


Abstract
We report on the characterization and target analysis of the small (s)RNA162 in the methanoarchaeon Methanosarcina mazei. Using a combination of genetic approaches, transcriptome analysis and computational predictions, the bicistronic MM2441-MM2440 mRNA encoding the transcription factor MM2441 and a protein of unknown function was identified as a potential target of this sRNA, which due to processing accumulates as three stabile 5′ fragments in late exponential growth. Mobility shift assays using various mutants verified that the non-structured single-stranded linker region of sRNA162 (SLR) base-pairs with the MM2440-MM2441 mRNA internally, thereby masking the predicted ribosome binding site of MM2441. This most likely leads to translational repression of the second cistron resulting in dis-coordinated operon expression. Analysis of mutant RNAs in vivo confirmed that the SLR of sRNA162 is crucial for target interactions. Furthermore, our results indicate that sRNA162-controlled MM2441 is involved in regulating the metabolic switch between the carbon sources methanol and methylamine. Moreover, biochemical studies demonstrated that the 5′ end of sRNA162 targets the 5′-untranslated region of the cis-encoded MM2442 mRNA. Overall, this first study of archaeal sRNA/mRNA-target interactions unraveled that sRNA162 acts as an antisense (as)RNA on cis- and trans-encoded mRNAs via two distinct domains, indicating that cis-encoded asRNAs can have larger target regulons than previously anticipated.
INTRODUCTION
In recent years, an increasing number of non-coding RNAs have been shown to participate in various regulatory cellular processes in both pro- and eukaryotes, mainly acting as post-transcriptional ribo-regulators. Although the abundant class of eukaryotic miRNAs mainly act by base-pairing to the 3′- untranslated region (UTR) or coding sequence (CDS) of the cognate target-mRNA, most of the functionally characterized bacterial small regulatory RNAs (sRNAs) target the 5′-UTR of mRNAs (1–3). Besides several sRNAs which directly modulate protein activity, the majority of functionally characterized sRNAs from bacteria belong to the class of trans-encoded antisense RNAs. In most cases, they bind with only partial sequence complementarities to the 5′-UTR of their target genes, and lead to translational repression by masking the ribosome binding site (RBS). Consequently, the association of the ribosomal 30S subunit to the RBS is inhibited, which is often coupled to target destabilization by RNases (2). However, repressive translational control and induction of mRNA degradation is not limited to direct base-pairing with the RBS, because some sRNA have been shown to target sequences that are located far upstream of the RBS, within the CDS or in the intergenic region of polycistronic mRNAs (4–9). Besides repression of target genes, bacterial sRNAs have also been demonstrated to up-regulate gene expression by disruption of inhibitory secondary structures which sequester the RBS, known as anti-antisense mechanism (10–12). The underlying sRNA-target interactions together with the coupled sRNA decay are often facilitated by the bacterial RNA chaperone Hfq, which contains a Sm-like domain (13–15). Additionally, bacterial sRNAs have been shown to target multiple, often functionally related targets, some of which are regulators themselves, and a single sRNA can encompass the expression of large regulons (reviewed in 16,17).
Although first examples of so-called cis-encoded antisense (as)RNAs, which are encoded on the opposite strand of their target genes, were studied already in the early 1980s on plasmids and transposons (18–20), asRNAs are less well characterized in comparison with their trans-acting counterparts. Such asRNAs generally act on their targets independently of Hfq, possibly due to the high extent of base-pairing between the sRNA and their target mRNAs. They overlap with their targets either at the 5′-UTR or 3′-UTR or within the center of the target mRNA. For asRNAs diverse regulatory mechanisms have been described, e.g. alteration of target stability, interplay with RNases, or interference with transcription (for details see 21 and 22). The massive detection of antisense transcription in various genome-wide transcriptome studies clearly suggests a more fundamental role in prokaryotic cells; e.g. in Helicobacter pylori for 46% of all ORFs antisense transcription was detected (23). Although a massive antisense transcription was also demonstrated in other human pathogens such as Listeria monocytogenis or Staphylococcus aureus, and also in the cyanobacterium Synechocystis sp. PCC 6803, the total impact of asRNA regulation is still not yet understood (24–27).
In contrast to bacteria until recently no sRNAs had been identified in archaea, disregarding the role of the extensively studied eukaryotic like small nuclear RNAs (snoRNAs), which participate in ribosomal RNA biogenesis and tRNA maturation (28–31). Currently, several archaea have been examined for the presence of sRNAs on a genome-wide scale either by RNomics approaches or using high throughput sequencing (HTPS) techniques, e.g. RNA-Seq approaches (HTPS of cDNAs), resulting in the detection of high numbers of sRNA candidates (32–35). To study the impact of sRNAs and to get a deeper insight into transcriptional and post-transcriptional regulation in the methanogenic archaeon Methanosarcina mazei Gö1, specifically in response to nitrogen (36), we have recently applied a newly developed differential RNA-Seq (dRNA-Seq) approach for the analysis of primary transcriptomes (23). This approach resulted in the identification 248 sRNA candidates, a high number of which were confirmed by northern blot analysis (36). Here, we report on the functional characterization and target analysis of the abundant sRNA162. Using genetic, biochemical and computational approaches, we demonstrate that sRNA162 interacts with both, a cis- and a trans-encoded target mRNAs via two distinct domains, a mechanism which has not been shown for any studied sRNA in prokaryotes so far.
MATERIALS AND METHODS
Strains and plasmids
Strains and plasmids, which were used in this study, are listed in Supplementary Table S1. Plasmid DNA was transformed M. mazei as described by Ehlers et al. (37).
Construction of M. mazei mutants and generation of plasmids
pRS699 was constructed by polymerase chain reaction (PCR) amplification of sRNA162 including its native promoter and terminator from genomic M. mazei DNA using primers s162-XhoI.for and s162-KpnI.rev (Supplementary Table S2). The 628 bp PCR-fragment was TA cloned into the pCR4-TOPO (Invitrogen Karlsruhe, Germany), resulting in pRS699. After restriction with XhoI and KpnI the fragment was inserted into the multiple cloning site of pWM321 (38). The resulting plasmid (pRS474) and mutant derivatives (discussed later) were transformed into M. mazei by liposome-mediated transformation as previously described (37,39). Puromycin-resistant transformants were selected as colonies that grew on minimal medium plates with trimethylamine as the carbon and energy source plus 5 µg puromycin/ml during incubation.
Using pRS699 as template the sRNA162 M1 and M2 mutants were generated by site-directed mutagenesis with the primer sets s162-Mut1-for/rev or s162-Mut2-for/rev resulting in pRS702 and pRS704. Further mutant derivatives were constructed by inverse PCR using pRS699 as PCR template: Replacement of the linker region between SL2 and SL3 by poly(T) with primers s162-Mut3-for/rev (pRS706) and the deletion of the 3′ end retaining the 5′ fragment (nucleotide 1–65, pRS701) and 3′ fragment (nucleotide 65–191, pRS708) of sRNA162 with s162-Mut4-for/rev and s162-Mut5-for/rev, respectively. The amplified products were religated and cloned into E. coli. Using this cloning strategy, the cognate sRNA variants kept their native sRNA162 promoter and terminator sequences. By using the aforementioned XhoI and KpnI restriction sites, the mutated sRNA162 variants were inserted into pWM321 as described earlier (pRS701, pRS703, pRS705, pRS707, pRS709).
For the construction of the chromosomal sRNA162, deletion mutant by homologous recombination in M. mazei, plasmid pRS650 was constructed as follows. The flanking upstream region (∼1 kbp) of sRNA162 was amplified by PCR using s162 Del1 XhoI and s162 Del2 EcoRI. The flanking downstream region (∼1 kbp) was amplified by PCR using s162 Del3 EcoRI and s162 Del4 XbaI. Both PCR products were restricted with the indicated restriction enzymes and separately ligated to correspondingly linearized pBluescript SK+ resulting in pRS648 and pRS649, respectively. The ∼1.8 kbp EcoRI fragment of pRS207 carrying the pac cassette from Methanococcus voltae (40) conferring puromycin resistance was ligated in a three body ligation together with the upstream fragment (XhoI/EcoRI restricted) into the XhoI/EcoRI restricted plasmid pRS649 generating plasmid pRS650. pRS650 was transformed into M. mazei using a liposome-mediated transformation protocol as described previously (37). Southern blot analyses of genomic DNA from puromycin-resistant transformants were used to verify pac insertion as described by Ehlers et al. (37).
The T7-hammerhead ribozyme fusion of sRNA162 and sRNA162Δ63-88 was generated by commercial gene synthesis (Eurofins MWG Operon, Ebersberg, Germany). TA cloning of both of the products into vector pCR4-TOPO (Invitrogen Karlsruhe, Germany) yielded plasmids pRS765 and pRS766, respectively.
To fuse a T7 promoter to MM2440-41, the operon was amplified with genomic DNA as template as described (see in vitro transcription). The PCR product was TA cloned into pCR4-TOPO (pRS767) and the obtained plasmid was subjected to site-directed mutagenesis with primers 2441-com_Mut1for/rev (pRS768) and 2441-com_Mut2for/rev (pRS769) to obtain compensatory mutants.
RNA isolation
Exponentially or stationary phase cultures (50–70 ml) were harvested at 4°C and RNA was isolated by phenol extraction as recently described (36) or using the RNeasy Midi Kit according to the manufacturer’s instructions (Qiagen, Hilden, Germany) and published for M. mazei (41,42).
Northern blot analysis
Cells were grown under different nitrogen (N) availabilities and harvested at different growth stages (N sufficiency: early exponential phase, OD600 = 0.2; mid-exponential phase, OD600 = 0.5, stationary phase, OD600 = 0.7; N limitation: mid-exponential phase, OD600 = 0.2). Northern blot analysis followed using the recently described protocol (36). sRNA162 and its potential processed derivatives were detected with a 5′-end radioactive labeled ssDNA oligo probe (Supplementary Table S2) and 5S rRNA as described in Jäger et al. (36). Full length riboprobe preparation followed the same procedure as described for T7 in vitro transcription, except that the MAXIscript T7 kit (Ambion; Applied Biosystems, Foster City, CA) was used according to the manufacturer. To amplify a T7 promoter containing PCR product which serve as template for antisense probe synthesis the primer pairs asPs162 for and asPs162rev, or asPs171 for and asPs171rev, were used, respectively (Supplementary Table S2).
