

Abstract
Transcriptional coactivator with PDZ-binding motif (TAZ) physically interacts with a variety of transcription factors and modulates their activities involved in cell proliferation and mesenchymal stem cell differentiation. TAZ is highly expressed in the kidney, and a deficiency of this protein results in multiple renal cysts and urinary concentration defects; however, the molecular functions of TAZ in renal cells remain largely unknown. In this study, we examined the effects of osmotic stress on TAZ expression and activity in renal cells. We found that hyperosmotic stress selectively increased protein phosphorylation at tyrosine 316 of TAZ and that this was enhanced by c-Abl activation in response to hyperosmotic stress. Interestingly, phosphorylated TAZ physically interacted with nuclear factor of activated T cells 5 (NFAT5), a major osmoregulatory transcription factor, and subsequently suppressed DNA binding and transcriptional activity of NFAT5. Furthermore, TAZ deficiency elicited an increase in NFAT5 activity in vitro and in vivo, which then reverted to basal levels following restoration of wild-type TAZ but not mutant TAZ (Y316F). Collectively, the data suggest that TAZ modulates cellular responses to hyperosmotic stress through fine-tuning of NFAT5 activity via tyrosine phosphorylation.
INTRODUCTION
Transcriptional coactivator with PDZ-binding motif (TAZ), also known as WW domain-containing transcription regulator 1 (WWTR1), was first identified as a 14-3-3-interacting phosphoprotein (16). Phosphorylation of TAZ at serine 89 is required for its interaction with 14-3-3 and for substantial retention in the cytoplasm (16). In addition to the 14-3-3-binding motif, TAZ contains multiple functional domains, including a WW domain, a coiled-coil domain, and a PDZ-binding motif, through which it modulates the activity of transcription factors involved in stem cell differentiation (13). For example, TAZ interacts with runt-related transcription factor 2 (RUNX2) and MyoD during osteoblast and myogenic differentiation and promotes cell lineage-specific gene transcription (6, 13, 15). During adipogenic differentiation, TAZ suppresses the activity of peroxisome proliferator-activated receptor (PPAR), thus inhibiting adipogenic differentiation from mesenchymal stem cells (13). TAZ also controls organ development, including thyroid, cardiac, and limb development, by association with thyroid transcription factor 1 (TTF-1), paired box gene 8 (PAX8), and PAX3 (7, 28–30). Furthermore, TAZ promotes cell proliferation through activation of TEAD transcription factors, resulting in tumor cell growth and epithelial-mesenchymal transition (4, 5, 19, 22, 37). Much of our understanding of the physiological function of TAZ in vivo has been derived from studies using TAZ knockout (KO) mice, which develop significant pathogenic phenotypes, such as lung emphysema and multiple kidney cysts (14, 24). Although the pathogenic mechanisms at play in TAZ KO mice largely remain to be explored, TAZ is known to suppress the expression and activity of the transmembrane protein polycystin 2 (PC2) in the kidney (8, 33). Increased PC2 expression in TAZ KO mice may be involved in the development of polycystic kidney disease (33). In addition, TAZ interacts with the transcription factor Glis-3 and enhances its transcriptional activity. It is evident that deficiency of either TAZ or Glis-3 in mice leads to abnormal cilium formation and to polycystic kidneys (17). Collectively, these studies suggest that TAZ plays critical roles in normal kidney development and function through different mechanisms.
Since hyperosmolar medullary interstitial fluid is essential for urinary concentration in the kidney, renal medullary cells are normally exposed to extracellular hyperosmotic stress, which can cause cell shrinkage, DNA damage, and cell apoptosis. To avoid hyperosmotic stress-induced damage, renal medullary cells exhibit rapid adjustment via a complex network of osmoprotective molecules, including kinases, heat shock proteins, p53, and osmolyte-accumulating genes (2). The osmolyte-accumulating genes include those encoding the betaine-GABA transporter (BGT1) (35) and the sodium/myo-inositol cotransporter (SMIT) (34, 36). Induction of these genes results in organic osmolyte accumulation, which prevents acute hyperosmotic stress-induced water loss and cell shrinkage. Expression of BGT1 and SMIT is transcriptionally controlled by the nuclear factor of activated T cells 5 (NFAT5)/tonicity-responsive enhancer-binding protein (TonEBP), referred to here as NFAT5 (10, 26, 27). NFAT5 deficiency blocks the expression of osmolyte-accumulating genes in the kidney medulla and produces a severe urinary concentration defect in mice, suggesting a critical role for NFAT5 in the hyperosmotic stress signaling pathway (21). However, recent studies indicate that NFAT5 is activated by various tonicity-independent mechanisms and exhibits tissue-specific functions, such as cell migration, vascular remodeling, and carcinoma invasion (12). Although the hyperosmolarity-dependent and -independent activation and functions of NFAT5 have been characterized extensively for numerous cell types, mechanisms underlying the suppression of NFAT5 activity remain unclear.
In this study, we investigated the effects of hyperosmotic stress on TAZ activity and its relationship with NFAT5 function in renal medullary cells. Interestingly, TAZ underwent tyrosine phosphorylation in response to hyperosmotic stress and specifically associated with NFAT5, suppressing its DNA-binding activity and transcriptional activity. Our results suggest that TAZ functions as a modulator of NFAT activity in response to hyperosmotic stress.
MATERIALS AND METHODS
Reagents.
