pot synthesis with sequential additions of (a) phosphate-citrate buffer (pH 5), (b) horseradish peroxidase enzyme and (c) 3% H2O2 produced the bicyclic scaffold (Scheme 1, Supplementary Scheme 2).[22] This method provides a concise, general synthetic route that can afford the benzotropolone derivatives with an overall yield of 15~60%. Compound 2 was selected as a representative for 2D NMR analysis for further characterizations (1H, 13C, HSQC, HMBC and COSY included in Supplementary Figure S6).
Scheme 1.

Representative synthesis for CU-CPT22: a) phosphate-citrate buffer (pH 5), 0.2 M Na2HPO4: 0.1 M citrate = 1:1; b) horseradish peroxidase enzyme catalyst; c) four aliquots of 3% H2O2, 42% overall yield.
Further screening of 26 structural analogs yielded additional hits, the most potent being CU-CPT22, which showed an IC50 of 0.58 ± 0.09 µM (Figure 1a). The improved IC50 of the top inhibitor, CU-CPT22, appears to be due to the addition of a six carbon aliphatic chain at the R6 position, which likely allows for hydrophobic contacts to the surface residues of the TLR1/2 complex (Figure 1b).
Figure 1.

a) Dose-dependent inhibition of NO production in RAW 264.7 macrophage cells by CU-CPT22 and the negative control compound 6. b) Binding site prediction of CU-CPT22 (show in the stick representation) to TLR1/2 performed by Glide 5.6 program[23]. The six-membered carbon chain fits well into the hydrophobic channel of hTLR1, having key hydrophobic interactions with Val311, Phe312, Pro315 and Val339.
The SAR for this series indicated that the total number of hydroxyl groups was critical. Methylation of one hydroxyl group at the R1 position had modest influence to its activity (4), while methylation of all four hydroxyl groups resulted in significant decrease of inhibition (6) (Table 1). Introduction of an F at the R1 position (3) decreased the activity about 20 folds, suggesting an electron-withdrawing group was not favored here. We also found that the seven-membered ring configuration in benzotropolone scaffold plays an important role for inhibitory activity as determined by the Diels-Alder [4+2]-cycloaddition products (6 vs 25, 9 vs 26).
We found that methylation of the R1 (–OH) eliminated the by-product formation, but still retained the activity of NCI35676 (Supplementary Scheme 2). Therefore, in the following SAR studies, the methoxy group was fixed at the R1 position. Meanwhile, the seven-membered ring in the benzotropolone scaffold was kept and substituent groups were introduced at the R6 position. The addition of a carboxyl group at the R6 position decreased the activity by approximately 5 folds (4 vs 10), while esterificaiton of this carboxyl group (11) returned the activity to the NCI35676 level, indicating that R6 may be critical for the inhibitory activity.
By introducing various aliphatic chains at the R6 position, we found that CU-CPT22 with a six carbon chain possessed the highest inhibitory activity for TLR1/2 (Table 1). This increased potency was likely caused by a good fit of the six membered carbon chain into the substrate tunnel of the TLR1 hydrophobic region (Figure 1b). When we replaced the ester with the amide group at R6 position in CU-CPT22, the activity slightly decreased (21). Reducing the carboxyl group to an alcohol (23) or introducing a large substitute at the R6 position (24) provided no significant change in activity. In summary, we identified compound CU-CPT22 as the lead structure, which shows dose-dependent inhibitory effects blocking Pam3CSK4-induced TLR1/2 activation with an IC50 of 0.58 ± 0.09 µM (Figure 1a).
Biophysical tests were carried out for CU-CPT22, along with the negative control compound 6, to demonstrate that CU-CPT22 directly binds to TLR1/2. The TLR2 protein was expressed in the baculovirus insect cell expression system using the methods described by Kuroki and co-workers.[24] The activities of the TLR2 and TLR1 protein were validated by the fluorescence anisotropy assay with the rhodamine-labeled, synthetic triacylated lipoprotein Pam3CSK4 as the probe (Figure 2a). It was demonstrated that CU-CPT22 was able to compete with Pam3CSK4 for binding to TLR1/2 with an inhibition constant (Ki) of 0.41 ± 0.07 µM, which is consistent with its potency observed in the whole cell assay. The anisotropy of rhodamine-labeled Pam3CSK4 showed a robust increase from 0.168 to 0.275 (Figure 2a) upon addition of TLR1/2 (excitation= 549 nm; emission= 566 nm). This increase is consistent with the anisotropy changes seen with ligand-receptor pairs of comparable sizes.[25] Increasing the concentration of CU-CPT22 to 6 µM decreased the anisotropy to background levels, presumably due to release of the fluorescently labeled Pam3CSK4 probe. This data was then fit to a one-site competition model. Good fitting (R2 > 0.98) inferred that CU-CPT22 and Pam3CSK4 compete for the same binding site on the TLR1/2 dimeric surface. By contrast, compound 6 was used as negative control in the anisotropy assay and demonstrated negligible binding up to 6 µM (Figure 2b). These results further support that CU-CPT22 can compete with Pam3CSK4 binding to TLR1/2.
