

Abstract
Serum retinol-binding protein 4 (RBP4) is the sole specific vitamin A (retinol) transporter in blood. Elevation of serum RBP4 in patients has been linked to cardiovascular disease and diabetic retinopathy. However, the significance of RBP4 elevation in the pathogenesis of these vascular diseases is unknown. Here we show that RBP4 induces inflammation in primary human retinal capillary endothelial cells (HRCEC) and human umbilical vein endothelial cells (HUVEC) by stimulating expression of proinflammatory molecules involved in leukocyte recruitment and adherence to endothelium, including vascular cell adhesion molecule 1 (VCAM-1), intercellular adhesion molecule 1 (ICAM-1), E-selectin, monocyte chemoattractant protein 1 (MCP-1), and interleukin-6 (IL-6). We demonstrate that these novel effects of RBP4 are independent of retinol and the RBP4 membrane receptor STRA6 and occur in part via activation of NADPH oxidase and NF-κB. Importantly, retinol-free RBP4 (apo-RBP4) was as potent as retinol-bound RBP4 (holo-RBP4) in inducing proinflammatory molecules in both HRCEC and HUVEC. These studies reveal that RBP4 elevation can directly contribute to endothelial inflammation and therefore may play a causative role in the development or progression of vascular inflammation during cardiovascular disease and microvascular complications of diabetes.
INTRODUCTION
Elevated levels of serum retinol-binding protein 4 (RBP4), the transport protein for vitamin A (retinol) in the blood circulation, correlate with insulin resistance and type 2 diabetes in several clinical studies (1, 13, 25, 32, 41, 49, 52, 68). Furthermore, studies in mice have shown that ectopic overexpression of serum RBP4 inhibits insulin signaling in muscle and adipose tissues and causes systemic insulin resistance, whereas Rbp4−/− mice have increased insulin sensitivity (8, 68). Type 2 diabetes is classically thought of as a metabolic disease, but it is also associated with a 2- to 4-fold-increased risk of cardiovascular disease (7, 15, 36, 37). In addition, a majority of patients with type 2 diabetes will develop some type of microvascular disease, such as retinopathy, within the first 2 decades of disease (26, 59). Together, cardiovascular disease and microvascular complications are the major causes of death and disability in patients with type 2 diabetes. Thus, understanding the factors that contribute to the development and progression of cardiovascular disease and microvascular complications is necessary to prolong survival and improve quality of life for patients with type 2 diabetes. Recent clinical studies have linked elevated serum RBP4 specifically to cardiovascular disease, including hypertension (58), stroke (34, 57), and atherosclerosis (10, 58). In addition, two studies have reported that patients with proliferative diabetic retinopathy, which is characterized by destructive retinal neovascularization and vision loss, have a significantly pronounced elevation of RBP4 compared to patients with mild or no retinopathy (43, 60). The finding that elevation of RBP4 is correlated with cardiovascular disease and diabetic retinopathy warrants investigation into the possible role of RBP4 in the pathogenesis of both types of vascular disease.
Chronic vascular inflammation is a major factor in the development of atherosclerosis as well as microvascular diseases such as diabetic retinopathy (7, 20, 24, 27, 39, 55, 62). Vascular inflammation begins with endothelial expression of proinflammatory cell surface adhesion molecules, such as vascular cell adhesion molecule 1 (VCAM-1), intercellular adhesion molecule 1 (ICAM-1), and endothelial-leukocyte adhesion molecule 1 (E-selectin), as well as endothelial secretion of soluble proinflammatory factors, such as monocyte chemoattractant protein 1 (MCP-1), and interleukin-6 (IL-6), which act in concert to promote the recruitment and adherence of leukocytes to the endothelium (24). Cellular oxidative stress is responsible for the activation of endothelial inflammation (24, 27). The primary source of oxidative stress in endothelial cells is NADPH oxidase, an enzyme that generates the free radical superoxide (24, 27). Superoxide reacts with other molecules to form a variety of reactive oxygen species, such as hydrogen peroxide and hydroxyl radicals, which promote proinflammatory cell signaling, including activation of nuclear factor kappa B (NF-κB), a transcription factor that induces expression of proinflammatory genes, such as the VCAM-1, ICAM-1, E-selectin, MCP-1, and IL-6 genes (19, 44, 56). In diabetic retinopathy, NADPH oxidase and NF-κB activation promote proinflammatory gene expression and induce leukostasis in retinal capillaries (2, 3, 16, 39, 61). In cardiovascular disease, NADPH oxidase activation and NF-κB-mediated upregulation of VCAM-1 expression in aortic endothelium are critical early events in the initiation of atherosclerotic lesions (11, 12, 21, 64).
Clinical studies have recently found a correlation between elevated serum RBP4 level and subclinical inflammation (5, 6, 69), and other studies have revealed that RBP4 induces inflammation in macrophages (22, 53). Macrophages are present in adipose tissue during obesity and contribute to chronic adipose inflammation and insulin resistance (23, 47, 48, 66). RBP4-activated macrophages secrete proinflammatory cytokines that impair insulin signaling in cocultured adipocytes (22, 53). The proinflammatory effects of RBP4 on macrophages are mediated in part through activation of toll-like receptor 4 (TLR4) and c-Jun N-terminal protein kinase (JNK) signaling and are retinol independent, since apo-RBP4 (retinol free) and holo-RBP4 (retinol bound) have the same proinflammatory effects (53). The finding that RBP4 stimulates proinflammatory pathways in macrophages, combined with previous studies that found that serum RBP4 levels correlate significantly with the presence of subclinical inflammation (5), hypertension (58), stroke (34, 57), atherosclerosis (10, 58), and diabetic retinopathy (43, 60), suggests that elevation of serum RBP4 may contribute to vascular disease by inducing endothelial inflammation; however, the effect of RBP4 on endothelial inflammation has not been studied.
In the current study, we investigated the direct effect of RBP4 on primary human macrovascular and microvascular endothelial cells. We show that RBP4 can directly induce production of proinflammatory molecules involved in leukocyte recruitment and adherence, including VCAM-1, ICAM-1, E-selectin, MCP-1, and IL-6 in both macro- and microvascular human endothelial cells. These novel effects of RBP4 are retinol independent and occur, in part, via activation of NADPH oxidase and NF-κB. These studies reveal that RBP4 elevation can directly contribute to endothelial inflammation and therefore may play a causative role in the development or progression of vascular inflammation during cardiovascular disease and diabetic microvascular complications.
MATERIALS AND METHODS
Expression and purification of recombinant human RBP4.