Rapid amplification of cDNA ends analysis
RACE (rapid amplification of cDNA ends) analysis was performed to determine the transcriptional start site (TSS) as well as the transcript termination site of sRNA162. The 5′-RACE system (Invitrogen, Karlsruhe, Germany) was used according to manufacturer’s instructions with oligonucleotide 5′-RACE 2441 as gene-specific primer to determine the TSS of sRNA162 (Supplementary Table S2). 3′-RACE analysis followed the 3′-RACE System (Invitrogen, Karlsruhe, Germany) according to manufacturer’s instructions after polyadenylation of RNA with Poly(A)-polymerase (NEB, Schwalbach, Germany). The precipitated RNA was resuspended in 11 -µl DEPC-H2O and directly used for 3′ RACE using the oligonucleotide 3′-RACE 2441 #2 as gene-specific primer (Supplementary Table S2). PCR products were subjected to TOPO-TA cloning using the TOPO-TA Cloning Kit (Invitrogen, Karlsruhe, Germany). 5′ end and 3′ end of sRNA162 were determined by DNA sequencing of both strands. For 5′-end analysis of the operon MM2440-41, the FirstChoice RLM-RACE kit (Ambion; Applied Biosystems) was used, according the manufacturer’s instructions. The oligonucleotides 5′-2441-RLM (RNA linker mediated)-out and 5′-2441-RLM-in were used as gene-specific primers to determine the TSS. In the same way, the oligonucleotides 5′-2442-RLM-out and 5′-2442-RLM-in or 5′-2446-RLM-out and 5′-2446-RLM-in were used to determine the TSS of MM2442 or MM2446, respectively (Supplementary Table S2).
In vitro T7 transcription, purification and 5′-end labeling of RNA
Templates for in vitro transcription were either amplified from genomic M. mazei wild-type (wt) DNA or from plasmids carrying the respective constructs (Supplementary Table S1). Primer sequences are summarized in Supplementary Table S2. Plasmids carrying sRNA162 (or derivatives, discussed earlier) were PCR amplified simultaneously fusing a T7 promoter to the PCR product. For the 3′ end of sRNA162, the primer pair T7-s162-SHORT-for and sRNA162-T7.rev was used, whereas the other T7 sRNA162 fusions were amplified with T7-HH-s162-for and sRNA162-T7.rev instead. Similar, T7-2440-41-for and T7-2440-41-rev1 or T7-2440-41-rev2 were used for the T7 fusions of the long and short versions of mRNA MM2440-2441, respectively. T3-MM2446-for and rev were used for the amplification and the incorporation of a T3 promoter to MM2446. In vitro transcription was performed using the MEGAscript T7 (or T3) kit (Ambion; Applied Biosystems) according to the manufacturer. Following extraction with phenol:chloroform:isopropanol (25:24:1 v/v) and purification with G-25 column, the RNA was precipitated overnight at −20°C with 3 vol. of ethanol and 20 µg glycogen (Applied Biosystems). RNA quality and integrity were checked on a denaturing polyacrylamide gel (6% PAA, 7 M Urea). The in vitro transcribed RNA was dephosphorylated with FastAP (Thermosensitive Alkaline Phosphatase, MWG Fermentas), according to the manufacturer and radioactively labeled at the 5′ end as described in Jäger et al. (36), additionally supplementing the labeling reaction with 40 U of RNasin (Promega, Mannheim, Germany).
A hammerhead ribozyme transcriptional fusion with sRNA162
Usually, in vitro transcripts were created by incorporating a T7 promotor into the oligonucleotides used for PCR of the template or by cloning of the respective PCR product into a T7 promotor containing vector. To ensure efficient transcription with high yield, the PCR-based T7 fusion usually introduces 3 Gs at the TSS (+1 site). To prevent the addition of non-sequence specific nucleotides into the transcripts and avoid the introduction of artificial structural changes, we used a method recently described by Holmquist et al. (43), based on the work of the Theobald-Dietrich and co-workers (44). Basically, we constructed and synthesized a DNA template of sRNA162 for T7 polymerase, where the promotor was fused to a hammerhead ribozyme (Supplementary Figure S3), further linked to the sequence of choice, here sRNA162. As soon as polymerization of the transcript starts, the RNA is simultaneously folded into its hammerhead shape. When folding is completed, the ribozyme is autocatalytically self-cleaved, whereas the transcription reaction of the downstream sequence continuous. Thus, the fusion guarantees transcription at the native +1 site of sRNA162, and can be engineered for any template choice, thereby providing an efficient method for the preparation of native RNA transcripts (Supplementary Figure S3).
In vitro structure analysis
Structure probing of sRNAs and mRNAs were conducted in total volume of 10 µl. Before RNase T1 and A cleavage the RNA (∼0.1 pmol) was denatured for 1 min at 95°C and chilled on ice for 5 min, then 10× Structure Buffer (Ambion; Applied Biosystems), 1 µg yeast RNA (Ambion; Applied Biosystems) were added. Following a renaturation step for 10 min at 37°C, 25 mM lead(II)acetate (Carl Roth, Karlsruhe), 0.01 U RNase T1 (Ambion; Applied Biosystems) or RNAse A (Ambion; Applied Biosystems) was added and incubated for to 2, 3 and 5 min. The reactions were either stopped with 0.2 M EDTA and precipitated, or by directly adding gel loading buffer II (Ambion; Applied Biosystems).
For RNase T1 ladders, the labeled RNA (∼0.2 pmol) was incubated for 1 min at 95°C in 1× Sequencing Buffer (supplied with RNase T1). Subsequently, 0.1 U RNase T1 was added to mix and further incubated for 5–10 min at 37°C. OH ladders were generated by incubating ∼0.2 pmol labeled RNA for 5 min at 95°C in 1× alkaline hydrolysis buffer (supplied with RNase T1). Reactions were stopped as described earlier. The samples were analysed on 8% polyacryl amide/7M urea sequencing gels and visualized with a phosphoimager (FLA-5000 Series, Fuji).
In vitro binding assays
Electro mobility shift assays were conducted in a total volume of 10 µl in the presence of 1X structure buffer (Ambion; Applied Biosystems) and 1 µg yeast RNA (Ambion; Applied Biosystems). 20 pmol of in vitro transcribed RNA were dephosphorylated and radioactively 5′ labeled as described earlier. 5 nM of the labeled RNA were incubated in presence with increasing amounts of the target RNA (8, 16, 32, 125, 250, 500, 1000, 2000 nM) for 15 min at 37°C and subsequently separated on native 6–8% poly acrylamide gel in a 0.5× Tris–borate buffer system (0.45 M, pH 8.0). Gels were analysed using a phosphoimager (FLA-5000 Series, Fuji).
Transcriptome analyses
For genome-wide expression profiling genome wide microarrays representing 97% of the ORFs were used (41,45). Total RNA was extracted from M. mazei sRNA162-overexpressing cultures and the wt containing pWM321 grown with 150 mM methanol as carbon and energy source as described in Veit et al. (41). In general, RNA was extracted at turbidities OD600 = 0.5. Purified RNA was converted to cDNA and labeled by fluorescent Cy-3 and Cy-5 dyes using the CyScribe first-strand cDNA labeling kit (GE Healthcare, Freiburg, Germany) as described (41,45). Microarray slides were incubated with aliquots of the cDNA preparations at 42°C overnight (Lucidea SlidePro hybridization chamber, GE Healthcare), for details of the hybridization and wash procedures see Hovey et al. (45). Signal intensities were analysed using a GenePix 4100A scanner and data normalization and evaluation was performed using the GenePix Pro software version 6.0 (Axon Instruments, Union City, USA) as described recently (41,42). RNA isolated from three independent cultures were used independently in pairs of labeling reactions and, for one pair, a dye-swap experiment was also performed. Only if a transcript had an abundance difference of at least 3-fold in the comparisons of wt RNA was the difference reported here to be significant.
Quantitative reverse transcriptase PCR (qRT-PCR) assays were performed with a QuantiTect Probe RT-PCR Kit (Qiagen, Hilden, Germany) using a 7300 real-time PCR system (ABI, Foster City, USA) and at least three independent RNA preparations for each strain as described (42,46). Primer sets used including the control genes are listed in the Supplementary Table S2. The fold change in transcript abundance for genes of interest was determined by comparison with the threshold cycle (Ct) of transcripts of three control genes (MM1621, MM2181, MM1215). The fold change in the abundance of a transcript was calculated using the formula fold change = 2−ΔΔCt as described (47).
Computational target predictions
The ribosome binding site (RBS) sequence positions for all annotated M. mazei genes were predicted following the approach of (48). Significant RBS locations were obtained for 55.2% of the genes. Putative base pair interactions with sRNA162 were predicted for all ORFs with at least 3-fold change in transcript level in the microarray analysis. For ORFs organized in an operon structure, the first ORF of the respective operon was also included. Sequences used for the interaction prediction contained the full length 5'-UTR (36) and additional 100 nt of the CDS. If the exact 5'-UTR start was unknown, 200 nt upstream of the annotated translation start were used instead. Interaction predictions were computed with the tool IntaRNA (49,50) requiring an interaction seed of eight consecutive base pairs. All mRNA subsequences were folded locally in a 100 nt window with 50 nt maximal base pair span. For those ORFs which showed reduced transcript levels in the sRNA162 overproducing mutant using the microarray, we selected all interactions that were predicted to be located in the mRNA region from −39 in the 5'UTR to +19 in the CDS, which is the maximal region covered by ribosomes (51). For all ORFs with increased transcript levels, we studied the influence of the putative interaction on the accessibility of the predicted RBS sequence. All interactions predicted to be located upstream of the RBS sequence were included. A measure for the accessibility of a subsequence is its probability to be unpaired, i.e. free of intra-molecular base pairings. We computed the unpaired probability of the RBS sequence before and after the putative interaction of the mRNA with the sRNA162 (49). When the change in this unpaired probability was greater than 0.001, we assumed that the structural rearrangement in the 5'-UTR caused by the interaction increases the accessibility of the RBS to activate the translation of the gene.