Figure 2.

Fluorescence anisotropy titration: a) titration of the TLR1/2 protein into the rhodamine-labeled Pam3CSK4 results in significant increase of fluoresence anistropy. Addition of CU-CPT22 competes with Pam3CSK4, resulting in lower fluoresence anistropy, demonstrating competitive binding between CU-CPT22 and Pam3CSK4 for TLR1/2. Data were fitted using a one-site competition model (R2 > 0.98). b) Normalized binding of CU-CPT22 compared with the negative control, compound 6.
One challenge of developing inhibitors to target TLRs is to engineer specificity and potency. There are at least 13 homologous TLRs present in murine macrophages, all sharing a ligand-binding domain with a double-horseshoe shape.[7] We therefore tested CU-CPT22 against a panel of homologous TLRs, including TLR1/2, TLR2/6, TLR3, TLR4, and TLR7 using TLR-specific ligands to selectively activate a particular TLR-mediated NO production. We found that CU-CPT22 inhibits TLR1/2 signaling without affecting other TLRs, showing it is highly selective in intact cells (Figure 3).
Figure 3.
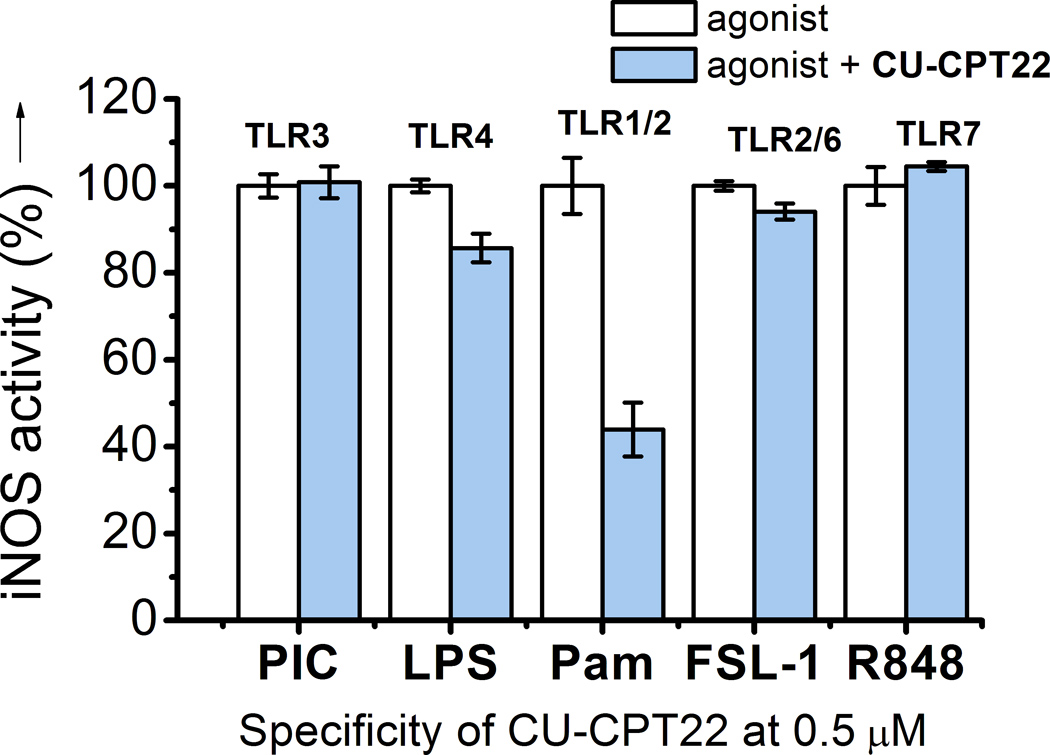
Specificity test for CU-CPT22 (0.5 µM) with with TLR-specific agonists used to selectively activate the respective TLRs: (1) TLR3: 15 µg/mL Poly (I:C), (2) TLR4: 10 ng/mL LPS, (3) TLR1/2: 200 ng/mL Pam3CSK4, (4) TLR2/6: 10 ng/mL FSL-1, and (5) TLR7: 100 nM R848 were used to selectively activate respective TLRs.