The open reading frame of human RBP4 was amplified by PCR from a cDNA clone purchased from Open Biosystems (Thermo Scientific, Lafayette, CO) (Clone ID LIFESEQ1949554, catalog no. IHS1380-97652535) using the following primers: forward, 5′-AACTCGAGGAGCGCGACTGCCGAGTGAGC-3′ (XhoI site underlined), and reverse, 5′-AAGGATCCCTACAAAAGGTTTCTTTCTGATCTG-3′ (BamHI site underlined). The PCR product was inserted into pET19b vector (Novagen) in frame with the vector N-terminal histidine tag sequence and confirmed by DNA sequencing. pET19b-His-RBP4 was then transformed into Escherichia coli strain BL21*, and His-RBP4 was purified as described previously (65) with modifications. LB media (2 × 1 liter) were each inoculated with 10 ml of an overnight culture and grown in flasks shaking at 37°C. When the optical density of the cultures reached 0.6 to 0.8 at 600 nm, IPTG (isopropyl-β-d-thiogalactopyranoside) was added at a final concentration of 1 mM to induce His-RBP4 protein expression, and the cultures were returned to shaking at 37°C overnight. Cells were harvested by centrifugation at 4,000 × g (20 min). The combined pellets were resuspended in a total of 50 ml lysis buffer [50 mM Tris, pH 7.5, 2 mM EDTA, 1 mM 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF), 0.1% Triton X-100]. The cell suspension was sonicated 5 times for 20 s, freeze-thawed twice, and centrifuged at 10,000 × g for 25 min to pellet the insoluble fraction. RBP4 remained in the insoluble pellet fraction and was resuspended in 14 ml of 7.5 M guanidine hydrochloride (Gd-HCl). The suspension was further diluted in 25 mM Tris, pH 9.0, to a final concentration of 5 M Gd-HCl. Dithiothreitol (DTT) (10 mM) was added, and the suspension was adjusted to pH 9.0. The suspension was stirred vigorously at room temperature for 4 to 6 h until the solution appeared translucent. Oxidative refolding was performed by combining the Gd-HCl protein suspension (∼21 ml) with 80 ml of redox refolding buffer (0.3 mM cystine, 3.0 mM cysteine, 1 mM EDTA, 25 mM Tris [pH 9.0], prepared fresh and degassed by nitrogen sparging). For purification of holo-RBP4, an ∼10-fold molar excess of retinol dissolved in ethanol was included to facilitate protein refolding and increase protein yield. For purification of apo-RBP4, retinol was not added, and the final protein yield was lower (∼50% of holo-RBP4 yield). When holo-RBP4 was purified, all steps after the addition of retinol in the refolding buffer were carried out in a darkroom lit by dim red light. When apo-RBP4 was purified, all steps were performed under normal lab light. The refolding mixture was stirred vigorously at 4°C overnight to facilitate RBP4 protein refolding. After refolding, the remaining insoluble protein was removed by centrifugation at 13,000 × g for 20 min. The supernatant containing the refolded protein was dialyzed in 25 mM Tris, pH 8.0, with 150 mM NaCl. After dialysis, the protein was incubated with nickel resin for 3 h at 4°C, and His-RBP4 was purified by nickel affinity chromatography. The eluted His-RBP4 was analyzed by the Bradford method of protein quantification and 250- to 350-nm absorbance spectra to confirm protein quality and the presence/absence of retinol in apo- and holo-RBP4 preparations. Purified His-RBP4 was dialyzed into phosphate-buffered saline (PBS), aliquoted, and stored at −80°C. The endotoxin level in purified His-RBP4 was quantified by Limulus amebocyte lysate assay test (ToxinSensor; Genscript, Piscataway, NJ) to be less than 0.001 endotoxin unit (EU) per μg protein (0.00043 EU/μg, which is similar to the level of endotoxin measured in ultrapure filtered water and generally considered to be nonimmunogenic). Native human RBP4, which was purified from normal human serum, was obtained from Athens Research Technology (Athens, GA). Native human RBP4 consisted primarily of holo-RBP4 according to the wavelength spectrum scan (see Fig. S2A in the supplemental material).
Human endothelial cell culture and treatment with RBP4 and chemical inhibitors.
Primary human retinal capillary endothelial cells (HRCEC) were obtained from Cell Systems Corporation (Kirkland, WA) and cultured in EGM-MV (Promocell, Germany). Primary human umbilical vein endothelial cells (HUVEC) were obtained from Life Technologies (Grand Island, NY) and cultured in Medium-200 with low serum growth supplement. HRCEC and HUVEC were plated and maintained on collagen I-coated tissue culture plates from BD Bioscience (San Jose, CA). THP-1 monocytes were obtained from the American Type Culture Collection (Manassas, VA) and cultured in RPMI 1640 medium with 10% fetal bovine serum (FBS) and 0.05 mM beta-mercaptoethanol. For experiments, HRCEC or HUVEC confluent monolayers were incubated in serum-starved medium with 2% FBS for the noted treatment durations. All experiments were conducted using HRCEC or HUVEC at passages 2 to 10. Cells were treated as indicated with either increasing concentrations of RBP4 (10 to 100 μg/ml), or bovine serum albumin (BSA) at a dose equimolar to the highest concentration of RBP4 (100 μg/ml, equivalent to 4.75 μM), or retinoids at equimolar concentrations to RBP4 for dose-response comparison or were left untreated. When cells were treated with holo-RBP4 or retinoids, experiments were performed in a darkroom under dim red light, and cells were maintained in the dark for the duration of the treatment. The NF-κB inhibitors pyrrolidine dithiocarbamate (PDTC) and JSH-23, the NADPH oxidase inhibitors diphenyleneiodonium chloride (DPI) and apocynin, lipopolysaccharide (LPS), and polymyxin B sulfate (PMB) were purchased from Sigma-Aldrich (St. Louis, MO). Studies with PDTC, JSH-23, DPI, and apocynin were conducted by preincubating cells with the inhibitor for 2 h prior to the addition of RBP4. Studies with PMB were conducted by adding PMB simultaneously with RBP4 or LPS.
qRT-PCR.
Cells were pelleted, and total RNA was isolated and purified using the TRIzol Plus RNA purification kit (Invitrogen Inc.). Reverse transcription was then performed using the Superscript III system (Invitrogen Inc.) to produce cDNA. PCR was then performed in triplicate on each cDNA sample (myIQ; Bio-Rad) for each gene (Hprt, Vcam-1, Icam-1, Mcp-1, E-selectin, Stra6, Lrp2, TLR2, and TLR4 genes), and threshold cycle (ΔCT) values were calculated against the housekeeping gene Hprt. Primers for all genes were designed to span introns so as to avoid amplification from genomic DNA (refer to Table S1 in the supplemental material for a complete list of primer pairs). Stratagene human reference total RNA (composed of total RNA from 10 human cell lines, with quantities of RNA from the individual cell lines optimized to maximize the representation of gene transcripts present in low, medium, and high abundance) was used as a positive control to validate quantitative reverse transcription-PCR (qRT-PCR) primer pairs. For reporting of results, all data were normalized to Hprt, which was assigned an arbitrary expression level of 10,000, and relative gene expression values were calculated by the following formula: relative expression = 10,000/2ΔCT, where ΔCT = (gene CT − Hprt CT). Melt curve analyses were conducted to verify the purity and size of resultant PCR products. At least 3 distinct biological samples were examined for each gene and treatment.
Western blot analysis.
Cells were lysed in Laemmli buffer (100 mM Tris, pH 6.8, 200 mM dithiothreitol, 4% sodium dodecyl sulfate, 0.2% bromophenol blue, 1 mM AEBSF, 20% glycerol) and subjected to immunoblotting with the indicated antibodies. Band densitometry measurements were performed using Image J software (NIH, Bethesda, MD). The following antibodies were used: from Cell Signaling Technology, P-Ser536-NF-κB p65 (catalog no. 3033) and fibrillarin (catalog no. 2639); from Santa Cruz Biotechnology, VCAM-1 (catalog no. sc-8304), E-selectin (catalog no. sc-14011), RBP4 (catalog no. sc-69795), Nox4 (catalog no. sc-30141), and Nox 2 (catalog no. sc-20782); from Abcam, total NF-κB p65 (catalog no. ab16502), STRA6 (catalog no. ab73490), and ICAM-1 (catalog no. ab27536); from Sigma-Aldrich, horseradish peroxidase (HRP)-conjugated beta-actin (catalog no. A3854); and from Novus Biologics, GAPDH (catalog no. NB300-221).