Antibodies (Abs) against phosphorylated serine 89 of TAZ (pS89), TAZ, Oct1, β-actin, phospho-Abl (pAbl), phosphotyrosine (pY; 4G10), c-Abl, and NFAT5 were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA), Cell Signaling Technology (Boston, MA), Upstate Biotechnology Inc. (Lake Placid, NY), and Abcam Inc. (Cambridge, MA). Flag Ab and Flag-M2 agarose beads were purchased from Sigma-Aldrich (St. Louis, MO). Recombinant c-Abl kinase and imatinib (STI-571) were obtained from Calbiochem (San Diego, CA) and Novartis Pharmaceuticals (Basel, Switzerland), respectively. Abs recognizing phosphotyrosine (pY118, pY141, pY300, and pY316) were generated by AbFrontier Inc. (Seoul, Republic of Korea).
Animals.
Wild-type (WT) C57BL/6 mice were purchased from Jackson Laboratories (Bar Harbor, ME). WT and TAZ KO mice were maintained under specific-pathogen-free conditions. All animal experiments were performed in accordance with IACUC guidelines and were approved by the IACUC at Ewha Womans University (protocols ELAGC-09-1017 and IACUC 2010-13-3).
Cell culture.
Cos7 and HEK293T cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and cultured in Dulbecco's modified Eagle medium (DMEM; Invitrogen, Carlsbad, CA). Cells were transiently transfected by calcium phosphate transfection methods. Tonicity-responsive immortalized mouse inner medullary collecting duct 3 cells (mIMCD-3 cells; ATCC) and mouse embryonic fibroblasts (MEF) of WT and TAZ KO mice were maintained in DMEM supplemented with 10% heat-inactivated fetal bovine serum (FBS; HyClone, Logan, UT). Cells were exposed to low-salt, normal, or hyperosmolar medium (final osmolarity, 150, 300, or 400 mosmol/kg H2O) for 2 to 4 h by use of NaCl. For hyperosmotic stimulation, cells were exposed to low-salt (150 mosmol/kg H2O) conditions for 2 h and subsequently cultured in normal or hyperosmolar medium for 4 h.
In situ PLA.
Renal mIMCD-3 cells were plated onto a glass slide and incubated in normal or hyperosmotic medium for 4 h. Cells were fixed and incubated with mouse anti-TAZ Ab (1:100; Abcam) and rabbit anti-NFAT5 Ab (27). The in situ proximity ligation assay (PLA) was performed according to the manufacturer's description (Duolink; Olink Bioscience, Uppsala, Sweden). Fluorescence was then examined by confocal fluorescence microscopy (LSM510 Meta; Carl Zeiss Inc., Germany).
Reporter gene assays.
293T cells were plated in a 6-well plate at 5 × 105 cells/well and transiently transfected with NFAT5 and TAZ expression vectors and an NFAT5/TonEBP-responsive element-linked luciferase vector (pTonE-luc), as well as with pCMVβgal as a transfection control. Luciferase activity was assayed using a Bright-Glo luciferase assay kit (Promega, Madison, WI). Relative luciferase units were calculated after normalization with β-galactosidase activity (Tropix, Bedford, MA).
Real-time PCR analysis.
Total RNA was isolated from cells by use of TRIzol (Gibco-BRL, Invitrogen) and used for reverse transcription for cDNA synthesis (Invitrogen). Real-time PCRs were performed with SYBR green premix buffer and an ABI Prism 7300 sequence detector (Perkin-Elmer Applied Biosystems, Foster City, CA). Relative expression levels were determined after normalization to the threshold cycle (CT) values for β-actin. Gene-specific primers were as follows: for BGT1, 5′-CTGGGAGAGACGGGTTTTGGGTATTACATC-3′ and 5′-GGACCCCAGGTCGTGGAT-3′; for SMIT, 5′-CCGGGCGCTCTATGACCTGGG-3′ and 5′-CAAACAGAGAGGCACCAATCG-3′; and for β-actin, 5′-AGAGGGAAATCGTGCGTGAC-3′ and 5′-CAATAGTGATGACCTGGCCGT-3′.
DNA pulldown assay.
Cells were lysed with HKMG buffer (10 mM HEPES, pH 7.9, 100 mM KCl, 5 mM MgCl2, 10% glycerol, 0.1% NP-40, and 1 mM dithiothreitol [DTT]) and incubated with biotinylated double-stranded DNA and subsequently with streptavidin-agarose beads. Precipitates were washed with HKMG buffer three times and applied to SDS-PAGE gels for immunoblot assay. The DNA sequences containing the NFAT5/TonEBP-binding element were as follows: 5′-CTTGGTGGAAAATTACCGCTGGT-3′ and 5′-AACCAGCGGTCCTTTTCCACCAA-3′.
Chromatin immunoprecipitation (ChIP) and quantitative real-time PCR.
Cells were formaldehyde cross-linked in a 1% solution for 10 min as described previously (15). Chromatin extracts were prepared and incubated with anti-NFAT5 Ab at 4°C overnight, followed by incubation with protein A/G-agarose beads (Pierce, Rockford, IL). Immune complexes were sequentially washed and then heated to reverse the formaldehyde cross-linking of the protein and DNA complexes. The DNA fragments were purified using a QIAquick PCR purification kit (Qiagen, Hilden, Germany) and then used for PCR. Primers used were as follows: SMIT-ChIP-F, 5′-TTGCGGCTTGGTGCAGCAGTC-3′; and SMIT-ChIP-R, 5′-TCGCTCGGCACGCACCCGCTC-3′.
Establishment of stable cell lines by retroviral transduction.
Virus-producing Phoenix cells were cultured in DMEM supplemented with 10% FBS and transfected with the TAZ-expressing retroviral vector by the calcium phosphate transfection method. Viral supernatants were harvested and added to mIMCD-3 or MEF cells in the presence of Polybrene (4 μg/ml; Sigma-Aldrich) for 24 h. Stable cells were established by puromycin resistance (Sigma-Aldrich).
Statistical analysis.