RESULTS
Characterizing sRNA162
We have recently identified sRNA162 in our dRNA-seq approach to study the transcriptome of M. mazei (36). Because northern blot analysis revealed growth phase-dependent expression and processing of this particular sRNA, we selected sRNA162 for further functional characterization and analysis of its target genes. The sRNA162 gene is located in the 739 bp intergenic region between MM2441, encoding a transcriptional regulator of the ArsR family, and MM2442, encoding a conserved protein of unknown function (Figure 1A). The TSS and termination site of sRNA162 were determined by 5′ RLM and 3′ RACE, respectively (see ‘Materials and Methods’ section), and revealed a primary transcript of 191 nucleotides (nt) (Figure 1B and C). In addition, the RACE approach revealed the presence of the homologous sRNA171 with exactly the same transcript length, which is encoded in the intergenic region between MM2448 and MM2449 (discussed later). Expression of both RNAs was confirmed by northern blot analysis, and further revealed that the constitutively transcribed full length sRNA162 is processed into three different stable 5′ fragments (nt 1–65, 1–60, 1–55). In stationary growth phase, complete processing of sRNA162 is observed independently of the supplied carbon source (Figure 1C; Supplementary Figure S1A). This is possibly achieved by exonucleases which degrade the 3′ fragment, since the corresponding, processed 3′ fragment(s) are neither detectable by northern blot analysis nor qRT-PCR (data not shown). In addition to the primary sRNA162 transcript and its respective 5′ processing fragments, other fragments were also detected using the probe directed against sRNA162 as evident in a sRNA162 deletion strain (Figure 1C). These are probably due to unspecific cross-hybridization (marked *) or cross-hybridization with sRNA171. To discriminate between the two homologous sRNAs, we additionally generated riboprobes specific for the respective full length sRNAs by in vitro transcription resulting in neglectable cross-hybridization in Northern blots (Supplementary Figure S1B and C). Hybridization with these riboprobes demonstrated that sRNA171 is also constitutively expressed and appears to be immediately processed into stable 5′ fragments, especially when cells enter stationary phase. However, additional processing fragments were detectable of ∼150 and 120 nt in length (Supplementary Figure S1C), indicating different processing of the two RNA species and/or different stabilities of the resulting 3′ fragments, though high conservation of their primary sequence (Figure 1D).
Figure 1.

Characterization of sRNA162. (A) Genomic context of sRNA162. (B) Promotor and terminator region of sRNA162. The 5′ end (+1) of sRNA162 was determined by 5′-RLM RACE, as well as the 3′ end (*) by 3′-RACE analysis. (C) Northern blot analysis of total RNA of wt and sRNA162 mutant strains grown with methanol from early exponential phase (OD600 of 0.2, lanes 1), mid exponential phase (OD600 of 0.5, lanes 2) and stationary phase (OD600 of 0.7, lanes 3). The three 5′-fragments of sRNA162 and the primary transcript are indicated by arrows. Cross-hybridization of the sRNA162 probe is indicated by an asterisk. The lower panel shows the expression of 5S rRNA of the respective RNA preparation. wt, wild-type strain; sRNA162 OE, sRNA162 overexpressed from pWM321 in wt; and ΔsRNA162, chromosomal sRNA162 deletion mutant. (D) Secondary structure alignment of sRNA162 homologues identified by computer-based searches of related Methanosarcina species performed with LocARNA (52), Ma, sRNA162 homologue of M. acetivorans C2A; Mb, M. barkeri strain fusaro. (E) Consensus secondary structure prediction by RNalifold (53). SL, stem loop.
BLAST searches for sRNA162 homologs in other prokaryotes detected the presence of homologous sequences exclusively in the two close relatives, M. acetivorans and M. barkeri. Secondary structure alignments of all sRNA162 homologs revealed not only highly conserved structural elements [stem loop (SL) 1–3] but also a 39 nt single stranded linker region (SLR) between SL2 and SL3 (Figure 1D), representing the most variable part within the predicted structural alignment. In vitro secondary structure probing using RNase T1, RNase A- or PbII-cleavage of in vitro synthesized 5′-end-labeled sRNA162, confirmed the in silico predicted secondary structure with only marginal variations in the organization of internal loops of SL3 (compare Figures 1E and 2). Analysis of the homologous sRNA171 revealed a similar secondary structure consisting of three stem loops (SL1–SL3), with the two structured elements SL2 and SL3 interrupted by a SLR of 39 nt (Supplementary Figure S2). Single nucleotide exchanges in comparison with sRNA162 are mostly present in the 5′ part of sRNA171 ranging from position 1 to 80. Of those located in the structured parts, the majority represents compensatory base-pair exchanges or nucleotide exchanges within bulge loops which do not affect base-pairing [e.g. G42 (sRNA162), A42 (sRNA171)]. Changes downstream of SL2 tend to be A to U substitutions, whereas at the cognate positions of sRNA162 Gs and Cs are dominating (Figures 1D and 2; Supplementary Figure S2).
Identification of potential sRNA162 targets by genetic and computational approaches
A deletion mutant of sRNA162 and a mutant expressing the sRNA162 gene from the low copy plasmid pWM321 under the control of its native promoter (further designated as ‘over-expressing mutant’) were generated (see ‘Materials and Methods’ section). Northern blot analysis confirmed deletion of sRNA162 and showed ∼30-fold higher transcript levels in the over-expressing mutant strain (Figure 1C). To identify potential target genes of sRNA162, genome-wide changes in transcript levels in this mutant in comparison with the wt strain were studied using established genomic microarrays for M. mazei (41,45). The transcriptome analysis demonstrated that approximately 185 of the ORFs showed significantly different transcript levels (≥3-fold) in the over-expressing mutant compared with the wt strain. These include 48 genes involved in energy and constructional metabolism, two transcriptional regulators, 14 genes encoding transport or membrane proteins and additional 51 genes with unknown function (Supplementary Table S3). Strikingly, elevated transcript levels of a high number of genes (7 operons) encoding soluble methyltransferases involved in degradation of methylamines were obtained in the mutant, although the strains were grown on methanol (Table 1). A transcriptome analysis of the sRNA162 deletion mutant was not performed. Because the first 65 nt of sRNA162 overlap with the 5′UTR of the flanking gene MM2442, we assumed polar effects in the deletion mutant on MM2442 (discussed later, second target).
Table 1.
Transcript levels of selected genes involved in TMA degradation and MM2441 of M. mazei sRNA162-overexpressing mutant versus M. mazei wt during growth on methanol as sole carbon and energy source identified by global expression profiling using genomic microarrays (41,45)
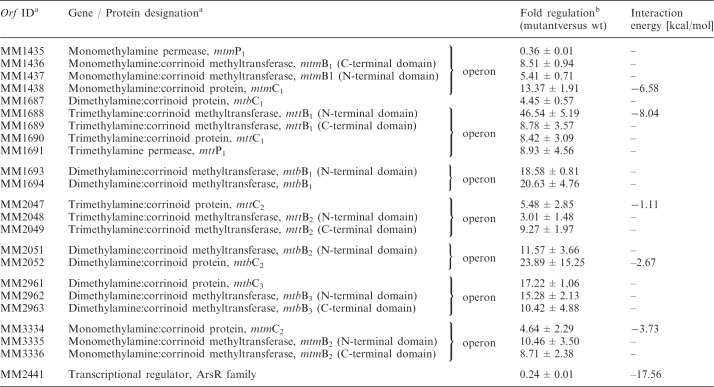 |
The in silico predicted interaction energy with sRNA162 is given.
aAs defined in (54).
bGene induction is represented as the ratio of median. Mean values obtained from five microarray experiments that satisfy the criteria defined in the ‘Material and Methods’ section are indicated.
All ORFs with significant changes in transcript levels in the overexpression strain (≥3-fold) were further analysed in silico to predict potential direct targets of sRNA162. The tool IntaRNA (49) was used to predict putative interactions between sRNA162 and each of the respective mRNAs, including either their native 5′UTR, if known (36), or if not known, including 200 nt upstream of the predicted translational start site (TLS) (see ‘Materials and Methods’ section). The obtained results for selected target candidates are summarized in Table 1. Additional potential interactions are listed in Supplementary Tables S4 and S5. The best scoring target was MM2441 encoded in reverse orientation upstream of the sRNA162 gene (Figure 1A). The predicted interaction between the two RNAs is centered on the potential RBS and the TLS of mRNA2441 as depicted in Figures 3A and B with a free energy score of −17.6 kcal/mol. This score is considerably better than the second best ranked target (MM2339) with a free energy score of −10.9 kcal/mol (Supplementary Table S4), or than any of the other methyltransferase genes with altered expression upon sRNA162 overexpression (see above and Table 1). Thus, the predicted primary target MM2441 was selected for further biochemical validation.
Figure 3.

sRNA162 interacts with the bicistronic mRNA MM2440-41 in vitro. (A) Overview of the transcriptional and translational features of MM2440-41. The boxed region indicates the predicted interacting site. Bold letters either represent the RBS or TLS of the respective ORFs. The in vitro transcribed target mRNAs are schematically drawn below. (B) In silico predictions of the sRNA162–MM2440-41 interaction performed with IntaRNA (49). The wild-type (wt), triplet mutations (M1 and M2) and compensatory mutations (M1′ and M2′) are indicated. (C) Cartoon of the proposed structure of sRNA162. Deletions or replacements of sRNA parts are indicated in red. (D–J) Electrophoretic mobility shift assays were performed using approximately 5 nM of radioactively 5'-end-labeled MM2440-41LONG (or compensatory mutants, respectively). The assays were performed as described in the ‘Materials and Methods’ section with increasing concentrations of unlabeled sRNA162 or mutated species from 0 to 2 µM. After 15 min incubation, samples were run on a native 6% PAA gel. The respective autoradiographs of the gels are shown.