Measurement of proinflammatory molecules in cell culture supernatants.
HRCEC and HUVEC culture media were collected after the indicated treatments, centrifuged at 10,000 × g for 2 min to remove cell debris, and stored at −20°C. Enzyme-linked immunosorbent assay (ELISA) kits from R&D Systems, Inc. (Minneapolis, MN) were used to measure soluble extracellular protein levels of VCAM-1 (catalog no. DY809), ICAM-1 (catalog no. DY720), MCP-1 (catalog no. DY279), and IL-6 (catalog no. DY206), and an ELISA kit from RayBiotech Inc. (Norcross, GA) was used to measure soluble E-selectin (catalog no. ELH-Eselectin-001). All ELISAs were performed according to the manufacturer's instructions.
Leukocyte adhesion assay.
HRCEC or HUVEC were grown to confluence in six-well plates and then treated with BSA or RBP4 for 18 h or tumor necrosis alpha (TNF-α) for 4 h in serum-starved 2% FBS medium. THP-1 monocytes were added at 1.25 million cells/well and cocultured with HRCEC or HUVEC for 3 h. Unbound THP-1 monocytes were removed by extensive washing with PBS (4 washes with shaking). Cells were fixed in 4% paraformaldehyde, bound THP-1 monocytes were visualized by phase-contrast microscopy at a magnification of ×20, and the numbers of adherent monocytes per field were determined and averaged from four random visual fields for each treatment group.
Immunocytochemistry.
HRCEC or HUVEC were cultured on collagen I-coated coverslips (BD Bioscience) and then treated with BSA, RBP4, or TNF-α in serum-starved 2% FBS medium for 12 h. The distribution pattern of total NF-κB p65 was visualized by fluorescent immunostaining using a Cellomics NF-κB activation HitKit HSC reagent kit from Thermo Fisher Scientific (Rockford, IL).
Quantification of nuclear NF-κB.
To quantify the nuclear level and relative transcriptional activity of NF-κB subunit p65 in HRCEC, nuclear extracts were prepared using a nuclear extract preparation kit from Active Motif (Carlsbad, CA), and the DNA binding capacity of NF-κB in nuclear extracts was quantified using an ELISA-based TransAM NF-κB kit from Active Motif. Briefly, HRCEC were treated in serum-starved 2% FBS medium containing either RBP4 (100 μg/ml) or BSA for 6 h, and nuclear extracts were prepared. To measure NF-κB DNA binding, nuclear extracts (10 μg/well) were applied to a 96-well plate coated with immobilized oligonucleotide containing the NF-κB consensus binding site (5′-GGGACTTTCC-3′). After a 1-h incubation period, nuclear extracts were removed, and a primary antibody for NF-κB p65 was added and incubated for 1 h, followed by incubation with a secondary antibody conjugated with HRP for 1 h and then in an HRP substrate solution. HRP-dependent color development was monitored and stopped with the addition of acid, and the plate absorbance was read at 450 nm using a 1420 microplate reader (PerkinElmer Life Sciences).
Retinoid treatment and quantification of intracellular retinoids.
HRCEC were grown to confluence in T75 flasks and then treated with holo-RBP4 (100 μg/ml) or an equimolar dose of each retinoid (retinol, retinal, or retinoic acid) for 6 h. All experiments involving the treatment of cells with holo-RBP4 or retinoids were performed in a darkroom under dim red light, and cells were maintained in the dark for the duration of the treatment. Cells were washed extensively in PBS and collected using a cell scraper. Cells were further washed by centrifugation and resuspension in fresh PBS. Each sample of cells was split in half to divide the sample evenly for analysis of retinoids on two separate high-performance liquid chromatography (HPLC) columns. Cells were pelleted, all PBS was removed, and cell pellets were stored at −80°C. For retinoid quantification, cells were resuspended in 200 μl PBS and sonicated. Neutrally charged retinoids were extracted with 300 μl ethanol and 300 μl hexane. For measurement of intracellular retinol, retinal, and retinyl ester, samples were analyzed by HPLC (Waters 515 HPLC pump and 2996 Photodiode Array Detector; Waters Corporation, Milford, MA) using a normal-phase 5-μm column (Lichrosphere SI-60; Alltech, Deerfield, IL) and isocratic solvent of 11.2% ethyl acetate, 2.0% dioxane, and 1.4% octanol in hexane. Retinoic acid was extracted as described previously (51). For measurement of intracellular retinoic acid, samples were analyzed by HPLC using a reverse-phase 3.5-μm column (Waters C18) and a solvent gradient of 0 to 80% methanol for 5 min, followed by 80 to 100% methanol for 5 to 10 min and finally 100% methanol for 10 to 14 min. The peak of each retinoid isomer was identified based on its absorption spectra and retention time on the column compared to pure retinoid standards. The quantification of each retinoid isomer was calculated from the area of each retinoid peak using synthetic purified retinoid standards for calibration with Empower software (Waters Corporation).
RESULTS
RBP4 induces expression of proinflammatory molecules in human endothelial cells.
Recombinant His-tagged human RBP4 was purified in the presence of retinol to generate holo-RBP4 (Fig. 1A and B). We first examined whether RBP4 induces expression of proinflammatory molecules in primary human retinal capillary endothelial cells (HRCEC) by quantitative reverse-transcription PCR. HRCEC were treated for 24 h with increasing concentrations of purified recombinant holo-RBP4 spanning the range of RBP4 levels measured in the serum of normal, obese, insulin-resistant, and type 2 diabetic patients, and the expression of proinflammatory genes was analyzed by qRT-PCR. We specifically evaluated the expression of VCAM-1, ICAM-1, E-selectin, and MCP-1, since these proinflammatory molecules have been shown to be significantly elevated in serum during cardiovascular disease and diabetic retinopathy (39, 55). RBP4 significantly and dose dependently upregulated the mRNA levels of VCAM-1, ICAM-1, E-selectin, and MCP-1 in HRCEC (Fig. 1C to F). Maximum effects were observed with 100 μg/ml RBP4, although significant induction of proinflammatory gene expression was also observed following treatment with as little as 10 to 25 μg/ml RBP4 (Fig. 1C to F). This suggests that even slight elevations of RBP4 can impact proinflammatory gene expression in endothelial cells.
Fig 1.

RBP4 induces expression of proinflammatory cell adhesion molecules and monocyte chemoattractant protein-1 in human endothelial cells in a dose-dependent manner. (A) Representative Coomassie blue staining and Western blot analysis of purified recombinant human holo-RBP4 (1 μg per lane). Lane 1, Coomassie blue-stained gel; lane 2, Western blot with RBP4 antibody. (B) UV spectrum analysis of purified recombinant holo-RBP4 demonstrates a retinol/RBP4 ratio of approximately 0.9, indicating that the majority of the purified RBP4 consists of holo-RBP4. (C to F) Quantitative RT-PCR analysis for mRNA expression of VCAM-1, ICAM-1, E-selectin, and MCP-1 in human retinal capillary endothelial cells (HRCEC) treated with increasing concentrations of holo-RBP4 (10 to 100 μg/ml) or BSA or left untreated (Unt.). Significant differences compared to BSA treatment: *, P < 0.05; **, P < 0.01; ***, P < 0.001 by one-way analysis of variance (ANOVA) with Tukey's post hoc test.