All data are expressed as means ± standard errors of the means (SEM). The statistical significance of the experimental results was calculated by unpaired Student's t test. Values of <0.05 were considered statistically significant (*, P < 0.05; **, P < 0.005; and ***, P < 0.0005).
RESULTS
Hyperosmotic stress induces tyrosine phosphorylation of TAZ through c-Abl activation.
In order to examine the effects of osmotic stress on TAZ expression and activity, mouse renal medullary cells, i.e., mIMCD-3 cells, were cultured under normal (300 mosmol/kg) and hyperosmotic (400 mosmol/kg) conditions. Hyperosmotic stimulation of mIMCD-3 cells had no effect on either expression of TAZ or phosphorylation of TAZ at serine 89 compared to that under normal conditions. However, phosphorylation of TAZ on tyrosine residues was strikingly elevated under hyperosmotic stress (observed using Ab 4G10) (Fig. 1A). This observation was confirmed by the finding that tyrosine phosphorylation of ectopically expressed TAZ proteins was selectively enhanced by hyperosmolarity (Fig. 1B). Interestingly, endogenous c-Abl tyrosine kinase was activated by hyperosmotic stimulation, as demonstrated by the increased detection of phosphorylated but not total c-Abl in response to hyperosmotic stress (Fig. 1C). To determine whether c-Abl was an upstream tyrosine kinase for TAZ, we performed an in vitro kinase assay using recombinant c-Abl kinase. Immunoprecipitated TAZ protein was directly phosphorylated by c-Abl in a cell-free system (Fig. 1D). Furthermore, coexpression of c-Abl with TAZ in 293T cells enhanced the tyrosine phosphorylation of TAZ (Fig. 1E). In contrast, Abl knockdown abolished TAZ phosphorylation induced by hyperosmotic stimulation (Fig. 1F). Furthermore, the c-Abl-induced tyrosine phosphorylation was inhibited by genistein, a nonselective tyrosine kinase inhibitor, and by STI-571, a c-Abl kinase-specific inhibitor (Fig. 1G). Hyperosmotic stress also promoted tyrosine phosphorylation of the endogenous TAZ protein along with c-Abl activation, but it failed to induce tyrosine phosphorylation of TAZ in the presence of STI-571 (Fig. 1H).
Fig 1.

Tyrosine phosphorylation of TAZ by hyperosmotic stress-induced c-Abl activation. (A) mIMCD-3 cells were exposed to 300 and 400 mosmol/kg NaCl for 4 h. Whole-cell extracts (WCE) were immunoprecipitated (IP) with anti-TAZ Ab, and immune complexes were immunoblotted (IB) with Abs against phosphotyrosine (4G10), phosphoserine TAZ (pS89), and TAZ. (B) Stable mIMCD-3 cells (mock and Flag-TAZ) were cultured under low-salt (150 mosmol/kg), normal (300 mosmol/kg), and hyperosmotic (400 mosmol/kg) conditions for 4 h and then incubated with Flag-M2 beads. TAZ immune complexes were analyzed by immunoblotting with 4G10 and anti-TAZ Abs. (C) Total proteins were extracted from mIMCD-3 cells for immunoblotting of phosphorylated Abl (pAbl), Abl, and actin. (D) Purified Flag-tagged TAZ was incubated with recombinant c-Abl kinase (rAbl) in the presence of radiolabeled ATP. The reaction mixture was then analyzed by SDS-PAGE and radiography. Separately, the TAZ level was assessed by immunoblotting. (E) 293T cells were transfected with Flag-TAZ, with (+) or without (−) the c-Abl expression vector. TAZ immune complexes were resolved by SDS-PAGE, followed by immunoblotting with 4G10. (F) mIMCD-3 cells were infected with viruses expressing pSRP-Abl (siAbl), and whole-cell extracts were used for TAZ immunoprecipitation, followed by immunoblotting with 4G10. (G) 293T cells were transfected with Flag-TAZ and c-Abl expression vectors and subsequently cultured in the presence of genistein (GNS; 50 μM) or STI-571 (STI; 10 μM) for 1 h. TAZ immune complexes were analyzed by immunoblotting. (H) mIMCD-3 cells were stimulated with hyperosmotic stress in the presence or absence of STI (10 μM). TAZ immune complexes were subsequently analyzed with 4G10 and anti-TAZ Abs.
Tyrosine 316 of TAZ is the c-Abl phosphorylation site.
We next attempted to identify the c-Abl phosphorylation sites in TAZ. Since TAZ contains 4 tyrosine residues (Y118, Y141, Y300, and Y316), we first generated TAZ-specific phosphotyrosine Abs (pY118, pY141, pY300, and pY316) by using synthetic TAZ peptides containing the appropriate phosphotyrosines. The c-Abl-induced tyrosine phosphorylation of TAZ was prominently detected by 4G10 and by immunoblotting with the pY316 TAZ Ab but not by immunoblotting with the other Abs (Fig. 2A). We also confirmed that hyperosmotic stress enhanced the tyrosine phosphorylation of endogenous TAZ selectively on Y316 (Fig. 2B). We next constructed tyrosine mutants of TAZ (Y118F, Y141F, Y300F, and Y316F) and performed the in vitro Abl kinase assay using purified TAZ proteins. TAZ proteins, including the WT and the Y118F, Y141F, and Y300F mutants, were phosphorylated by c-Abl, but Abl phosphorylation was not observed in the Y316F mutant (Fig. 2C). Overexpression of c-Abl with WT and Y316F TAZ also increased the phosphorylation of WT TAZ but failed to phosphorylate the Y316F mutant, as demonstrated by immunoblotting with the 4G10 and pY316 TAZ Abs (Fig. 2D). In addition, stable mIMCD-3 cells expressing Flag-tagged WT TAZ and its tyrosine mutants were established and exposed to hyperosmotic stress. Hyperosmotic stimulation induced tyrosine phosphorylation in WT, Y118F, Y141F, and Y300F TAZ proteins, but this hyperosmotic stress-induced phosphorylation was impaired in the Y316F mutant (Fig. 2E), indicating that tyrosine 316 of TAZ is the c-Abl phosphorylation site.