In vitro verification of a direct sRNA162/mRNA 2441 interaction
Transcript analysis by 5′-RLM RACE revealed that MM2441 is encoded as the second gene in a bicistronic operon together with MM2440 (encoding a hypothetical protein), which is preceded by a short 5′-UTR of 36 nt (Figure 3A). Furthermore, the two ORFs overlap because the predicted TLS of MM2441 is located within the 3′ coding region of MM2440, 37 nt upstream of the translational stop of MM2440 (Figure 3A). Electrophoretic mobility shift assays using a 5′ labeled in vitro synthesized fragment of the bicistronic mRNA MM2440-41 (+1 to +469 relative to the TSS) including the predicted binding site (MM2440-41LONG; Figure 3A) and in vitro synthesized non-labeled full length sRNA162 (see ‘Materials and Methods’ section), demonstrated that binding to 5 nmol MM2440-41LONG occurred at concentrations ≥32 nM of sRNA162 (Figure 3D). The reverse assay, using 5′-labeled sRNA162 and non-labeled MM2440-41LONG, further verified a direct interaction between the two RNAs (Supplementary Figure S4A). The interaction was strongly diminished when equal amounts of non-labeled sRNA162 were used as competitor RNA, confirming specificity of the binding (compare lines 9 and 10, Supplementary Figure S4A). In contrast, up to 2 µM of sRNA162 did not affect the mobility of a shorter 5′ fragment of the target mRNA, which did not include the predicted binding site (MM2440-41SHORT, ranging from+1 to 248; Supplementary Figures S4B and C), demonstrating that the predicted interaction site is compulsory for binding. The absence of the predicted, non-structured binding site in sRNA162 (sRNA162Δ63-88) or changing the region into polyT (sRNA162 M3), resulted in a complete loss of the mobility shift of MM2440-41LONG (Figures 3E and F; reverse assays Supplementary Figure S4D and E). Triple point mutations within the potential interacting site changing either position 64–66 in sRNA162 from CAC antistart codon to GUG (M1 mutant) or position 79–81 from CAU to GUA (M2 mutant), further demonstrated that the M2 mutation totally abolished binding (Figure 3G; reverse assay Supplementary Figure S4F), whereas binding of the M1 mutant is only slightly reduced (Figure 3H and Supplementary Figure S4G reverse assay). Introducing compensatory mutations at position 461 to 463 (M2′) or 446 to 448 (M1′) of MM2440-41LONG, totally restored the interaction with the M2 mutant sRNA, whereas the interaction with sRNA162 M1 is significantly improved (Figure 3I and J; Supplementary Figure S4H and I). These findings strongly indicate a specific binding of sRNA162 to the mRNA (MM2440-41), which most likely leads to masking of the RBS and the TLS of MM2441, as depicted in Figure 3B (wt). The binding affinity appears to be relatively low in comparison with a variety of bacterial trans-acting sRNAs (e.g. GcvB; (4)), indicating that a chaperone such as Hfq might be required to facilitate annealing of the two RNAs. Thus, the effect of Lsm-like proteins (MM339, MM2383) on binding has been tested by gel shift experiments in the presence of up to 75 µM purified his-tagged proteins (calculated for the hexamers); however, no significant effect was observed (data not shown).
Gel shift assays using the 5′ fragment (nt 1–65) of sRNA162 (resembling the largest detectable stabile 5′ fragment in vivo) or the 3′ fragment (nt 65–191) containing most of the binding site (nt 60–88) but missing the structured loops SL1 and 2 (Figure 3C) further demonstrated, that both of the truncated fragments are not able to interact with the target mRNA (Supplementary Figure S4J–M). Because both mutants lack parts of the binding site, this strongly suggests that the entire interacting site is essential for effective target binding, and/or the structured 5′ part of sRNA162 (SL1 and 2), though not directly involved in the interaction, is highly required for a stable mRNA MM2440-41 target interaction in vivo.
In vivo target validation by genetic approaches
The mutant overexpressing sRNA162 showed a partial growth defect when grown on methanol as sole carbon and energy source (Supplementary Figure S5). To gain a deeper insight, whether the growth reduction is due to interaction of sRNA162 with the bicistronic operon MM2440-41, or due to the interaction with a yet unknown target (or a combination of both), additional mutant strains were generated. Mutant derivatives of sRNA162 identical to the variants used for EMSAs (Figure 3C), were ectopically expressed in the wt background from pWM321 under the control of the native promoter and with the native terminator, including the separated 5′ (1–65 nt) and 3′ fragment (65–191 nt, containing most of the predicted binding site), and the M1 to M3 derivatives of sRNA162. Neither overexpression of the fragments nor the mutant derivatives of the full length sRNA162 effected growth (Figure 4A); solely overexpressing full length wt sRNA162 led to the observed significant change in growth rate. This finding strongly indicates that exclusively the primary transcript, containing the intact wt SLR, is capable to interact with the proposed target(s) and is crucial for the obtained growth defect, most likely due to down regulation of MM2441. However, at the current experimental status we cannot rule out the possibility that the phenotype is due to interactions with other potential targets, since the chromosomal compensatory mutations in MM2440-2441 nor a double deletion of sRNA162 and MM2441 have not been generated due to experimental bottle necks (e.g. the lack of a second selection marker).
Figure 4.

Growth and northern blot analysis of sRNA162 overexpressing mutants. (A) Growth of wt with additional sRNA162 (sRNA162 OE; empty diamond), 5' end of sRNA162 (5' end; empty circles), 3' end of sRNA162 (3' end; filled squares), M1 mutant (M1; empty squares), M2 mutant (M2; filled triangles) and M3 mutant (M3; filled circles), each ectopically overexpressed from pWM321 in wt background. Cells were grown on methanol to mid-exponential phase. (B) Northern blot analysis of the respective M. mazei strains using in vitro synthesized full-length RNA probes.
To test whether processing of the sRNA is altered in case of the sRNA162 derivatives northern blot analysis was performed (Figure 4B). Compared with the wt background, overexpression and subsequent processing of full length sRNA162 results in significant higher transcript levels of the primary transcript, as well as the three 5′ fragments. A similar increase of the three 5′ fragments was also detected upon overexpression of the 5′ fragment (1–65 nt) of sRNA162 (Figure 4B, lane 3) indicating that this extended 5′ fragment is further processed into similar fragments and ratio as for the processing of the full length sRNA162. Remarkably, overexpression of the 3′ fragment (nt 65–191), results in stable expression of the 3′ portion without detectable processing pointing toward a crucial role of the structured 5′ end of sRNA162 for processing. However, the increased stability of the 3′ fragment might also result from the artificial truncation. Mutant derivatives of full length sRNA, M1 and M2, showed higher accumulation of the primary transcript compared with the wt, strongly indicating altered processing. In contrast, when replacing the SLR by poly(T) (derivative M3), the primary transcript appears less stable compared with the M1 and M2 derivatives and the defined 5′ fragments are still produced in high amounts, although as well some additional processing products are visible (Figure 4B).
Considering that several soluble methyltransferases involved in degradation of methylamines showed elevated transcript levels upon sRNA162-overexpression (Table 1), growth was studied with trimethylamine (TMA) as sole energy and carbon (C) source. No difference in expression or processing of sRNA162 was obtained when cells were grown on TMA (Supplementary Figure S1A). Moreover, when grown on TMA the sRNA162 overexpression strain displayed the same growth phenotype as observed with methanol (data not shown). However, when shifting cells from methanol to TMA, sRNA162-overexpression appears to result in significant faster adaptation following the C source shift compared with the wt (Figure 5) probably due to already synthesized soluble methyltransferases. To further support this suggestion, transcript levels of soluble methyltransferases in exponential growth phase with methanol were evaluated. Because of high sequence similarities of methyltransferase genes reliable transcript quantification using the PCR-fragment based microarray is not possible (45); thus, we performed qRT-PCR analysis using specific primers (46,55). A variety of methyltransferases with different substrate specificities showed significantly reduced mRNA levels of their respective operons, including methanol-dependent methyltransferases (mtaC1B1, MM0174-75; mtaC3B3, MM1648-47), TMA-dependent methyltransferases (mttB1C1 MM1688-90; mttB2C2, MM2049-47), dimethylamine-dependent methyltransferases (mtbC1B1, MM1687, 1693-94; mtbC2B2, MM2052-50) and monomethylamine-dependent methyltransferases (mtmC1B1, MM1438-36) (Figure 6 and Supplementary Figure S6), explaining the reduced growth rates on methanol and TMA. Solely, mtmC2B2, encoding a monomethylamine-dependent methyltransferase (MM3334-36), showed approximately 5-fold increased transcript levels upon sRNA162-overexpression, most likely leading to the observed faster adaptation of the sRNA162 overproducing mutant strain after a shift from methanol to TMA (Figure 6).
Figure 5.

Growth behavior of the mutant overexpressing sRNA162 when shifted from methanol to trimethylamine in comparison with the wt. Cells were precultured as described in the ‘Materials and Methods’ section with methanol as sole carbon and energy source. After reaching exponential growth phase, cells were inoculated in fresh media either containing methanol (control) or trimethylamine (shift) as carbon and energy source. Growth was monitored by measuring the turbidity at 600 nm. Wt; sRNA162 OE, sRNA162 overexpressed from pWM321 in wt. Depicted are mean values and standard deviations of three biological replicates.
Figure 6.