We next examined the effects of RBP4 treatment on the protein levels of proinflammatory factors. HRCEC were treated either with increasing concentrations of holo-RBP4 for 24 h to determine the dose dependency of changes in protein levels or with the highest dose of holo-RBP4 (100 μg/ml) for 4, 8, 24, and 48 h to examine the time dependency of changes in protein. Cell lysates were analyzed by Western blotting, and cell culture media were analyzed by ELISA to quantify changes in extracellular soluble forms of proinflammatory proteins. RBP4 increased the cellular and extracellular level of VCAM-1 protein in a dose- and time-dependent manner (Fig. 2A, D, G, and K). RBP4 also increased E-selectin protein levels in a dose-dependent manner (Fig. 2B and H), although the increase in cellular E-selectin protein peaked at 4 h post-RBP4 treatment (Fig. 2E), while extracellular soluble E-selectin continued to accumulate with increasing RBP4 incubation time (Fig. 2L). MCP-1, a secreted proinflammatory molecule, was increased in response to RBP4 treatment in a dose- and time-dependent manner (Fig. 2J and N). RBP4 treatment did not increase the steady-state cellular protein levels of ICAM-1 (Fig. 2C and F); however, extracellular soluble ICAM-1 levels were increased in a dose- and time-dependent manner by RBP4 treatment (Fig. 2I and M). IL-6 secretion was also dose-dependently increased by RBP4 treatment (see Fig. S7A in the supplemental material). The highest dose of RBP4 (100 μg/ml) increased extracellular levels of sVCAM-1, E-selectin, sICAM-1, MCP-1, and IL-6 approximately 400-fold, 30-fold, 3-fold, 5-fold, and 3-fold, respectively. However, a concentration of 25 μg/ml was sufficient to increase levels of sVCAM-1, sICAM-1, MCP-1, and IL-6 by 150-fold, 2-fold, 4-fold, and 2-fold, respectively.
Fig 2.

RBP4 increases cellular and extracellular protein levels of proinflammatory molecules in HRCEC in a dose- and time-dependent manner. (A to C) Western blots of VCAM-1 (A), E-selectin (B), and ICAM-1(C) from HRCEC treated with BSA or increasing concentrations of RBP4 for 24 h. (D to F) Western blots of VCAM-1 (D), E-selectin (E), and ICAM-1 (F) from HRCEC treated with RBP4 at 100 μg/ml for the indicated times of 0 to 48 h. (G to J) ELISA-based quantification of soluble extracellular levels of sVCAM-1 (G), E-selectin (H), sICAM-1 (I), and MCP-1 (J) from HRCEC media following treatment with increasing concentrations of holo-RBP4 or BSA or no treatment (Unt.). (K to N) ELISA-based quantification of soluble extracellular levels of sVCAM-1 (K), E-selectin (L), sICAM-1 (M), and MCP-1 (N) in HRCEC media following treatment with holo-RBP4 at 100 μg/ml for the indicated times of 0 to 48 h. Significant differences compared to BSA treatment: *, P < 0.05; **, P < 0.01; ***, P < 0.001 by one-way ANOVA with Tukey's post hoc test.
We confirmed that RBP4-induced changes in protein expression were not due to endotoxin (lipopolysaccharide) contamination of purified recombinant RBP4, which contained less than 0.001 EU/μg protein (data not shown). HRCEC were treated with holo-RBP4 at 100 μg/ml for 24 h in the absence or presence of increasing concentrations of polymyxin B, which binds to and neutralizes endotoxin. In a control experiment, HRCEC were treated with purified LPS (100 ng/ml) for 24 h in the absence or presence of increasing concentrations of polymyxin B. Polymyxin B blocked LPS-mediated increase of cellular VCAM-1 protein (see Fig. S1A in the supplemental material) and secreted MCP-1 (see Fig. S1B). In contrast, polymyxin B had no significant effect on RBP4-mediated increase of cellular VCAM-1 or secreted MCP-1 (see Fig. S1A and B in the supplemental material). Moreover, native RBP4 purified from human serum had the same proinflammatory potency as recombinant RBP4 (see Fig. S2C through H in the supplemental material), demonstrating that no other bacterially derived immunogenic components are responsible for the effects of recombinant RBP4.
RBP4 induces endothelial inflammation.
To determine if RBP4-induced expression of proinflammatory genes was sufficient to promote monocyte adherence to endothelial cells, we performed leukocyte adherence assays to quantify monocyte adherence to HRCEC. Cells were either left untreated or treated with BSA/RBP4 for 18 h, and then THP-1 monocytes were cocultured with HRCEC for 3 h. Cells were washed in PBS with gentle agitation to remove unbound monocytes and imaged by phase-contrast microscopy. We observed that RBP4 significantly increased monocyte adherence to HRCEC monolayers, demonstrating that RBP4 stimulates in vitro leukostasis (Fig. 3A and B).
Fig 3.
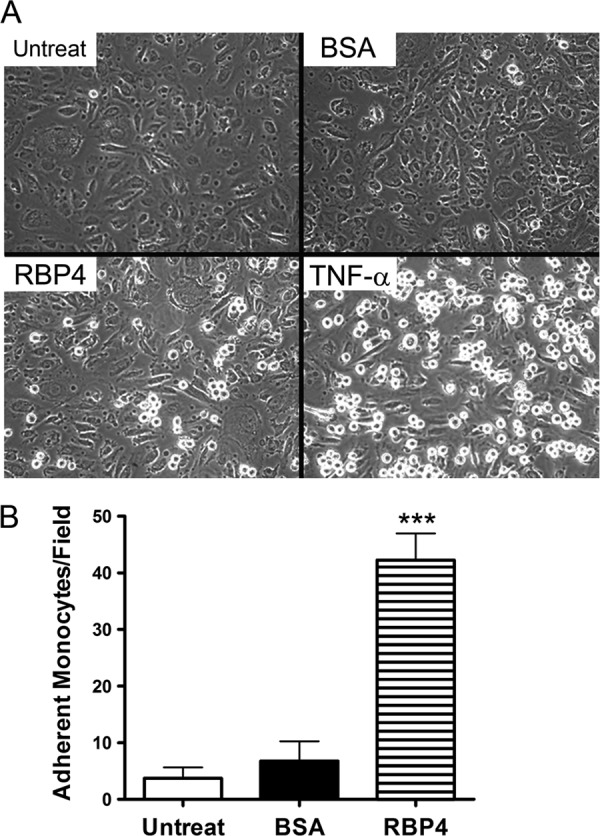
RBP4 increases leukocyte adherence to human endothelial cells. Confluent monolayers of HRCEC were treated with either holo-RBP4 (100 μg/ml), BSA, or TNF-α (100 ng/ml) for 18 h. THP-1 monocytes were then added and cocultured for 3 h. (A) Representative phase-contrast images (magnification, ×20) of monocyte adherence to HRCEC after the indicated treatment. Adherent monocytes are apparent as small bright circular bodies above the HRCEC layer. (B) Adherent monocytes were counted per visual field at magnification of ×20. The graph shows the means ± standard deviations from 4 different visual fields for each treatment group (adherent leukocytes in TNF-α positive control were 205 ± 25 per visual field). ***, P < 0.001 versus BSA treatment by one-way ANOVA with Tukey's post hoc test. Untreat, untreated cells.
RBP4 induces proinflammatory molecules through activation of NF-κB.