Importanly, CU-CPT22 was found to have no significant cytotoxicity at various concentrations up to 100 µM in RAW 264.7 cells using the established MTT methodology (Supplementary Figure S7). Furthermore, kinase profiling showed that compound CU-CPT22 demonstrated minimal non-specific inhibition against a panel of 10 representative kinases (PDGFRB, MET, DDR2, SRC, MAPK1, PAK1, AKT1, PKC-γ, CAMK1, and PLK4) (Supplementary Figure S8). Lastly, we used a secondary cellular assay to confirm that CU-CPT22 also inhibits the downstream signaling transduction. In addition to NO production suppression, the release of the proinflammatory cytokines, TNF-α and IL-1β, that are also regulated by the TLR1/2 singaling cascade was investigated. The result demonstrated that CU-CPT22 can inhibit about 60% of TNF-α (Figure 4a) and 95% of IL-1β (Figure 4b) at 8 µM. These results further supported that compound CU-CPT22 suppresses TLR1/2-mediated inflammation response.
Figure 4.

Downstream signaling inhibition. a) ELISA assay results showed that CU-CPT22 inhibits the TNF-α and b) IL-1β production activated by 300 ng/ml Pam3CSK4 in the RAW 264.7 cells.
It is worth noting that CU-CPT22 can inhibit TLR1/2, while no significant inhibition to TLR2/6 is observed (Figure 3). Based on these experimental observations, we attempted to obtain insights into the possible binding modes of CU-CPT22 (Figure 1b). In the absence of an X-ray crystal structure (structural analysis of the TLR/ligand complexes is highly challenging due to difficult protein preparation), we carried out computational modeling to illustrate the potential binding mode of CU-CPT22 with TLR1/2. Comparing the crystal structures of TLR1/TLR2/Pam3CSK4[11] (Supplementary Figure S1a) and TLR2/TLR6/Pam2CSK4[26] (Supplementary Figure S1b), we observed two lipid chains of Pam3CSK4 and Pam2CSK4 (Supplementary Figure S1a and S1b, yellow) interact with a hydrophobic channel in TLR2, and the amide-bound lipid chain of Pam3CSK4 (Supplementary S1a, red) lies in a second hydrophobic channel of TLR1 that does not exist between Pam2CSK4 and TLR6.[11, 25] The remarkable selectivity for TLR1/2 is likely due to the specific contacts between the aliphatic chain at the R6 position of CU-CPT22 and the hydrophobic channel of TLR1 (Figure 1b), as this hydrophobic channel is absent in TLR6[26] (Supplementary Figure S1b).
In conclusion, we identified a novel small molecule, CU-CPT22 that can compete with the synthetic triacylated lipoprotein (Pam3CSK4) binding to TLR1/2 with potency and specificity. The downstream signaling experiments further supported that CU-CPT22 suppresses TLR1/2-mediated inflammation response. This novel, small molecule agent provides a much needed molecular probe for studying ligand interactions of the TLR1/2 protein complex.
Supplementary Material
Footnotes
We thank the National Institutes of Health (DA026950, DA025740 and NS067425) for financial support of this work. The sTLR2 DNA plasmid was kindly provided by Dr. Chiaki Nishitani and Dr. Yoshio Kuroki.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.Lee CC, Avalos AM, Ploegh HL. Nat. Rev. Immunol. 2012;12:168–179. doi: 10.1038/nri3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hennessy EJ, Parker AE, O’Neill LAJ. Nat. Rev. Drug Discov. 2010;9:293–307. doi: 10.1038/nrd3203. [DOI] [PubMed] [Google Scholar]
- 3.Zaka DE, Schmitza F, Golda ES, Diercksa AH, Peschona JJ, Valvoa JS, Niemistöb A, Podolskya I, Fallena SG, Suena R, Stolyara T, Johnsona CD, Kennedya KA, Hamiltonc MK, Siggsd OM, Beutlerd B, Aderem A. Proc. Natl. Acad. Sci. USA. 2011;108:11536–11541. doi: 10.1073/pnas.1107577108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kawai T, Akira S. Nat. Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 5.Imler JL, Hoffmann J. Nat. Immunol. 2003;4:105–106. doi: 10.1038/ni0203-105. [DOI] [PubMed] [Google Scholar]