Transcriptional profile of selected genes in exponential growth phase. For selected genes, the transcriptional levels were determined in exponential growth phase by qRT-PCR (for primers see Supplementary Table S5). Fold changes in the sRNA162 overexpressing mutant (vs. wt) are given by mean values and standard deviation of three biologically independent experiments. MM0174, mtaC1 encoding methanol corrinoid protein; MM1073, mtaC2 encoding methanol corrinoid protein; MM1070, mtaA1 encoding methylcobalamin-coenzyme M methyltransferase; MM1438, mtmC1 encoding monomethylamine corrinoid protein; MM1687, mtbC1 encoding dimethylamine corrinoid protein; MM1690, mttC1 encoding dimethylamine corrinoid protein; MM2047, mttC2 encoding trimethylamine corrinoid protein; MM2052, mtbC2 encoding dimethylamine corrinoid protein; MM2961, mtbC3 encoding dimethylamine corrinoid protein; MM3334, monomethylamine corrinoid protein; MM2440, hypothetical protein; MM2441,: transcriptional regulator, ArsR family; MM2442, hypothetical protein; MM2446, conserved protein.
On the basis of these findings, we hypothesize that MM2441 is most likely post-transcriptionally regulated by sRNA162 and encodes a transcriptional regulator, which represses transcription of mtmB2C2 encoding an essential soluble methyltransferase involved in TMA degradation. However, transcription of several other methyltransferase genes appears to be activated by MM2441 (see discussion).
MM2442 as the second potential target of sRNA162
Although short UTRs or even leaderless mRNAs have been predominantly found in (hyper-) thermophilic archaea (35,56), in methanoarchaea long 5′ UTRs with an average size of ∼150 nt are more the rule than the exception (36). Because sRNA162 is located 150 nt upstream of the TLS of MM2442 in opposite orientation (Figure 1A), we determined the TSS of MM2442 encoding a conserved protein of unknown function. 5′-RLM RACE analysis identified the TSS of MM2442 to be 184 nt upstream of the predicted TLS resulting in an overlap of 65 nt in antisense orientation with the 5′ end of sRNA162 (Figure 7A). This strongly suggests that besides targeting mRNA2440-41 in trans, sRNA162 very likely represents also a cis-encoded antisense regulator of MM2442 mRNA. Gel mobility shift experiments with full length sRNA162 and its 3′ and 5′ fragments demonstrated that full length sRNA162 indeed binds with its 5′ region to the 5′-UTR of MM2442 mRNA, whereas the isolated stable 5′ fragment appears to have even a significant higher binding affinity (Figure 7B–D; reverse assays Supplementary Figure S7). Moreover, in vitro foot printing analysis confirmed that sRNA162 binds to the leader of MM2442 by its first 65 nt as depicted in Supplementary Figure S8.
Figure 7.

The 5'-UTR of MM2442 as a sRNA162 target. (A) Promotor region of MM2442 and sRNA162. The 5′ end (+1) of MM2442 was determined by 5′-RLM RACE. The TATA box and BRE are indicated for both, MM2442 and the cis-encoded antisense sRNA162. The predicted interacting site is boxed. (B–D) Electrophoretic mobility shift assays were performed using ∼5 nM of radioactively 5'-end-labeled MM2442. The reactions were performed as described in the ‘Materials and Methods’ section with increasing concentrations of unlabeled sRNA162 or mutated species from 0 to 2 µM. After 15 min incubation, samples were run on a native 6% PAA gel. The respective autoradiographs of the gels are shown.
Characterizing sRNA171 and its potential targets
The homologous sRNA171 is encoded in the IGR between MM2448 encoding a transposase and MM2449, encoding a protein of unknown function, which however shows some homology to MM2442. Upstream of MM2448 a second transposase gene (MM2447) is present, which is further upstream flanked by MM2446, encoding a transcriptional regulator of the ArsR family with MM2441 as the closest homolog (Supplementary Figure S9). Overall, the high conservation of the two sRNAs (including their promoters) as well as the conservation of the flanking regions of sRNA162 and sRNA171 and their respective genomic organization suggests that sRNA171 has been generated by a duplication of the complete gene locus MM2440–MM2442 most likely via a transposition event (Supplementary Figure S10). This is further supported by the finding that in the close relatives M. acetivorans and M. barkeri only a single copy of the sRNA is present in similarly organized genetic loci (Supplementary Figure S10).
Even though sRNA171 generally showed high sequence conservation to sRNA162 4 nt changes are obvious within the SLR, which might alter the binding properties of the sRNA (Figure 1D). However, due to the similar genomic organization potential cross interaction between sRNA171 and the respective target mRNAs of sRNA162 and vice versa were investigated by in silico predictions (Supplementary Figure S9C) and gel shift experiments. This demonstrated a significantly lower binding affinity of sRNA171 to the 2440–41 mRNA than observed for sRNA162 (Supplementary Figure S9D; reverse assay Supplementary Figure S7) and no binding to MM2446 for both sRNAs (Supplementary Figure S9F and G; reverse assays Supplementary Figure S7). Most interestingly, the second target of sRNA162, the 5′ UTR of MM2442, was bound by sRNA171 (Supplementary Figure S9E; reverse assay Supplementary Figure S7) with similar affinity as observed for the 5′ fragment of sRNA162 (Figure 7D). Consequently, the targets of both sRNAs generated by duplication might formerly have been the same, but following the transposition event both sRNA-genes and targets might have picked up several different mutations leading to diversification of targets and functions.
DISCUSSION
In the last two decades, a continuously growing number of sRNAs has been identified in prokaryotes. However, most of the functional characterization of sRNAs has been carried out in enterobacteria. Although expression of sRNAs has also been verified in several archaeal species (32–36,57–63), neither a potential target mRNA nor a molecular mechanism of target regulation or physiological role has been elucidated in archaea so far. Considering that cellular processes in archaea in general show many mechanistic features more similar to their eukaryotic than bacterial counterparts, e.g. transcription and translation machineries (64–68), 3′ targeting of mRNAs similar to eukaryotic miRNAs has been considered to be very likely for archaeal sRNAs. In this study, however, we present the first detailed characterization of an archaeal sRNA interacting with the 5′ UTR of two mRNAs, a cis- and a trans-encoded target.
An archaeal sRNA masks the ribosome-binding site of its target
We provide several lines of evidence, that sRNA162 acts in trans on the neighboring operon MM2440-41 by internally binding to the bicistronic mRNA. The interacting site of sRNA162 was narrowed down to the SLR between SL2 and 3 (Figure 2) by in vitro binding assays and in vivo studies, clearly demonstrating that the SLR is crucial for functionality (Figures 3 and 4A). However, the observed binding affinity of sRNA162 to its target MM2441 is comparably low (for comparison see refs. 4, 69, 70). No further enhancement was obtained in the presence of the two heterologously expressed Lsm-like proteins of M. mazei, although we currently cannot exclude non-physiological folding or incorrect assembly of Lsm oligomers due to heterologous expression. The finding that sRNA162 binds to the predicted RBS as well as the TLS of MM2441, strongly argues that the interaction inhibits translation initiation most likely resulting in dis-coordinated operon expression. Because we do not see any changes at the mRNA level, regulation probably most likely occurs at the translational level. The discovery that sRNA162 targets the RBS within the 5′ region of MM2441 mRNA elucidated an unexpected regulatory mechanism of an archaeal sRNA, which was up to now exclusively described for bacterial sRNAs.
Figure 2.

Structure mapping of 5′-end-labeled sRNA162 and proposed secondary structure of sRNA162. (A) 5 nM 5′-end-labeled sRNA162 was subjected to RNase T1, lead(II) and RNase A cleavage. The cleavage was performed for 2, 3 or 5 min, respectively. Lane OH: alkaline hydrolysis laddar. Lane T1: RNase T1 laddar under denaturing conditions. The position of the cleaved Gs is given on the left of the gel. Lane C: untreated sRNA162. The approximate positions of stem-loop structures SL1, SL2 and SL3 according to the sRNA162 structure shown in (B) are depicted by vertical bars on the right of the gel. (B) Proposed secondary structure of sRNA162 determined by in vitro structure mapping. Cleavages according to (A) are indicated by circles for RNase T1 cleavage under both native and denaturing conditions, by squares for native RNase A cleavage and by black arrows for native lead(II) cleavage. The 3′ ends of the three small processed fragments of sRNA162 detected by northern blot analysis are indicated as well as the predicted interacting site with MM2441 (black bar). Processing sites (PS1-3) are indicated.
Homologs of RNase E and RNase III that are frequently involved in sRNA-mediated target regulation in bacteria (71–75) are not yet described in the archaeal domain. However, it is tempting to speculate that the increased turnover of sRNA162 into the stable 5′ fragments observed in stationary phase (Figure 1C and D) might be facilitated by orthologous archaeal RNases. This growth-phase dependent processing of sRNA162 leads to the accumulation of high amounts of stable 5′ fragments, indicating that processing may result in another (regulatory) outcome. However, ectopical overexpression of both, the longest 5′ fragment (65 nt) and the 3′ fragment of sRNA162 (including most of the predicted binding site) does not affect growth as observed upon overexpression of the full length sRNA162 (Figure 4A), strongly suggesting an essential (stabilizing) effect of the 5′ fragment on sRNA162, which appears crucial for functionality. Furthermore, it remains to be shown whether the resulting 5′ fragments of sRNA162 have additional regulatory functions.
MM2441 most likely participates in the metabolic switch from methanol to (tri)methylamine
In vivo evidence was obtained that sRNA162 is involved in the adaptation process in response to different C-sources (e.g. shift from methanol to trimethylamine). Several soluble methyltransferases involved in methanol and methylamine-driven methanogenesis have been demonstrated to be differentially expressed upon overexpression of sRNA162 but were not predicted as direct targets by bioinformatic predictions (see Supplementary Tables S3–S5). Based on our finding that sRNA162 most likely interferes with MM2441 translation, we hypothesize that the indirect down-stream effects on transcription of several soluble methyltransferase genes in response to sRNA162 overexpression results from the reduced synthesis of the ArsR-type transcriptional regulator MM2441. We further propose that during growth on methanol MM2441 represses transcription of the MM3334-3335 operon, encoding a monomethylamine-dependent methyltransferase (mtmB2) and the cognate corrinoid protein (mtmC2). However, other methyltransferase operons [e.g. MM1438-34 (mtmC1B1P1) and MM1687, 1693-94 (mtbC1B1)] are most likely activated by MM2441. In line with this, significantly elevated transcript levels for the mtmB2C2 operon and reduced transcript levels for mtmC1 and mtbC1 were detected by qRT-PCR in the sRNA162 overexpressing mutant (Figure 6), probable due to decreased amounts of MM2441. This change in methyltransferase expression patterns upon sRNA162 overexpression most likely results in faster growth adaptation after a switch from methanol to TMA (Figures 6 and Supplementary Figure S6).