Fig 2.
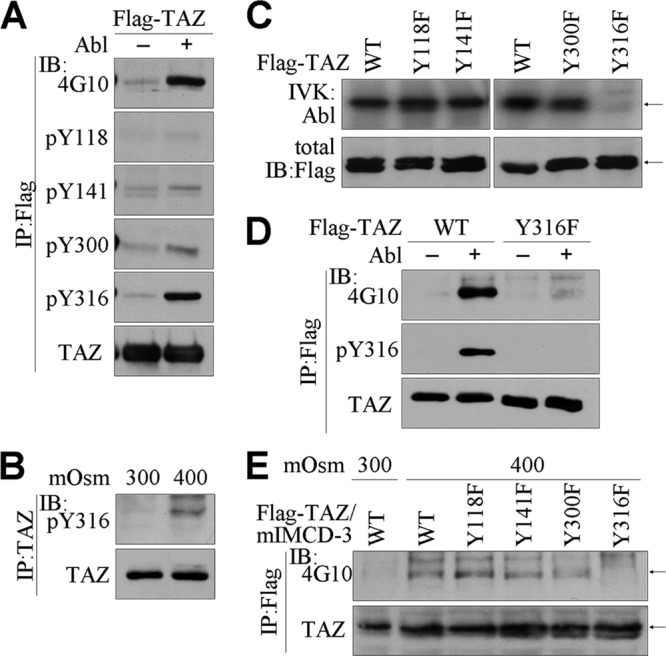
c-Abl-induced phosphorylation of TAZ at tyrosine 316. (A) 293T cells were transfected with Flag-TAZ and c-Abl expression vectors. TAZ immune complexes were resolved by SDS-PAGE and immunoblotted with 4G10 and Abs against pY116, pY141, pY300, pY316 TAZ, and TAZ. (B) Parental mIMCD-3 cells were incubated with hyperosmolar medium for 4 h. Endogenous TAZ protein was immunoprecipitated and immunoblotted with pY316 TAZ or TAZ Ab. (C) Purified TAZ proteins were incubated with recombinant c-Abl kinase in the presence of radiolabeled ATP. The reaction mixture was analyzed by SDS-PAGE and radiography. (D) 293T cells were transfected with WT TAZ and Y316F TAZ, with or without the c-Abl expression vector. Immune complexes were resolved and immunoblotted with 4G10, pY316 TAZ, and TAZ Abs. (E) mIMCD-3 cells were stably transfected with the TAZ expression vector, and stable cells were selected in the presence of puromycin. The cells were incubated under hyperosmotic stress for 4 h, followed by immunoprecipitation and immunoblot analysis.
TAZ physically interacts with NFAT5 via hyperosmotic stress-induced phosphorylation.
To identify the functions of phosphorylated TAZ in the hyperosmotic stress-mediated signaling pathway, we examined the relationship between TAZ and NFAT5, a key regulator of the hyperosmotic stress signaling pathway (3, 27). To determine the interaction between TAZ and NFAT5, we transiently expressed Flag-TAZ and NFAT5 together with c-Abl in 293T cells. Interestingly, coimmunoprecipitation and immunoblotting analysis demonstrated a physical interaction between TAZ and NFAT5 (Fig. 3A). However, Y316F mutant TAZ was incapable of interacting with NFAT5 (Fig. 3A). In addition, treatment of cells with STI-571 reduced the physical interaction between TAZ and NFAT5 and diminished the tyrosine phosphorylation of TAZ (Fig. 3B), indicating a tyrosine phosphorylation-dependent interaction of TAZ with NFAT5. Since NFAT5 is also phosphorylated at tyrosine 143 by c-Abl (11), we tested whether tyrosine phosphorylation of NFAT5 was also essential for its interaction with TAZ. The interaction of NFAT5 with TAZ was sustained in Y143F mutant NFAT5, as indicated by the comparable presence of WT and mutant NFAT5 in TAZ immune complexes (Fig. 3C). Most importantly, analysis of endogenous TAZ immune complexes verified that TAZ interacted with NFAT5 under normal conditions and interacted more strongly under hyperosmotic stress, in a tyrosine phosphorylation-dependent manner (Fig. 3D). However, treatment of cells with STI-571 attenuated the interaction between TAZ and NFAT5 under hypertonic conditions (Fig. 3E). Furthermore, the in situ PLA system visualized the individual TAZ-NFAT5 interaction in the nuclei of mIMCD-3 cells cultured in normal or hyperosmolar medium but not in those cultured in low-salt (150 mosmol/kg) medium (Fig. 3F).
Fig 3.

Physical interaction between phosphorylated TAZ and NFAT5. (A to C) 293T cells were transfected with c-Abl, Myc-tagged WT or mutated (Y143F or MT) NFAT5, and Flag-tagged WT or Y316F TAZ, as indicated above each panel. (A) NFAT5 immune complexes were precipitated by incubation using anti-Myc Ab and analyzed by SDS-PAGE and immunoblotting. (B) Cells were additionally treated with STI-571 (10 μM) for 1 h before harvest. TAZ immune complexes were analyzed by immunoblotting with Abs against Myc, pY316, and TAZ. (C) TAZ immune complexes were resolved by SDS-PAGE and subjected to immunoblotting. (D to F) mIMCD-3 cells were stimulated with different tonicities for 4 h. (D) Endogenous TAZ immune complexes were precipitated with TAZ Ab, followed by SDS-PAGE and immunoblotting with NFAT5 and pY316 Abs. (E) Separately, mIMCD-3 cells were cultured either in the presence or in the absence of STI, and TAZ immune complexes were immunoblotted with NFAT Ab. (F) Tonicity-stimulated mIMCD-3 cells were fixed and incubated with TAZ and NFAT5 Abs, followed by incubation with specific secondary Abs with PLA probes, using a Duolink in situ PLA kit. Samples were observed under a confocal microscope, and the number of spots per cell was measured. nd, not determined.