- 6.Botos I, Segal DM, Davies DR. Structure. 2011;19:447–459. doi: 10.1016/j.str.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Akira S, Uematsu S, Takeuchi O. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 8.Rolls A, Shechter R, London A, Ziv Y, Ronen A, Levy R, Schwartz M. Nat. Cell Biol. 2007;9:1081–1088. doi: 10.1038/ncb1629. [DOI] [PubMed] [Google Scholar]
- 9.O’neill LAJ, Bryant CE, Doyl SL. Pharmacol. Rev. 2009;61:177–197. doi: 10.1124/pr.109.001073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takeuchi O, Hoshino K, Akira S. J. Immunol. 2000;165:5392–5396. doi: 10.4049/jimmunol.165.10.5392. [DOI] [PubMed] [Google Scholar]
- 11.Jin MS, Kim SE, Heo JY, Lee ME, Kim HM, Paik SG, Lee H, Lee JO. Cell. 2007;130:1071–1082. doi: 10.1016/j.cell.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 12.Kurt-Jones EA, Chan M, Zhou S, Wang J, Reed G, Bronson R, Arnold MM, Knipe DM, Finberg RW. Proc. Natl. Acad. Sci. USA. 2004;101:1315–1320. doi: 10.1073/pnas.0308057100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou S, Cerny AM, Bowen G, Chan M, Knipe DM, Kurt-Jones EA, Finberg RW. Antivir. Res. 2010;87:295–306. doi: 10.1016/j.antiviral.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang HZ, Cui B, Liu HZ, Mi S, Yan J, Yan HM, Hua F, Lin H, Cai WF, Xie WJ, Lv XX, Wang XX, Xin BM, Zhan QM, Hu ZW. PLoS One. 2009;4:e6520. doi: 10.1371/journal.pone.0006520. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.a) Liang Q, Wu Q, Jiang J, Duan J, Wang C, Smith MD, Lu H, Wang Q, Nagarkatti P, Fan D. J. Biol. Chem. 2011;286:26470–26479. doi: 10.1074/jbc.M111.227934. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kalluri1 MD, Datla P, Bellary A, Basha K, Sharma A, Sharma A, Singh S, Upadhyay S, Rajagopal V. FEBS J. 2010;277:1639–1652. doi: 10.1111/j.1742-4658.2010.07589.x. [DOI] [PubMed] [Google Scholar]; c) Abdel-Aal AM, Al-Isae K, Zaman M, Toth I. Bioorg. Med. Chem. Lett. 2011;21:5863–5865. doi: 10.1016/j.bmcl.2011.07.102. [DOI] [PubMed] [Google Scholar]
- 16.Cheng K, Wang XH, Yin H. Small molecule inhibitors of the TLR3/dsRNA complex. J. Am. Chem. Soc. 2011;133:3764–3767. doi: 10.1021/ja111312h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nussler AK, Glanemann M, Schirmeier A, Liu L, Nüssler N. Nat. Protoc. 2006;1:2223–2226. doi: 10.1038/nprot.2006.341. [DOI] [PubMed] [Google Scholar]
- 18.Wu TW, Zeng LH, Wu J, Fung KP, Weisel RD, Hempel A, Camerman N. Biochem. Pharmacol. 1996;52:1073–1080. doi: 10.1016/0006-2952(96)00447-9. [DOI] [PubMed] [Google Scholar]
- 19.Leone M, Zhai D, Sareth S, Kitada S, Reed JC, Pellecchia M. Cancer Res. 2003;63:8118–8121. [PubMed] [Google Scholar]
- 20.Kitada S, Leone M, Sareth S, Zhai D, Reed JC, Pellecchia M. J. Med. Chem. 2003;46:4259–4264. doi: 10.1021/jm030190z. [DOI] [PubMed] [Google Scholar]
- 21.Sang S, Lambert JD, Tian S, Hong J, Hou Z, Ryu JH, Stark RE, Rosen RT, Huang MT, Yang CS, Ho CT. Bioorg. Med. Chem. 2004;12:459–467. doi: 10.1016/j.bmc.2003.10.024. [DOI] [PubMed] [Google Scholar]
- 22.Kerschensteiner L, Löbermann F, Steglich W, Trauner D. Tetrahedron. 2011;67:1536–1539. [Google Scholar]
- 23.Glide, version 5.6. New York, NY: Schrödinger, LLC; 2010. [Google Scholar]
- 24.Iwaki D, Mitsuzawa H, Murakami S, Sano H, Konishi M, Akino T, Kuroki Y. J. Biol. Chem. 2002;277:24315–24320. doi: 10.1074/jbc.M107057200. [DOI] [PubMed] [Google Scholar]
- 25.Xu HQ, Zhang AH, Auclair C, Xi XG. Nucleic Acids Res. 2003;31:e70. doi: 10.1093/nar/gng070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kang JY, Nan X, Jin MS, Youn SJ, Ryu YH, Mah S, Han SH, Lee H, Paik SG, Lee JO. Immunity. 2009;31:873–884. doi: 10.1016/j.immuni.2009.09.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.