Soluble methyltransferase genes involved in the degradation of methanol and methylamines are in general among the most highly regulated genes in methanoarchaea (46,55,76–78). Several studies clearly demonstrated growth phase and carbon- and energy source dependent regulation of the respective enzymes in M. acetivorans and M. mazei and several overlapping regulatory circuits (55,76–79). A number of putative transcriptional regulators have been discovered, which impact on expression of the corresponding genes (76). For some of those, a dual functional role has been predicted, i.e. they act as activators for transcription of several methyltransferase genes as well as repressors for others (76), which is in agreement with our findings.
Overall, sRNA162 appears to be involved in regulating the metabolic switch from methanol to methylamines during the transition of exponential to stationary growth phase, when cellular proteins and methylamines are degraded, most likely by post-transcriptional regulation of MM2441. We propose the following regulation (Figure 8): During exponential growth with methanol as carbon source, sRNA162 is constitutively transcribed and subsequently binds to mRNA MM2440-2441. Thereby, sRNA162 sequesters the translation initiation region (RBS and TLS) of MM2441, and controls its translation rates to ensure low MM2441 protein levels that are still sufficient to repress the mtmB2C2 operon expression. Upon binding to its target, sRNA162 might be simultaneously cleaved by RNases. However, in stationary growth phase the turnover of the sRNA162 primary transcript is reinforced by a yet unknown factor that reflects entering the stationary growth phase (or artificially by overexpressing sRNA162). This results in full translational repression of MM2441 and consequently de-repression of mtmB2C2 transcription as well as reduced transcription of other methyltransferase operons (e.g. mtmC1B1P1 and mtbC1B1) due to the absence of MM2441.
Figure 8.

Proposed model of the sRNA162 regulatory network. (A) sRNA162 is constitutively expressed during exponential growth with methanol as sole carbon and energy source. When sRNA162 is transcribed, it subsequently binds to mRNA MM2440-2441, sequestering the RBS and TLS of MM2441, thereby controlling translation rates to ensure constantly low MM2441 levels. The low MM2441 protein levels are sufficient to repress the mtmBC2 operon expression. When the turnover of the primary transcript of sRNA162 is reinforced (e.g. in stationary phase or by overexpressing sRNA162) by a yet unknown factor, this results in enhanced translational repression of MM2441. At the same time, transcription of mtmB2C2 is de-repressed by reduced MM2441 protein levels. The 5′- UTR of MM2442 encodes two potential small oligopeptides (sORF, black)). The regulatory outcome of the interaction with sRNA162 is unknown.
sRNA162 interacts with cis- and trans-encoded targets via two distinct domains
Despite acting as a trans-encoded antisense (as)RNA targeting MM2441, we further demonstrated that sRNA162 also represents a cis-acting asRNA. While the SLR domain is essential for the proposed interaction with MM2441, we provided evidence that sRNA162 targets the 5′-UTR of MM2442 by its very 5′ end (nt 1–65). Because the transcript level of MM2442 is slightly negatively affected by sRNA162 overexpression (qRT-PCR, Figure 6), translational repression and subsequent degradation of the transcript appears most likely, but transcriptional inference or promoter occlusion by sRNA162 is also possible. Upstream of the TLS of MM2442 (encoding a protein of so far unknown function) no RBS is present. A more detailed inspection of the long 5′-UTR of MM2442, further demonstrated that the 5′ UTR sequence contains two small (s)ORFs, potentially encoding an oligopeptide of 23 or 30 amino acids (Supplementary Figure S11). This might indicate a coupled translation of the sORF and MM2442, and consequently, sRNA162 could act by targeting an upstream sORF of the bicistronic mRNA. Since sRNA162 is masking the preceding RBS and CDS of the putative oligopeptide, it could lead to simultaneous translational repression of the sORF and MM2442. A similar mechanism has been described for RyhB sRNA in E. coli (5), which represses the translation of the ferric uptake regulator (Fur) by binding to the RBS of a translationally coupled small ORF (designated as ‘uORF′) upstream of the Fur CDS. Moreover, translational activation of downstream genes linked to a sORFs is mechanistically also possible, as exemplified by PhrS sRNA in Pseudomonas aeruginosa (80). The accumulation of the sRNA162 5′-fragments during stationary growth probably maintains or even enhances translational repression of MM2442 and the potential small peptide. However, ectopic overproduction of the 5′ fragment of sRNA162 did not result in an obvious phenotype. Therefore, the role of the sORF and MM2442 and the regulation by sRNA162 is still elusive and remains to be investigated.
Together these findings strongly suggest that sRNA162 acts on cis- and trans-encoded targets. Regulation of trans-encoded target mRNAs by cis-encoded asRNAs has been only proposed lacking any experimental evidence. Very recently Sayed et al. (81) described two RNAs organized as a type I toxin–antitoxin system consisting of SprA1, which encodes a small cytolytic peptide, and SprA1AS the cis-encoded riboregulator of SprA1 expression. Although SprA1AS overlaps with the 3′ end of SprA1 RNA, translation inhibition by SprA1 is achieved by a functional domain outside of the complementary target sequence by forming a pseudoknot (81). However, sRNA162, differs from this system as it is not encoded in the same genomic locus as its trans-encoded target mRNA, thus represents the first cis-encoded asRNA, additionally regulating a second target in trans.
In conclusion, we have demonstrated for the first time that an archaeal sRNA, sRNA162, has a regulatory function in archaea by interacting with the translation initiation region (RBS and / or TLS) of its trans-encoded target(s), indicating that this archaeal sRNA acts similar as its bacterial counterparts. Moreover, we obtained evidence that sRNA162 also targets a second, cis-encoded target. These obtained insights into the archaeal sRNA162 and its interaction with its targets blur the paradigm of a border between cis- and trans- encoded sRNAs.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–5 and Supplementary Figures 1–11.
FUNDING
Funding for open access charge: German Research Council (DFG) priority program (SPP) 1258 ‘Sensory and Regulatory sRNAs in Prokaryotes’ [Schm1052/9-1, Schm1052/9-2].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Jörg Vogel (University of Würzburg) for helpful comments and discussion on the manuscript. Further, they thank the members of their laboratory for useful discussions on the manuscript, as well as Claudia Kießling and Cornelia Goldberg for technical assistance.
REFERENCES
- 1.Carthew RW, Sontheimer EJ. Origins and Mechanisms of miRNAs and siRNAs. Cell. 2009;136:642–655. doi: 10.1016/j.cell.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Waters LS, Storz G. Regulatory RNAs in bacteria. Cell. 2009;136:615–628. doi: 10.1016/j.cell.2009.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Storz G, Vogel J, Wassarman KM. Regulation by small RNAs in bacteria: expanding frontiers. Mol. Cell. 2011;43:880–891. doi: 10.1016/j.molcel.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sharma CM, Darfeuille F, Plantinga TH, Vogel J. A small RNA regulates multiple ABC transporter mRNAs by targeting C/A-rich elements inside and upstream of ribosome-binding sites. Genes Dev. 2007;21:2804–2817. doi: 10.1101/gad.447207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vecerek B, Moll I, Blasi U. Control of Fur synthesis by the non-coding RNA RyhB and iron-responsive decoding. Embo J. 2007;26:965–975. doi: 10.1038/sj.emboj.7601553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pfeiffer V, Papenfort K, Lucchini S, Hinton JCD, Vogel J. Coding sequence targeting by MicC RNA reveals bacterial mRNA silencing downstream of translational initiation. Nat. Struct. Mol. Biol. 2009;16:840–846. doi: 10.1038/nsmb.1631. [DOI] [PubMed] [Google Scholar]
- 7.Desnoyers G, Morissette A, Prevost K, Masse E, Prévost K, Massé E. Small RNA-induced differential degradation of the polycistronic mRNA iscRSUA. EMBO J. 2009;28:1551–1561. doi: 10.1038/emboj.2009.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Darfeuille F, Unoson C, Vogel J, Wagner EGH. An antisense RNA inhibits translation by competing with standby ribosomes. Mol. Cell. 2007;26:381–392. doi: 10.1016/j.molcel.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 9.Bouvier M, Sharma CM, Mika F, Nierhaus KH, Vogel J. Small RNA binding to 5′ mRNA coding region inhibits translational initiation. Mol. Cell. 2008;32:827–837. doi: 10.1016/j.molcel.2008.10.027. [DOI] [PubMed] [Google Scholar]