TAZ suppresses DNA-binding and transcriptional activities of NFAT5.
Next, we determined the effects of the TAZ-NFAT5 interaction on NFAT5 activity. Transcriptional activity of NFAT5 was assessed with an NFAT5/TonEBP-responsive luciferase reporter gene (pTonE-luc) in the presence or absence of TAZ. Ectopic expression of NFAT5 increased the luciferase activity of pTonE-luc, but coexpression of TAZ significantly suppressed this activity, in a dose-dependent manner (Fig. 4A). Furthermore, the NFAT5-induced reporter activity was inhibited by overexpression of WT or Y118F TAZ, whereas Y316F TAZ did not suppress NFAT5 activity (Fig. 4B), consistent with the inability of Y316F TAZ to interact with NFAT5. The TAZ-mediated suppression of NFAT5 transcriptional activity led us to examine the DNA-binding activity of NFAT5 in the presence of TAZ. A DNA pulldown assay revealed that NFAT5 bound directly to a DNA sequence containing the NFAT5/TonEBP-binding element, and this DNA-NFAT5 association was markedly inhibited by overexpression of WT TAZ but not by overexpression of the Y316F mutant (Fig. 4C). More significantly, ChIP assay demonstrated that NFAT5 bound to the SMIT promoter region in response to hyperosmolarity. The NFAT5-DNA association was suppressed by the overexpression of WT TAZ but not by overexpression of Y316F TAZ (Fig. 4D). More significantly, NFAT5 binding to the SMIT promoter was augmented in TAZ-deficient cells, as evidenced by ChIP and real-time PCR analysis (Fig. 4E and F). These results indicate that the TAZ-NFAT5 interaction suppresses the transcriptional activity of NFAT5 by inhibiting the DNA-binding activity of NFAT5.
Fig 4.

Suppression of NFAT5 activity by TAZ. (A) 293T cells were transfected with NFAT5 and different amounts of TAZ expression vector together with pTonE-luc. pCMVβgal was used as a control. NFAT5 and TAZ expression was determined by immunoblot analysis. Relative luciferase activity was calculated after normalization to β-galactosidase activity. (B) 293T cells were transfected with NFAT5 and TAZ (WT or Y316F) expression vectors together with pTonE-luc and pCMVβgal. Reporter activity was calculated from 3 independent experiments and expressed as the mean. (C) NFAT5 was overexpressed in 293T cells together with WT or Y316F TAZ. Whole-cell extracts were incubated with biotinylated double-stranded DNA containing the NFAT5/TonEBP-binding element for 1 h and subsequently incubated with streptavidin-agarose beads for an additional 2 h, followed by SDS-PAGE and immunoblotting. DNA-binding complexes were analyzed with NFAT5 and TAZ Abs. (D) mIMCD-3 or stable mIMCD-3 cells (mock [con], TAZ, and Y316F TAZ) were stimulated with either 150 or 400 mosmol/kg for 4 h. Whole-cell extracts were immunoprecipitated with NFAT5 Ab, and chromatin was analyzed by a PCR using specific primers for the NFAT5-binding element within the SMIT gene promoter. (E) mIMCD-3 cells (shcon and shTAZ) were cultured under 400 mosmol/kg and harvested for ChIP and real-time PCR. (F) WT and KO MEF cells were stimulated with 400 mosmol/kg for 4 h, and cells were analyzed by ChIP and quantitative PCR. *, P < 0.05; **, P < 0.005; ***, P < 0.0005.
Expression of BGT1 and SMIT is negatively regulated by TAZ expression.
Since TAZ suppressed the DNA-binding and transcriptional activities of NFAT5, we investigated the effects of TAZ overexpression on the expression of endogenous NFAT5 target genes, such as the BGT1 and SMIT genes. The TAZ protein level was increased in stable TAZ cells compared to control mIMCD-3 cells, whereas NFAT5 expression was comparable between control and stable TAZ cells and unchanged by tonicity challenges (Fig. 5A). Interestingly, the relative expression of BGT1 and SMIT was drastically increased by hyperosmotic stimulation in control cells, but this increase was significantly attenuated by the overexpression of TAZ (Fig. 5B). Conversely, we measured the levels of BGT1 and SMIT in TAZ knockdown cells (shTAZ cells) (Fig. 5C). Hyperosmotic stress consistently increased the expression of BGT1 and SMIT in control (shcon) cells. However, the levels of BGT1 and SMIT were substantially elevated by reductions in TAZ expression under both normal and hyperosmolar conditions (Fig. 5D). These results suggest that the TAZ expression level may be important for fine-tuning NFAT5 activity induced by hyperosmotic stimulation.
Fig 5.