Previous studies have shown that NF-κB can induce expression of VCAM-1, ICAM-1, MCP-1, E-selectin, and IL-6 (19, 56), the same proinflammatory genes induced by RBP4 treatment. Thus, we reasoned that RBP4 induction of proinflammatory genes may be mediated through activation of NF-κB. To investigate this, we determined if RBP4 treatment induced NF-κB activation in HRCEC by evaluating the nuclear translocation of NF-κB subunit p65. We observed a marked increase in the nuclear localization of NF-κB in response to holo-RBP4 treatment (Fig. 4A). We also quantified the nuclear level and DNA-binding activity of NF-κB subunit p65 in RBP4- versus BSA-treated HRCEC using an ELISA-based NF-κB transcription factor assay. We found that holo-RBP4 significantly increased nuclear levels of NF-κB subunit p65 protein and its DNA-binding activity (Fig. 4B). RBP4 also caused a significant increase in activation of NF-κB subunit p65 through phosphorylation at Ser536 (Fig. 4C). To determine if RBP4-mediated induction of proinflammatory molecules is dependent on activation of NF-κB transcription, we pretreated HRCEC with two different chemical inhibitors of NF-κB, pyrrolidine dithiocarbamate (PDTC) and JSH-23, for 2 h prior to the addition of holo-RBP4. Pretreatment with either PDTC or JSH-23 significantly reduced RBP4-mediated induction of cellular VCAM-1 protein (Fig. 4D and E) and extracellular soluble protein levels of VCAM-1, E-selectin, ICAM-1, and MCP-1 (Fig. 4F through I) and IL-6 (see Fig. S7B in the supplemental material). These data demonstrate that RBP4 induction of proinflammatory molecules is at least partially dependent on NF-κB activation.
Fig 4.

RBP4 activates NF-κB and stimulates proinflammatory protein expression at least partially through an NF-κB-dependent mechanism in HRCEC. (A) Nuclear translocation of NF-κB p65 detected by immunocytochemistry in HRCEC after treatment with holo-RBP4 (100 μg/ml), BSA, or TNF-α for 12 h; untreat, untreated cells. (B) The relative nuclear transcriptional activity of NF-κB p65 was quantified using the ELISA-based TransAM NF-κB assay. HRCEC were treated with BSA or holo-RBP4 (100 μg/ml) for 6 h, and nuclear extracts were prepared and analyzed. Western blots of beta-actin, GAPDH, and fibrillarin demonstrate the enrichment and quality of nuclear extract preparations. Lane C, cytosolic extract; lane N, nuclear extract (10 μg protein/lane). (C) HRCEC were treated with either BSA or holo-RBP4 (100 μg/ml) for 24 h, and phosphorylation of NF-κB p65 at Ser536 was quantified by Western blotting and band densitometry analysis. (D through I) HRCEC were pretreated with NF-κB inhibitors PDTC (100 or 500 μM) or JSH-23 (10 or 50 μM) for 2 h prior to the addition of holo-RBP4 at 100 μg/ml for 24 h. (D through E) Western blots of VCAM-1 following treatment with RBP4 in the absence or presence of PDTC (D) or JSH-23 (E). (F through I) ELISA-based quantification of soluble extracellular levels of sVCAM-1 (F), sICAM-1 (G), MCP-1 (H), and E-selectin (I) in HRCEC media. Significant differences compared to RBP4 treatment alone: *, P < 0.05; **, P < 0.01; ***, P < 0.001 by one-way ANOVA with Tukey's post hoc test. Significant difference compared to BSA treatment (panels B and C): †, P < 0.01 by Student's t test.
RBP4 induces proinflammatory molecules through an NADPH oxidase-dependent mechanism.
Since NADPH oxidase activation is the main source of oxidative stress in endothelial cells and contributes to NF-κB activation (27), we investigated whether RBP4-induced changes in gene expression could be the result of NADPH oxidase activation. We observed that Nox2 and Nox4 protein expression was increased in a dose-dependent manner by treatment with holo-RBP4 in HRCEC (Fig. 5A and B). To determine if RBP4-mediated induction of proinflammatory molecules is dependent upon NADPH oxidase activation, we pretreated HRCEC with two specific chemical inhibitors of NADPH oxidase, diphenyleneiodonium (DPI) and apocynin, for 2 h prior to the addition of holo-RBP4. Pretreatment with either DPI or apocynin significantly reduced RBP4-mediated induction of cellular VCAM-1 protein (Fig. 5C and D) and extracellular soluble VCAM-1, E-selectin, ICAM-1, and MCP-1 (Fig. 5E through H) and IL-6 (see Fig. S7B in the supplemental material). This demonstrates that RBP4 activates NADPH oxidase as a mechanism to increase oxidative stress and proinflammatory gene expression.
Fig 5.

RBP4 activates NADPH oxidase protein expression and stimulates proinflammatory protein expression via an NADPH oxidase-dependent mechanism in HRCEC. (A and B) HRCEC were treated with BSA or increasing concentrations of holo-RBP4 for 24 h, and cell lysates were analyzed by Western blotting for Nox2 (A) and Nox4 (B). (C through H) HRCEC were pretreated with NADPH oxidase inhibitors DPI (20 μM) or apocynin (500 or 1,000 μM) for 2 h prior to the addition of holo-RBP4 at 100 μg/ml for 24 h. (C and D) Western blots of VCAM-1 following treatment with RBP4 in the absence or presence of DPI (C) or apocynin (D). (E through H) ELISA-based quantification of soluble extracellular levels of sVCAM-1 (E), sICAM-1 (F), MCP-1 (G), and E-selectin (H) in HRCEC media. Significant differences compared to RBP4 treatment alone: *, P < 0.05; **, P < 0.01; ***, P < 0.001 by one-way ANOVA with Tukey's post hoc test.
RBP4-induced endothelial inflammation is retinol independent.
The classically studied and well-defined function of serum RBP4 is to deliver retinol to target tissues (9, 38). Retinol and its enzymatically derived retinoid derivatives, most notably retinoic acid, are known to exert many effects on cellular biology, including the ability to bind nuclear receptors and activate gene expression (9). Thus, it is important to discern whether the effects of RBP4 on endothelial cells are retinol dependent. To investigate this question, we first measured the expression of the RBP4 receptor, stimulated by retinoic acid gene 6 (STRA6), in HRCEC by qRT-PCR and Western blotting. We found an extremely low level of STRA6 mRNA present in HRCEC (20-fold less than the HPRT housekeeping gene), and STRA6 mRNA expression level was unchanged by treatment with holo-RBP4 (Fig. 6A). Moreover, STRA6 protein expression was undetectable by Western blotting (Fig. 6B). To determine if holo-RBP4 delivered retinoids to HRCEC, we performed HPLC to quantify the level of retinoid isomers in HRCEC following treatment with holo-RBP4 or an equimolar concentration of BSA, retinol, retinal, or retinoic acid. No intracellular retinoids were detected after treatment with BSA (data not shown). Treatment with each retinoid isomer resulted in a significant increase in intracellular retinoids (Fig. 6C). In contrast, treatment with holo-RBP4 did not significantly increase intracellular retinoids, and only a small amount of retinol was detected in HRCEC treated with holo-RBP4 (Fig. 6C). This confirms that the RBP4 receptor STRA6, which functions to take up retinol from holo-RBP4, is not present in HRCEC and that RBP4-mediated inflammation in HRCEC is STRA6 independent.
Fig 6.