- 10.Hammer BK, Bassler BL. Regulatory small RNAs circumvent the conventional quorum sensing pathway in pandemic Vibrio cholerae. Proc. Natl Acad. Sci. USA. 2007;104:11145–11149. doi: 10.1073/pnas.0703860104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Podkaminski D, Vogel J. Small RNAs promote mRNA stability to activate the synthesis of virulence factors. Mol. Microbiol. 2010;78:1327–1331. doi: 10.1111/j.1365-2958.2010.07428.x. [DOI] [PubMed] [Google Scholar]
- 12.Fröhlich KS, Vogel J. Activation of gene expression by small RNA. Curr. Opin. Microbiol. 2009;12:674–682. doi: 10.1016/j.mib.2009.09.009. [DOI] [PubMed] [Google Scholar]
- 13.Valentin-Hansen P, Eriksen M, Udesen C. The bacterial Sm-like protein Hfq: a key player in RNA transactions. Mol. Microbiol. 2004;51:1525–1533. doi: 10.1111/j.1365-2958.2003.03935.x. [DOI] [PubMed] [Google Scholar]
- 14.Aiba H. Mechanism of RNA silencing by Hfq-binding small RNAs. Curr. Opin. Microbiol. 2007;10:134–139. doi: 10.1016/j.mib.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 15.Brennan RG, Link TM. Hfq structure, function and ligand binding. Curr. Opin. Microbiol. 2007;10:125–133. doi: 10.1016/j.mib.2007.03.015. [DOI] [PubMed] [Google Scholar]
- 16.Papenfort K, Vogel J. Multiple target regulation by small noncoding RNAs rewires gene expression at the post-transcriptional level. Res. Microbiol. 2009;160:278–287. doi: 10.1016/j.resmic.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 17.Beisel CL, Storz G. Base pairing small RNAs and their roles in global regulatory networks. FEMS Microbiol. Rev. 2010;34:866–882. doi: 10.1111/j.1574-6976.2010.00241.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tomizawa J, Itoh T, Selzer G, Som T. Inhibition of ColE1 RNA primer formation by a plasmid-specified small RNA. Proc. Natl. Acad. Sci. USA. 1981;78:1421–1425. doi: 10.1073/pnas.78.3.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stougaard P, Molin S, Nordstrom K. RNAs involved in copy-number control and incompatibility of plasmid R1. Proc. Natl. Acad. Sci. USA. 1981;78:6008–6012. doi: 10.1073/pnas.78.10.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gerdes K, Wagner EGH. RNA antitoxins. Curr. Opin. Microbiol. 2007;10:117–124. doi: 10.1016/j.mib.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 21.Georg J, Hess WR. Cis-antisense RNA, another level of gene regulation in bacteria. Microbiol. Mol. Biol. Rev. 2011;75:286–300. doi: 10.1128/MMBR.00032-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomason MK, Storz G. Bacterial antisense RNAs: How many are there and what are they doing? Annu. Rev. Genet. 2010;44:167. doi: 10.1146/annurev-genet-102209-163523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharma CM, Hoffmann S, Darfeuille F, Reignier J, Findeiss S, Sittka A, Chabas S, Reiche K, Hackermüller J, Reinhardt R, et al. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature. 2010;464:250–255. doi: 10.1038/nature08756. [DOI] [PubMed] [Google Scholar]
- 24.Georg J, Voss B, Scholz I, Mitschke J, Wilde A, Hess WR. Evidence for a major role of antisense RNAs in cyanobacterial gene regulation. Mol. Syst. Biol. 2009;5:305. doi: 10.1038/msb.2009.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lasa I, Toledo-Arana A, Dobin A, Villanueva M, de los Mozos IR, Vergara-Irigaray M, Segura V, Fagegaltier D, Penadés JR, Valle J, et al. Genome-wide antisense transcription drives mRNA processing in bacteria. Proc. Natl Acad. Sci. USA. 2011;108:20172–20177. doi: 10.1073/pnas.1113521108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Toledo-Arana A, Dussurget O, Nikitas G, Sesto N, Guet-Revillet H, Balestrino D, Loh E, Gripenland J, Tiensuu T, Vaitkevicius K, et al. The Listeria transcriptional landscape from saprophytism to virulence. Nature. 2009;459:950–956. doi: 10.1038/nature08080. [DOI] [PubMed] [Google Scholar]
- 27.Mitschke J, Georg J, Scholz I, Sharma CM, Dienst D, Bantscheff J, Voss B, Steglich C, Wilde A, Vogel J, et al. An experimentally anchored map of transcriptional start sites in the model cyanobacterium Synechocystis sp. PCC6803. Proc. Natl Acad. Sci. USA. 2011;108:2124–2129. doi: 10.1073/pnas.1015154108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Omer AD, Lowe TM, Russell AG, Ebhardt H, Eddy SR, Dennis PP. Homologs of small nucleolar RNAs in Archaea. Science. 2000;288:517–522. doi: 10.1126/science.288.5465.517. [DOI] [PubMed] [Google Scholar]
- 29.Dennis PPP, Omer A, Lowe T. A guided tour: small RNA function in Archaea. Mol. Microbiol. 2001;40:509–519. doi: 10.1046/j.1365-2958.2001.02381.x. [DOI] [PubMed] [Google Scholar]
- 30.Rozhdestvensky TS, Tang TH, Tchirkova IV, Brosius J, Bachellerie JP, Huttenhofer A. Binding of L7Ae protein to the K-turn of archaeal snoRNAs: a shared RNA binding motif for C/D and H/ACA box snoRNAs in Archaea. Nucleic Acids Res. 2003;31:869–877. doi: 10.1093/nar/gkg175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weisel J, Wagner S, Klug G. The Nop5-L7A-fibrillarin RNP complex and a novel box C/D containing sRNA of Halobacterium salinarum NRC-1. Biochem. Biophys. Res. Commun. 2010;394:542–547. doi: 10.1016/j.bbrc.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 32.Tang TH, Rozhdestvensky TS, d’ Orval BC, Bortolin ML, Huber H, Charpentier B, Branlant C, Bachellerie JP, Brosius J, Huttenhofer A. RNomics in Archaea reveals a further link between splicing of archaeal introns and rRNA processing. Nucleic Acids Res. 2002;30:921–930. doi: 10.1093/nar/30.4.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tang TH, Bachellerie JP, Rozhdestvensky T, Bortolin ML, Huber H, Drungowski M, Elge T, Brosius J, Huttenhofer A. Identification of 86 candidates for small non-messenger RNAs from the archaeon Archaeoglobus fulgidus. Proc. Natl Acad. Sci. USA. 2002;99:7536–7541. doi: 10.1073/pnas.112047299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Straub J, Brenneis M, Jellen-Ritter A, Heyer R, Soppa J, Marchfelder A. Small RNAs in haloarchaea: identification, differential expression and biological function. RNA Biol. 2009;6:281–292. doi: 10.4161/rna.6.3.8357. [DOI] [PubMed] [Google Scholar]
- 35.Wurtzel O, Sapra R, Chen F, Zhu Y, Simmons Ba, Sorek R. A single-base resolution map of an archaeal transcriptome. Genome Res. 2009;20:133–141. doi: 10.1101/gr.100396.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jäger D, Sharma CM, Thomsen J, Ehlers C, Vogel J, Schmitz RA. Deep sequencing analysis of the Methanosarcina mazei Go1 transcriptome in response to nitrogen availability. Proc. Natl Acad. Sci. USA. 2009;106:21878–21882. doi: 10.1073/pnas.0909051106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ehlers C, Weidenbach K, Veit K, Deppenmeier U, Metcalf WW, Schmitz RA. Development of genetic methods and construction of a chromosomal glnK(1) mutant in Methanosarcina mazei strain Go1. Mol. Genet. Genomics. 2005;273:290–298. doi: 10.1007/s00438-005-1128-7. [DOI] [PubMed] [Google Scholar]
- 38.Metcalf WW, Zhang JK, Apolinario E, Sowers KR, Wolfe RS. A genetic system for Archaea of the genus Methanosarcina: liposome-mediated transformation and construction of shuttle vectors. Proc. Natl Acad. Sci. USA. 1997;94:2626–2631. doi: 10.1073/pnas.94.6.2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weidenbach K, Gloer J, Ehlers C, Sandman K, Reeve JN, Schmitz RA. Deletion of the archaeal histone in Methanosarcina mazei Go1 results in reduced growth and genomic transcription. Mol. Microbiol. 2008;67:662–671. doi: 10.1111/j.1365-2958.2007.06076.x. [DOI] [PubMed] [Google Scholar]
- 40.Gernhardt P, Possot O, Foglino M, Sibold L, Klein A. Construction of an integration vector for use in the archaebacterium Methanococcus voltae and expression of a eubacterial resistance gene. Mol. Gen. Genet. 1990;221:273–279. doi: 10.1007/BF00261731. [DOI] [PubMed] [Google Scholar]
- 41.Veit K, Ehlers C, Ehrenreich A, Salmon K, Hovey R, Gunsalus RP, Deppenmeier U, Schmitz RA. Global transcriptional analysis of Methanosarcina mazei strain Go1 under different nitrogen availabilities. Mol. Genet. Genomics. 2006;276:41–55. doi: 10.1007/s00438-006-0117-9. [DOI] [PubMed] [Google Scholar]
- 42.Weidenbach K, Ehlers C, Kock J, Ehrenreich A, Schmitz RA. Insights into the NrpR regulon in Methanosarcina mazei Go1. Arch. Microbiol. 2008;190:319–332. doi: 10.1007/s00203-008-0369-3. [DOI] [PubMed] [Google Scholar]
- 43.Holmqvist E, Reimegard J, Sterk M, Grantcharova N, Romling U, Wagner EGH, Reimegård J, Römling U. Two antisense RNAs target the transcriptional regulator CsgD to inhibit curli synthesis. EMBO J. 2010;29:1840–1850. doi: 10.1038/emboj.2010.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fechter P, Rudinger J, Giegé R, Théobald-Dietrich a. Ribozyme processed tRNA transcripts with unfriendly internal promoter for T7 RNA polymerase: production and activity. FEBS Lett. 1998;436:99–103. doi: 10.1016/s0014-5793(98)01096-5. [DOI] [PubMed] [Google Scholar]
- 45.Hovey R, Lentes S, Ehrenreich A, Salmon K, Saba K, Gottschalk G, Gunsalus RP, Deppenmeier U. DNA microarray analysis of Methanosarcina mazei Go1 reveals adaptation to different methanogenic substrates. Mol. Genet. Genomics. 2005;273:225–239. doi: 10.1007/s00438-005-1126-9. [DOI] [PubMed] [Google Scholar]