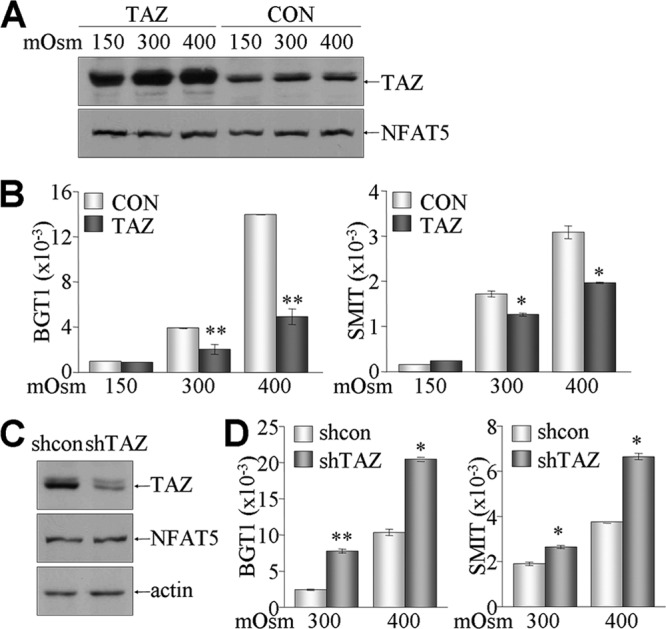
Inhibitory effects of TAZ on NFAT5-induced gene expression. (A and B) Stable TAZ transfectants (TAZ) and control cells (CON) were cultured under different tonicity conditions for 4 h. (A) Expression of TAZ and NFAT5 was determined by immunoblotting. (B) Total RNA was isolated from cells and subjected to reverse transcription and real-time PCR for measuring BGT1 and SMIT1 levels. Relative expression levels were calculated after normalization against β-actin levels. (C and D) TAZ knockdown (shTAZ) and control (shcon) cells were established and stimulated with normal and hyperosmolar conditions for 4 h. (C) Protein levels of TAZ, NFAT5, and actin were confirmed by immunoblotting. (D) Relative mRNA levels of BGT1 and SMIT were determined by real-time PCR. *, P < 0.05; **, P < 0.005.
Restoration of TAZ, but not Y316F TAZ, in TAZ-deficient cells leads to suppression of enhanced NFAT5 activity.
We wanted to confirm that the increase in NFAT5 activity caused by the lack of TAZ could be reversed directly by TAZ restoration. For the reintroduction of TAZ or the Y316F mutant, MEF cells were isolated from WT and KO mice, and cellular responses were examined under different osmotic stresses. TAZ expression in the WT and KO MEF cells was verified by immunoblot analysis (Fig. 6A). MEF cells were then transfected with the pTonE-luc reporter gene and challenged with media of different osmolarities. Hyperosmotic stimulation increased the reporter activity in WT MEF cells but had a much greater effect on the TAZ KO cells (Fig. 6B). Consistently, the BGT1 mRNA level was much greater in hyperosmolarity-stimulated KO cells than in WT cells (Fig. 6C). Next, the increased pTonE-luc activity in TAZ KO cells reverted to basal levels upon restoration of WT TAZ. However, introduction of Y316F TAZ was much less effective at inhibiting reporter activity (Fig. 6D). The endogenous mRNA level of SMIT was accordingly regulated by the restoration of WT TAZ or the Y316F mutant (Fig. 6E). Lastly, we attempted to measure the in vivo effect of TAZ deficiency on expression levels of NFAT5-mediated osmoregulatory genes, as TAZ suppressed NFAT5 activity in vitro. Despite severe morphological defects in the kidneys of TAZ KO mice, expression of SMIT and BGT1 was significantly increased in the renal medulla of TAZ KO kidneys compared to that in WT kidneys (Fig. 6F).
Fig 6.

Resuppression of NFAT5 activity by restored TAZ, but not Y316F TAZ, in TAZ KO cells. (A) TAZ expression in WT and KO MEF cells was determined by immunoblotting. (B) WT and KO MEF cells were transfected with reporter genes (pTonE-luc and pCMVβgal) and treated with different tonicities for 24 h. (C) Total RNAs were harvested from WT and KO MEF cells incubated under hyperosmolar conditions and used for measuring mRNA levels of BGT1 and SMIT. (D) TAZ KO MEF cells were transfected with WT or Y316F TAZ expression vector together with reporter gene vectors (pTonE-luc and pCMVβgal). Cells were then challenged with normal or hyperosmolar medium for 24 h. Reporter activities (relative light units [RLU]) in panels B and D were calculated after normalization with β-galactosidase activity and are expressed as means ± SEM for three independent experiments. (E) KO MEF cells were restored with WT and Y316F TAZ expression vectors and subsequently treated with hyperosmolar medium. Total RNA was used for reverse transcription and real-time PCR analysis. (F) Total RNAs were isolated from the kidneys of WT and KO mice (10 to 12 weeks old; n = 4), followed by reverse transcription and real-time PCR analysis. *, P < 0.05; **, P < 0.005; ***, P < 0.0005.
DISCUSSION
Our results demonstrate that TAZ undergoes tyrosine phosphorylation at tyrosine 316 by c-Abl kinase upon hyperosmotic stimulation and then specifically interacts with and suppresses the transcriptional activity of NFAT5. While ectopic TAZ overexpression suppresses the transcription of NFAT5 target genes, such as the SMIT and BGT1 genes, TAZ deficiency increases the expression of SMIT and BGT1 in vitro and in vivo. Collectively, our data are the first to demonstrate that TAZ functions as a suppressor of NFAT5 activity in response to hyperosmotic stress.