RBP4-mediated activation of NF-κB, NADPH oxidase, and proinflammatory molecules is retinol independent. (A) Quantitative RT-PCR analysis of STRA6 mRNA expression in HRCEC following 24 h of treatment with RBP4 (100 μg/ml) or BSA or no treatment (Untr.). STRA6 mRNA expression is shown relative to HPRT housekeeping gene expression and is 20-fold less abundant than HPRT. (B) STRA6 protein expression was undetectable by Western blotting of HRCEC. HEK-293A cells stably expressing STRA6 were also analyzed as a positive control. (C) HRCEC intracellular retinoid content following treatment with holo-RBP4 (100 μg/ml) or an equimolar amount (4.75 μM) of retinol, retinal, or retinoic acid. HRCEC did not uptake a significant amount of retinol from holo-RBP4. (D and E) Western blots of VCAM-1 and Nox2 (D) or Nox4 (E) in HRCEC treated with increasing concentrations of apo-RBP4 for 24 h. (F) HRCEC were treated with either BSA or apo-RBP4 (100 μg/ml) for 24 h, and phosphorylation of NF-κB p65 at Ser536 was quantified by Western blotting and band densitometry analysis. (G through J) ELISA-based quantification of soluble extracellular levels of MCP-1 (G), sVCAM-1 (H), sICAM-1 (I), and E-selectin (J) in HRCEC media following 24 h of treatment with increasing concentrations of either apo- or holo-RBP4 as indicated. Significant differences compared to BSA treatment: *, P < 0.05; **, P < 0.01; ***, P < 0.001 by one-way ANOVA with Tukey's post hoc test.
Since the treatment of HRCEC with retinoids alone in the cell culture media caused an increase in intracellular retinoids, we were able to investigate the possibility that retinoids directly contribute to the induction of proinflammatory molecules. To test this, we evaluated the expression of proinflammatory molecules in endothelial cells following treatment with retinol, retinoic acid, holo-RBP4, or BSA alone. Retinol treatment was unable to increase cellular VCAM-1 protein levels (see Fig. S3A in the supplemental material). Likewise, treatment with retinol or retinoic acid did not significantly increase the extracellular soluble protein levels of VCAM-1, ICAM-1, E-selectin, or MCP-1 (see Fig. S3C through F in the supplemental material).
To further investigate if retinol is responsible for the effects of holo-RBP4 in endothelial cells, we independently purified apo-RBP4 (retinol-free RPB4) and confirmed the absence of retinol by spectrophotometric wavelength scan analysis (data not shown). We tested the effects of treating HRCEC with apo-RBP4 versus holo-RBP4. Similar to treatment with holo-RBP4 (Fig. 2), treatment with apo-RBP4 increased VCAM-1, Nox2, and Nox4 protein in a dose-dependent manner (Fig. 6D and E) and also increased activation of NF-κB through phosphorylation at Ser536 (Fig. 6F). In addition, apo-RBP4 induced a dose-dependent increase in extracellular soluble MCP-1, VCAM-1, ICAM-1, E-selectin, and IL-6 that was comparable to the level of induction by holo-RBP4 (Fig. 6G through J; see Fig. S7A in the supplemental material). Similarly, pretreatment with NF-κB or NADPH oxidase inhibitors also blocked apo-RBP4-mediated induction of proinflammatory factors (see Fig. S4 and S7 in the supplemental material).
RBP4-induced endothelial inflammation occurs in HUVEC via NF-κB and NADPH oxidase-dependent pathways.
Previous studies have shown that endothelial cells can be divided into at least two distinct subtypes, microvascular and macrovascular, which often differ in their cellular homeostatic properties and responses to stimuli (28–30, 35, 63), yet both micro- and macrovascular reactivities are impaired in patients with type 2 diabetes (14, 20). Since elevation of serum RBP4 has been linked to insulin resistance in type 2 diabetes (25, 32, 68) as well as in microvascular and macrovascular diseases (10, 34, 43, 57, 58, 60), it is important to determine how RBP4 affects both subtypes of endothelial cells. To establish if RBP4 exerts similar proinflammatory effects in macrovascular endothelial cells, we analyzed the effects of RBP4 on the expression of proinflammatory molecules and the activation of NF-κB and NADPH oxidase in human umbilical vein endothelial cells (HUVEC).
Similar to our results in HRCEC, upregulation of VCAM-1, ICAM-1, and MCP-1 mRNA was observed in HUVEC treated with holo-RBP4 (see Fig. S5A through C in the supplemental material). In HUVEC, RBP4 induced a dose-dependent increase in VCAM-1, E-selectin, Nox2, and Nox4 protein (Fig. 7D) and a dose-dependent increase in extracellular soluble protein levels of VCAM-1, ICAM-1, MCP-1, and IL-6 (Fig. 7A through C; see Fig. S7D in the supplemental material). Maximum effects were observed with 100 μg/ml of apo- or holo-RBP4, although significantly increased expression of proinflammatory factors was also observed following treatment with as little as 10 to 25 μg/ml RBP4 (Fig. 7A through C; see Fig. S7D in the supplemental material). RBP4-mediated induction of MCP-1 was especially robust, as a concentration of 10 μg/ml increased MCP-1 levels 6-fold compared to cells treated with BSA alone (Fig. 7C). The RBP4-mediated increase in proinflammatory factors was sufficient to induce leukocyte adherence to HUVEC (see Fig. S5D and E in the supplemental material). RBP4 activated NF-κB subunit p65 in HUVEC, as demonstrated by increased nuclear translocation and phosphorylation at Ser536 (Fig. 7E and F). Similar to what was observed in HRCEC, RBP4-mediated induction of proinflammatory molecules in HUVEC was dependent on NF-κB and NADPH oxidase (Fig. 8). Moreover, the proinflammatory effects of RBP4 on HUVEC were retinol independent, since apo-RBP4 and holo-RBP4 had similar potency levels (Fig. 7A through C; see Fig. S7D in the supplemental material) and STRA6 protein expression was undetectable in HUVEC by Western blotting (see Fig. S6B in the supplemental material).
Fig 7.

RBP4 induces NF-κB, NADPH oxidase, and proinflammatory molecules in human umbilical vein endothelial cells. (A through C) ELISA-based quantification of soluble extracellular levels of sVCAM-1 (A), sICAM-1 (B), and MCP-1 (C) in HUVEC media following 24 h of treatment with increasing concentrations of either apo- or holo-RBP4 as indicated. (D) Western blots of E-selectin and Nox2, VCAM-1, and Nox4 in HUVEC treated with increasing concentrations of holo-RBP4 for 24 h. (E) Nuclear translocation of NF-κB p65 detected by immunocytochemistry in HUVEC after treatment with holo-RBP4 (100 μg/ml), BSA, or TNF-α for 12 h. Untreat, untreated cells. (F) HUVEC were treated with either BSA or apo-RBP4 (100 μg/ml) for 24 h, and phosphorylation of NF-κB p65 at Ser536 was quantified by Western blotting and band densitometry analysis. Significant differences compared to BSA treatment: *, P < 0.05; **, P < 0.01; ***, P < 0.001 by one-way ANOVA with Tukey's post hoc test; †, P < 0.01 by Student's t test.
Fig 8.