- 46.Veit K, Ehlers C, Schmitz RA. Effects of nitrogen and carbon sources on transcription of soluble methyltransferases in Methanosarcina mazei strain Gö1. J. Bacteriol. 2005;187:6147–6154. doi: 10.1128/JB.187.17.6147-6154.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Talaat AM, Lyons R, Howard ST, Johnston SA. The temporal expression profile of Mycobacterium tuberculosis infection in mice. Proc. Natl. Acad. Sci. USA. 2004;101:4602–4607. doi: 10.1073/pnas.0306023101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Starmer J, Stomp A, Vouk M, Bitzer D. Predicting Shine-Dalgarno sequence locations exposes genome annotation errors. PLoS Comput. Biol. 2006;2:e57. doi: 10.1371/journal.pcbi.0020057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Busch A, Richter AS, Backofen R. IntaRNA: efficient prediction of bacterial sRNA targets incorporating target site accessibility and seed regions. Bioinformatics. 2008;24:2849–2856. doi: 10.1093/bioinformatics/btn544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Richter AS, Schleberger C, Backofen R, Steglich C. Seed-based INTARNA prediction combined with GFP-reporter system identifies mRNA targets of the small RNA Yfr1. Bioinformatics. 2010;26:1–5. doi: 10.1093/bioinformatics/btp609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huttenhofer A, Noller HF. Footprinting mRNA-ribosome complexes with chemical probes. EMBO J. 1994;13:3892–3901. doi: 10.1002/j.1460-2075.1994.tb06700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Heyne S, Will S, Beckstette M, Backofen R. Lightweight comparison of RNAs based on exact sequence-structure matches. Bioinformatics. 2009;25:2095–2102. doi: 10.1093/bioinformatics/btp065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bernhart SH, Hofacker IL, Will S, Gruber AR, Stadler PF. RNAalifold: improved consensus structure prediction for RNA alignments. BMC Bioinformatics. 2008;9:474. doi: 10.1186/1471-2105-9-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Deppenmeier U, Johann A, Hartsch T, Merkl R, Schmitz RA, Martinez-Arias R, Henne A, Wiezer A, Baumer S, Jacobi C, et al. The genome of Methanosarcina mazei: evidence for lateral gene transfer between bacteria and archaea. J. Mol. Microbiol. Biotechnol. 2002;4:453–461. [PubMed] [Google Scholar]
- 55.Krätzer C, Carini P, Hovey R, Deppenmeier U, Kratzer C. Transcriptional profiling of methyltransferase genes during growth of Methanosarcina mazei on trimethylamine. J. Bacteriol. 2009;191:5108–5115. doi: 10.1128/JB.00420-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brenneis M, Soppa J. Regulation of translation in haloarchaea: 5′- and 3′-UTRs are essential and have to functionally interact in vivo. PLoS One. 2009;4:e4484. doi: 10.1371/journal.pone.0004484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Phok K, Moisan A, Rinaldi D, Brucato N, Carpousis AJ, Gaspin C, Clouet-d’Orval B. Identification of CRISPR and riboswitch related RNAs among novel noncoding RNAs of the euryarchaeon Pyrococcus abyssi. BMC Genomics. 2011;12:312. doi: 10.1186/1471-2164-12-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Klein RJ, Misulovin Z, Eddy SR. Noncoding RNA genes identified in AT-rich hyperthermophiles. Proc. Natl Acad. Sci. USA. 2002;99:7542–7547. doi: 10.1073/pnas.112063799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Starostina NG, Marshburn S, Johnson LS, Eddy SR, Terns RM, Terns MP. Circular box C/D RNAs in Pyrococcus furiosus. Proc. Natl Acad. Sci. USA. 2004;101:14097–14101. doi: 10.1073/pnas.0403520101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Speckmann WA, Li Z, Lowe TM, Eddy SR, Terns RM, Terns MP. Archaeal guide RNAs function in rRNA modification in the eukaryotic nucleus. Curr. Biol. 2002;12:199–203. doi: 10.1016/s0960-9822(02)00655-3. [DOI] [PubMed] [Google Scholar]
- 61.Gaspin C, Cavaille J, Erauso G, Bachellerie JP, Cavaillé J. Archaeal homologs of eukaryotic methylation guide small nucleolar RNAs: lessons from the Pyrococcus genomes. J. Mol. Biol. 2000;297:895–906. doi: 10.1006/jmbi.2000.3593. [DOI] [PubMed] [Google Scholar]
- 62.Danan M, Schwartz S, Edelheit S, Sorek R. Transcriptome-wide discovery of circular RNAs in Archaea. Nucleic Acids Res. 2011;40:3131–3142. doi: 10.1093/nar/gkr1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Soppa J, Straub J, Brenneis M, Jellen-Ritter A, Heyer R, Fischer S, Granzow M, Voss B, Hess WR, Tjaden B, et al. Small RNAs of the halophilic archaeon Haloferax volcanii. Biochem. Soc. Trans. 2009;37:133–136. doi: 10.1042/BST0370133. [DOI] [PubMed] [Google Scholar]
- 64.Langer D, Hain J, Thuriaux P, Zillig W. Transcription in archaea: similarity to that in eucarya. Proc. Natl Acad. Sci. USA. 1995;92:5768–5772. doi: 10.1073/pnas.92.13.5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Geiduschek EP, Ouhammouch M. Archaeal transcription and its regulators. Mol. Microbiol. 2005;56:1397–1407. doi: 10.1111/j.1365-2958.2005.04627.x. [DOI] [PubMed] [Google Scholar]
- 66.Brinkman AB, Dahlke I, Tuininga JE, Lammers T, Dumay V, de Heus E, Lebbink JH, Thomm M, de Vos WM, van Der Oost J. An Lrp-like transcriptional regulator from the archaeon Pyrococcus furiosus is negatively autoregulated. J. Biol. Chem. 2000;275:38160–38169. doi: 10.1074/jbc.M005916200. [DOI] [PubMed] [Google Scholar]
- 67.Bell SD. Archaeal transcriptional regulation—variation on a bacterial theme? Trends Microbiol. 2005;13:262–265. doi: 10.1016/j.tim.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 68.Garrett RA, Klenk H-P. Archaea. Evolution, Physiology, and Molecular Biology. Oxford, UK: Blackwell Publishing; 2007. [Google Scholar]
- 69.Pulvermacher SC, Stauffer LT, Stauffer GV. The role of the small regulatory RNA GcvB in GcvB/mRNA posttranscriptional regulation of oppA and dppA in Escherichia coli. FEMS Microbiol. Lett. 2008;281:42–50. doi: 10.1111/j.1574-6968.2008.01068.x. [DOI] [PubMed] [Google Scholar]
- 70.Papenfort K, Pfeiffer V, Mika F, Lucchini S, Hinton JC, Vogel J. SigmaE-dependent small RNAs of Salmonella respond to membrane stress by accelerating global omp mRNA decay. Mol. Microbiol. 2006;62:1674–1688. doi: 10.1111/j.1365-2958.2006.05524.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Afonyushkin T, Vecerek B, Moll I, Blasi U, Kaberdin VR, Bläsi U. Both RNase E and RNase III control the stability of sodB mRNA upon translational inhibition by the small regulatory RNA RyhB. Nucleic Acids Res. 2005;33:1678–1689. doi: 10.1093/nar/gki313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Morita T, Maki K, Aiba H. RNase E-based ribonucleoprotein complexes: mechanical basis of mRNA destabilization mediated by bacterial noncoding RNAs. Genes Dev. 2005;19:2176–2186. doi: 10.1101/gad.1330405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ikeda Y, Yagi M, Morita T, Aiba H. Hfq binding at RhlB-recognition region of RNase E is crucial for the rapid degradation of target mRNAs mediated by sRNAs in Escherichia coli. Mol. Microbiol. 2011;79:419–432. doi: 10.1111/j.1365-2958.2010.07454.x. [DOI] [PubMed] [Google Scholar]
- 74.Opdyke JA, Fozo EM, Hemm MR, Storz G. RNase III participates in GadY-dependent cleavage of the gadX-gadW mRNA. J. Mol. Biol. 2011;406:29–43. doi: 10.1016/j.jmb.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Condon C, Putzer H. The phylogenetic distribution of bacterial ribonucleases. Nucleic Acids Res. 2002;30:5339–5346. doi: 10.1093/nar/gkf691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bose A, Metcalf WW. Distinct regulators control the expression of methanol methyltransferase isozymes in Methanosarcina acetivorans C2A. Mol. Microbiol. 2008;67:649–661. doi: 10.1111/j.1365-2958.2007.06075.x. [DOI] [PubMed] [Google Scholar]
- 77.Bose A, Pritchett MA, Metcalf WW. Genetic analysis of the methanol- and methylamine-specific methyltransferase 2 genes of Methanosarcina acetivorans C2A. J. Bacteriol. 2008;190:4017–4026. doi: 10.1128/JB.00117-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Opulencia RB, Bose A, Metcalf WW. Physiology and posttranscriptional regulation of methanol:coenzyme M methyltransferase isozymes in Methanosarcina acetivorans C2A. J. Bacteriol. 2009;191:6928–6935. doi: 10.1128/JB.00947-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bose A, Pritchett MA, Rother M, Metcalf WW. Differential regulation of the three methanol methyltransferase isozymes in Methanosarcina acetivorans C2A. J. Bacteriol. 2006;188:7274–7283. doi: 10.1128/JB.00535-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sonnleitner E, Gonzalez N, Sorger-Domenigg T, Heeb S, Richter AS, Backofen R, Williams P, Hüttenhofer A, Haas D, Bläsi U. The small RNA PhrS stimulates synthesis of the Pseudomonas aeruginosa quinolone signal. Mol. Microbiol. 2011;80:868–885. doi: 10.1111/j.1365-2958.2011.07620.x. [DOI] [PubMed] [Google Scholar]
- 81.Sayed N, Jousselin A, Felden B. A cis-antisense RNA acts in trans in Staphylococcus aureus to control translation of a human cytolytic peptide. Nat. Struct. Mol. Biol. 2011;19:105–112. doi: 10.1038/nsmb.2193. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.