Interestingly, TAZ selectively associates with and modulates the activity of NFAT5 in a tyrosine phosphorylation-dependent manner. Hyperosmotic stress activates multiple mitogen-activated kinases (1), protein kinase A (9), and tyrosine kinases such as Syk and Yes (25, 31), which then modulate the expression of genes required for osmotic regulation. TAZ is also phosphorylated by Ser/Thr protein kinases at serine residues (19, 20), but its S89 phosphorylation was not affected by hyperosmotic stimulation. Recently, c-Abl was identified as the kinase responsible for hyperosmotic stress signaling via NFAT5 phosphorylation (11). Gallazzini et al. observed that c-Abl was activated within 1 h upon stimulation at 500 mosmol/kg and that the activated c-Abl phosphorylated NFAT5 on tyrosine 143 and stimulated its nuclear localization and transcriptional activity (11). Our results demonstrate that TAZ, like NFAT5, is a novel substrate for c-Abl in kidney cells stimulated with hyperosmolarity for 4 h. However, phosphorylated TAZ subsequently suppresses NFAT5 activity through a phosphorylation-dependent physical interaction. Interestingly, c-Abl-induced tyrosine phosphorylation of TAZ is a prerequisite for suppressing NFAT5 activity, whereas c-Abl-mediated NFAT5 phosphorylation is not essential for interaction of NFAT5 with TAZ. These findings suggest that c-Abl may have dual roles in timely regulation of renal tonicity by accelerating and decelerating NFAT5 activity through sequential phosphorylation of NFAT5 and TAZ. The detailed mechanisms and in vivo implications of c-Abl-mediated NFAT5 and TAZ regulation remain to be uncovered. In addition, a few punctate spots of TAZ-NFAT5 complexes were visualized in the nucleus, indicating the fine-tuning of NFAT5 activity by phosphorylated TAZ.
Our results suggest that TAZ deficiency may cause constitutive activation of NFAT5 in renal medullary cells and thus lead to cellular resistance to hyperosmotic stress-induced damage. However, TAZ KO mice die shortly after birth, due to developmental defects in multiple organs; the few viable TAZ KO mice display multicystic kidneys with polyuria (24). Despite abnormal renal structure or function, the renal medullas of TAZ KO mice express significantly higher levels of BGT1 and SMIT than those of WT mice. Most importantly, however, the in vivo importance of TAZ-mediated NFAT5 suppression after exposure to hyperosmotic stress should be verified in viable mice with kidney-specific knockout of TAZ. On the other hand, NFAT5 has been characterized extensively as having hyperosmolarity-independent and tissue-specific functions in a variety of cell types (12, 18, 32). NFAT5 KO mice are embryonic lethal due to impaired cardiac development and function, and the embryos display renal atrophy (21, 23); thus, it appears that NFAT5 is essential for heart and kidney development. Therefore, TAZ-mediated NFAT5 regulation may also be important in regulating heart and kidney development, but its detailed molecular functions and mechanisms are poorly understood. Together, our results suggest that TAZ functions in fine-tuning NFAT5 activity through tyrosine phosphorylation and contributes to cellular adaptation to hyperosmotic stress.
ACKNOWLEDGMENTS
This work was supported by an Ewha Global Top 5 Grant 2011 of the Ewha Womans University, by the R&D Program (A080908) of the Korea Health Industry Development Institute, and partly by a National Research Foundation grant funded by the Ministry of Education and Science Technology of the Republic of Korea (2012-0000952).
Footnotes
Published ahead of print 8 October 2012
REFERENCES
- 1. Berl T, Siriwardana G, Ao L, Butterfield LM, Heasley LE. 1997. Multiple mitogen-activated protein kinases are regulated by hyperosmolality in mouse IMCD cells. Am. J. Physiol. 272:F305–F311 [DOI] [PubMed] [Google Scholar]
- 2. Burg MB, Ferraris JD, Dmitrieva NI. 2007. Cellular response to hyperosmotic stresses. Physiol. Rev. 87:1441–1474 [DOI] [PubMed] [Google Scholar]
- 3. Cha JH, et al. 2001. Hydration status affects nuclear distribution of transcription factor tonicity responsive enhancer binding protein in rat kidney. J. Am. Soc. Nephrol. 12:2221–2230 [DOI] [PubMed] [Google Scholar]
- 4. Chan SW, et al. 2008. A role for TAZ in migration, invasion, and tumorigenesis of breast cancer cells. Cancer Res. 68:2592–2598 [DOI] [PubMed] [Google Scholar]
- 5. Chan SW, et al. 2009. TEADs mediate nuclear retention of TAZ to promote oncogenic transformation. J. Biol. Chem. 284:14347–14358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cui CB, Cooper LF, Yang X, Karsenty G, Aukhil I. 2003. Transcriptional coactivation of bone-specific transcription factor Cbfa1 by TAZ. Mol. Cell. Biol. 23:1004–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Di Palma T, et al. 2009. TAZ is a coactivator for Pax8 and TTF-1, two transcription factors involved in thyroid differentiation. Exp. Cell Res. 315:162–175 [DOI] [PubMed] [Google Scholar]
- 8. Duning K, et al. 2010. Polycystin-2 activity is controlled by transcriptional coactivator with PDZ binding motif and PALS1-associated tight junction protein. J. Biol. Chem. 285:33584–33588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ferraris JD, Persaud P, Williams CK, Chen Y, Burg MB. 2002. cAMP-independent role of PKA in tonicity-induced transactivation of tonicity-responsive enhancer/osmotic response element-binding protein. Proc. Natl. Acad. Sci. U. S. A. 99:16800–16805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ferraris JD, et al. 1996. ORE, a eukaryotic minimal essential osmotic response element. The aldose reductase gene in hyperosmotic stress. J. Biol. Chem. 271:18318–18321 [DOI] [PubMed] [Google Scholar]