RBP4-mediated induction of proinflammatory proteins in HUVEC is via activation of NADPH oxidase and NF-κB. (A through C) HUVEC were pretreated with NF-κB inhibitors PDTC (100 or 500 μM) or JSH-23 (10 or 50 μM) or with NADPH oxidase inhibitors DPI (20 μM) or apocynin (500 or 1,000 μM) for 2 h prior to the addition of holo-RBP4 at 100 μg/ml for 24 h. ELISA-based quantification of soluble extracellular levels of sICAM-1 (A), sVCAM-1 (B), and MCP-1 (C) in HUVEC media. Significant differences compared to RBP4 treatment alone: *, P < 0.05; **, P < 0.01; ***, P < 0.001 by one-way ANOVA; †, P < 0.01 by Student's t test. (D and E) Western blots of VCAM-1 in HUVEC following RBP4 treatment in the presence or absence of PDTC (D) or DPI (E).
DISCUSSION
Clinical studies have linked elevation of serum RBP4 to vascular inflammation and vascular disease (10, 34, 43, 57, 58, 60). However, the significance of RBP4 elevation in the pathogenesis of vascular disease is unknown. Endothelial inflammation is a key component in the pathogenesis of both atherosclerosis and microvascular complications of diabetes (20, 39, 55, 62). Endothelial inflammation is initiated by the production of proinflammatory molecules, including cell adhesion molecules, VCAM-1, ICAM-1, and E-selectin, which serve as docking proteins for leukocyte adherence, and soluble proinflammatory factors such as MCP-1 and the cytokine IL-6 that attract circulating leukocytes to inflamed endothelium (24, 39, 55). The present study is the first to show that elevation of serum RBP4 induces expression of all of these key proinflammatory factors in both primary human microvascular and macrovascular endothelial cells, resulting in a significant increase in leukocyte adherence. Furthermore, we establish that RBP4 induces inflammation in endothelial cells by a novel mechanism that is unrelated to its role as a carrier of retinoids and involves activation of NADPH oxidase and NF-κB, two key signaling molecules known to be involved in the induction of endothelial inflammation during diabetic retinopathy and atherosclerosis (2, 11, 12, 16, 27, 39, 64). These data suggest that RBP4 elevation may directly contribute to vascular inflammation and the development and/or progression of vascular diseases such as atherosclerosis and diabetic retinopathy.
Several clinical studies have found a significant increase in serum RBP4 levels in patients with obesity, insulin resistance, type 2 diabetes, or vascular disease compared to normal subjects; however, the absolute level of RBP4 measured in patient serum varies between different clinical studies since there is no standardized method for measuring serum RBP4 levels (reviewed in reference 31). This has resulted in a rather broad definition of normal/healthy versus disease serum RBP4 levels, so that RBP4 levels in normal/healthy subjects range from 10 to 50 μg/ml and levels in patients with obesity, insulin resistance, type 2 diabetes, or vascular disease can range from 17 to 150 μg/ml (10, 25, 31, 32, 34, 41, 43, 54, 57, 58, 60). Therefore, in the present study we chose to test a wide range of concentrations of RBP4 (10 to 100 μg/ml) encompassing the full spectrum of serum RBP4 levels reported in various clinical studies. This approach allowed us to determine how incremental changes in total RBP4 levels may cause significant changes in production of proinflammatory molecules. The most significant induction of proinflammatory molecules was achieved with the maximum concentration of RBP4 (100 μg/ml), although a much lower concentration of only 10 to 25 μg/ml RBP4 was often sufficient to cause a significant increase in proinflammatory molecules compared to controls (Fig. 1, 2, 6, and 7; see Fig. S2 and S7 in the supplemental material). This suggests that endothelial cells are responsive to small increases in RBP4 levels, which may explain why clinical studies have found that a relatively incremental increase in serum RBP4 level (as low as 2.7 to 8 μg/ml increase) is associated with vascular disease (43, 57, 60).
The best-characterized function of RBP4 is to deliver retinol to tissues via binding to the RBP4 cell membrane receptor STRA6 (9, 38). A previous study (8) showed that RBP4 binding to STRA6 can stimulate JAK2/STAT5 signaling to mediate changes in gene expression and inhibit insulin signaling in adipocytes; however, that study found that only holo-RBP4 and not apo-RBP4 was capable of inducing such STRA6-mediated cell signaling. In contrast, we demonstrate that RBP4-mediated induction of endothelial inflammation was independent of retinol and STRA6, since (i) STRA6 protein expression was undetectable in HRCEC and HUVEC; (ii) treatment with holo-RBP4 did not result in cellular uptake of retinoids; and (iii) most importantly, apo-RBP4 induced production of proinflammatory molecules to the same extent as holo-RBP4. Our results are similar to those of a recent study (53), which showed that RBP4 induces production of proinflammatory cytokines in macrophages through a retinol- and STRA6-independent mechanism involving the activation of NF-κB. Thus, RBP4 is capable of exerting retinol-dependent effects via activation of STRA6 and retinol-independent effects via activation of different receptors and cell signaling pathways in different cell types.
The receptor stimulated by RBP4 in endothelial cells has not been determined. In macrophages, RBP4-mediated induction of proinflammatory cytokines is mediated in part by activation of toll-like receptor 4 (TLR4) signaling (22, 53). TLR4 is expressed in endothelial cells (4, 46), and TLR4 signaling has the potential to activate downstream NF-κB signaling (22, 46, 53), so it is possible that RBP4 acts through TLR4 to mediate NF-κB-dependent inflammation in endothelial cells. Other candidate receptors for RBP4 in endothelial cells include other TLR family members and a member of the low-density lipoprotein-related receptor family, megalin (also known as LRP2), which has previously been shown to bind to RBP4 in kidney epithelium (17) and was reported to be expressed in endothelial cells (18). However, megalin mRNA is undetectable by qRT-PCR in HRCEC and HUVEC (see Fig. S6D in the supplemental material), whereas TLR4 mRNA is present in both cell types (see Fig. S6D). Future studies will investigate the cell surface receptor and signaling pathways activated by RBP4 in endothelial cells and examine vascular inflammation in RBP4 transgenic (RBP4-Tg) mice.
Our finding that apo-RBP4 and holo-RBP4 similarly induce inflammation in endothelial cells is in agreement with previous clinical studies (25, 50, 53), which found that increases in apo-RBP4 (or the ratio of RBP4 to retinol) are most significantly associated with insulin resistance and type 2 diabetes. This implies that the association of elevated serum RBP4 with vascular diseases is likely independent of serum retinol levels, so that elevation of RBP4 specifically, and not a coincident increase in serum retinol, is associated with vascular disease. Therefore, it may be possible to diagnose subclinical vascular inflammation by simply measuring total serum RBP4 levels and disregarding the level of serum retinol. Furthermore, if RBP4-induced endothelial inflammation is reversible upon reduction of RBP4 levels, this strategy could potentially be applied as a therapy for patients at risk for developing atherosclerosis or microvascular disease. Previous studies have already found that genetically or pharmacologically lowering RBP4 levels in mice can improve insulin resistance (68). Moreover, clinical studies have shown that exercise or other weight loss interventions that resulted in improved insulin sensitivity also significantly reduced serum RBP4 levels (5, 32, 33, 45).