- 11. Gallazzini M, Yu MJ, Gunaratne R, Burg MB, Ferraris JD. 2010. c-Abl mediates high NaCl-induced phosphorylation and activation of the transcription factor TonEBP/OREBP. FASEB J. 24:4325–4335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Halterman JA, Kwon HM, Wamhoff BR. 2012. Tonicity-independent regulation of the osmosensitive transcription factor TonEBP (NFAT5). Am. J. Physiol. Cell Physiol. 302:C1–C8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hong JH, et al. 2005. TAZ, a transcriptional modulator of mesenchymal stem cell differentiation. Science 309:1074–1078 [DOI] [PubMed] [Google Scholar]
- 14. Hossain Z, et al. 2007. Glomerulocystic kidney disease in mice with a targeted inactivation of Wwtr1. Proc. Natl. Acad. Sci. U. S. A. 104:1631–1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jeong H, et al. 2010. TAZ as a novel enhancer of MyoD-mediated myogenic differentiation. FASEB J. 24:3310–3320 [DOI] [PubMed] [Google Scholar]
- 16. Kanai F, et al. 2000. TAZ: a novel transcriptional co-activator regulated by interactions with 14-3-3 and PDZ domain proteins. EMBO J. 19:6778–6791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kang HS, Beak JY, Kim YS, Herbert R, Jetten AM. 2009. Glis3 is associated with primary cilia and Wwtr1/TAZ and implicated in polycystic kidney disease. Mol. Cell. Biol. 29:2556–2569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kojima R, et al. 2010. Hypertonicity-induced expression of monocyte chemoattractant protein-1 through a novel cis-acting element and MAPK signaling pathways. J. Immunol. 184:5253–5262 [DOI] [PubMed] [Google Scholar]
- 19. Lei QY, et al. 2008. TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol. Cell. Biol. 28:2426–2436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu CY, et al. 2010. The hippo tumor pathway promotes TAZ degradation by phosphorylating a phosphodegron and recruiting the SCFbeta-TrCP E3 ligase. J. Biol. Chem. 285:37159–37169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lopez-Rodriguez C, et al. 2004. Loss of NFAT5 results in renal atrophy and lack of tonicity-responsive gene expression. Proc. Natl. Acad. Sci. U. S. A. 101:2392–2397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mahoney WM, Jr, Hong JH, Yaffe MB, Farrance IK. 2005. The transcriptional co-activator TAZ interacts differentially with transcriptional enhancer factor-1 (TEF-1) family members. Biochem. J. 388:217–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mak MC, et al. 2011. Embryonic lethality in mice lacking the nuclear factor of activated T cells 5 protein due to impaired cardiac development and function. PLoS One 6:e19186 doi:10.1371/journal.pone.0019186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Makita R, et al. 2008. Multiple renal cysts, urinary concentration defects, and pulmonary emphysematous changes in mice lacking TAZ. Am. J. Physiol. Renal Physiol. 294:F542–F553 [DOI] [PubMed] [Google Scholar]
- 25. Miah SM, et al. 2004. Activation of Syk protein tyrosine kinase in response to osmotic stress requires interaction with p21-activated protein kinase Pak2/gamma-PAK. Mol. Cell. Biol. 24:71–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Miyakawa H, et al. 1998. Cis- and trans-acting factors regulating transcription of the BGT1 gene in response to hypertonicity. Am. J. Physiol. 274:F753–F761 [DOI] [PubMed] [Google Scholar]
- 27. Miyakawa H, Woo SK, Dahl SC, Handler JS, Kwon HM. 1999. Tonicity-responsive enhancer binding protein, a rel-like protein that stimulates transcription in response to hypertonicity. Proc. Natl. Acad. Sci. U. S. A. 96:2538–2542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Murakami M, Nakagawa M, Olson EN, Nakagawa O. 2005. A WW domain protein TAZ is a critical coactivator for TBX5, a transcription factor implicated in Holt-Oram syndrome. Proc. Natl. Acad. Sci. U. S. A. 102:18034–18039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Murakami M, et al. 2006. Transcriptional activity of Pax3 is co-activated by TAZ. Biochem. Biophys. Res. Commun. 339:533–539 [DOI] [PubMed] [Google Scholar]
- 30. Park KS, et al. 2004. TAZ interacts with TTF-1 and regulates expression of surfactant protein-C. J. Biol. Chem. 279:17384–17390 [DOI] [PubMed] [Google Scholar]
- 31. Reinehr R, Becker S, Hongen A, Haussinger D. 2004. The Src family kinase Yes triggers hyperosmotic activation of the epidermal growth factor receptor and CD95. J. Biol. Chem. 279:23977–23987 [DOI] [PubMed] [Google Scholar]
- 32. Roth I, et al. 2010. Osmoprotective transcription factor NFAT5/TonEBP modulates nuclear factor-kappaB activity. Mol. Biol. Cell 21:3459–3474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tian Y, et al. 2007. TAZ promotes PC2 degradation through an SCFbeta-Trcp E3 ligase complex. Mol. Cell. Biol. 27:6383–6395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Uchida S, et al. 1993. Molecular cloning of the cDNA for an MDCK cell Na(+)- and Cl(−)-dependent taurine transporter that is regulated by hypertonicity. Proc. Natl. Acad. Sci. U. S. A. 90:7424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yamauchi A, et al. 1992. Cloning of a Na(+)- and Cl(−)-dependent betaine transporter that is regulated by hypertonicity. J. Biol. Chem. 267:649–652 [PubMed] [Google Scholar]
- 36. Yamauchi A, Uchida S, Preston AS, Kwon HM, Handler JS. 1993. Hypertonicity stimulates transcription of gene for Na(+)-myo-inositol cotransporter in MDCK cells. Am. J. Physiol. 264:F20–F23 [DOI] [PubMed] [Google Scholar]
- 37. Zhang H, et al. 2009. TEAD transcription factors mediate the function of TAZ in cell growth and epithelial-mesenchymal transition. J. Biol. Chem. 284:13355–13362 [DOI] [PMC free article] [PubMed] [Google Scholar]