In conclusion, we have identified a novel mechanism whereby elevation of serum RBP4 induces significant endothelial inflammation in primary human microvascular and macrovascular endothelial cells. We show that RBP4-mediated endothelial inflammation is retinol and STRA6 independent and acts via NADPH oxidase- and NF-κB-dependent pathways. These are the first studies to provide mechanistic insights into the clinical correlation of elevated serum RBP4 with macro- and microvascular diseases, such as atherosclerosis and diabetic retinopathy, which develop in part due to chronic low-grade vascular inflammation. Thus, RBP4 may contribute to cardiovascular disease and diabetic microvascular complications by inducing endothelial inflammation. These findings could lead to new diagnostic and therapeutic approaches to predict and reduce morbidity and mortality in patients with cardiovascular disease and diabetes.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a grant from the Oklahoma Center for the Advancement of Science and Technology (HR10-150S, to K.M.F.), a Fight for Sight Grant-in-Aid award (to K.M.F.), and National Institutes of Health NEI grants EY018659, EY012231, and EY019309 (to J.-X.M.) and NCRR grant P20RR024215 (to J.-X.M.).
Footnotes
Published ahead of print 15 October 2012
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1. Aeberli I, et al. 2007. Serum retinol-binding protein 4 concentration and its ratio to serum retinol are associated with obesity and metabolic syndrome components in children. J. Clin. Endocrinol. Metab. 92:4359–4365 [DOI] [PubMed] [Google Scholar]
- 2. Al-Shabrawey M, et al. 2005. Inhibition of NAD(P)H oxidase activity blocks vascular endothelial growth factor overexpression and neovascularization during ischemic retinopathy. Am. J. Pathol. 167:599–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Al-Shabrawey M, et al. 2008. Role of NADPH oxidase in retinal vascular inflammation. Invest. Ophthalmol. Vis. Sci. 49:3239–3244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Andonegui G, et al. 2009. Mice that exclusively express TLR4 on endothelial cells can efficiently clear a lethal systemic Gram-negative bacterial infection. J. Clin. Invest. 119:1921–1930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Balagopal P, et al. 2007. Reduction of elevated serum retinol binding protein in obese children by lifestyle intervention: association with subclinical inflammation. J. Clin. Endocrinol. Metab. 92:1971–1974 [DOI] [PubMed] [Google Scholar]
- 6. Barazzoni R, et al. 2011. High plasma Rbp4 is associated with systemic inflammation independently of low Rbp4 adipose expression and is normalized by transplantation in non-obese, non-diabetic patients with chronic kidney disease. Clin. Endocrinol. (Oxf.) doi:10.1111/j.1365-2265.2011.03990.x [DOI] [PubMed] [Google Scholar]
- 7. Beckman JA, Creager MA, Libby P. 2002. Diabetes and atherosclerosis: epidemiology, pathophysiology, and management. JAMA 287:2570–2581 [DOI] [PubMed] [Google Scholar]
- 8. Berry DC, Jin H, Majumdar A, Noy N. 2011. Signaling by vitamin A and retinol-binding protein regulates gene expression to inhibit insulin responses. Proc. Natl. Acad. Sci. U. S. A. 108:4340–4345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Blomhoff R, Blomhoff HK. 2006. Overview of retinoid metabolism and function. J. Neurobiol. 66:606–630 [DOI] [PubMed] [Google Scholar]
- 10. Bobbert T, et al. 2010. Relation between retinol, retinol-binding protein 4, transthyretin and carotid intima media thickness. Atherosclerosis 213:549–551 [DOI] [PubMed] [Google Scholar]
- 11. Brand K, et al. 1996. Activated transcription factor nuclear factor-kappa B is present in the atherosclerotic lesion. J. Clin. Invest. 97:1715–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brand K, Page S, Walli AK, Neumeier D, Baeuerle PA. 1997. Role of nuclear factor-kappa B in atherogenesis. Exp. Physiol. 82:297–304 [DOI] [PubMed] [Google Scholar]
- 13. Bremer AA, Devaraj S, Afify A, Jialal I. 2011. Adipose tissue dysregulation in patients with metabolic syndrome. J. Clin. Endocrinol. Metab. 96:E1782–E1788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Caballero AE, et al. 1999. Microvascular and macrovascular reactivity is reduced in subjects at risk for type 2 diabetes. Diabetes 48:1856–1862 [DOI] [PubMed] [Google Scholar]
- 15. Cersosimo E, DeFronzo RA. 2006. Insulin resistance and endothelial dysfunction: the road map to cardiovascular diseases. Diabetes Metab. Res. Rev. 22:423–436 [DOI] [PubMed] [Google Scholar]
- 16. Chen P, Guo AM, Edwards PA, Trick G, Scicli AG. 2007. Role of NADPH oxidase and ANG II in diabetes-induced retinal leukostasis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 293:R1619–R1629 [DOI] [PubMed] [Google Scholar]
- 17. Christensen EI, et al. 1999. Evidence for an essential role of megalin in transepithelial transport of retinol. J. Am. Soc. Nephrol. 10:685–695 [DOI] [PubMed] [Google Scholar]
- 18. Chun JT, Wang L, Pasinetti GM, Finch CE, Zlokovic BV. 1999. Glycoprotein 330/megalin (LRP-2) has low prevalence as mRNA and protein in brain microvessels and choroid plexus. Exp. Neurol. 157:194–201 [DOI] [PubMed] [Google Scholar]
- 19. Collins T, et al. 1995. Transcriptional regulation of endothelial cell adhesion molecules: NF-kappa B and cytokine-inducible enhancers. FASEB J. 9:899–909 [PubMed] [Google Scholar]
- 20. Creager MA, Luscher TF, Cosentino F, Beckman JA. 2003. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy. Part I. Circulation 108:1527–1532 [DOI] [PubMed] [Google Scholar]
- 21. Cybulsky MI, et al. 2001. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J. Clin. Invest. 107:1255–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Deng ZB, et al. 2009. Adipose tissue exosome-like vesicles mediate activation of macrophage-induced insulin resistance. Diabetes 58:2498–2505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. De Taeye BM, et al. 2007. Macrophage TNF-alpha contributes to insulin resistance and hepatic steatosis in diet-induced obesity. Am. J. Physiol. Endocrinol. Metab. 293:E713–E725 [DOI] [PubMed] [Google Scholar]
- 24. Endemann DH, Schiffrin EL. 2004. Endothelial dysfunction. J. Am. Soc. Nephrol. 15:1983–1992 [DOI] [PubMed] [Google Scholar]
- 25. Erikstrup C, et al. 2009. RBP-to-retinol ratio, but not total RBP, is elevated in patients with type 2 diabetes. Diabetes Obes. Metab. 11:204–212 [DOI] [PubMed] [Google Scholar]
- 26. Fong DS, et al. 2004. Retinopathy in diabetes. Diabetes Care 27(Suppl 1):S84–S87 [DOI] [PubMed] [Google Scholar]
- 27. Frey RS, Ushio-Fukai M, Malik AB. 2009. NADPH oxidase-dependent signaling in endothelial cells: role in physiology and pathophysiology. Antioxid. Redox Signal. 11:791–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Geiger M, Stone A, Mason SN, Oldham KT, Guice KS. 1997. Differential nitric oxide production by microvascular and macrovascular endothelial cells. Am. J. Physiol. 273:L275–L281 [DOI] [PubMed] [Google Scholar]
- 29. Glassberg MK, et al. 1992. Microvascular and macrovascular endothelial cells produce different constrictor substances. J. Appl. Physiol. 72:1681–1686 [DOI] [PubMed] [Google Scholar]
- 30. Grafe M, et al. 1998. Human cardiac microvascular and macrovascular endothelial cells respond differently to oxidatively modified LDL. Atherosclerosis 137:87–95 [DOI] [PubMed] [Google Scholar]
- 31. Graham TE, Wason CJ, Bluher M, Kahn BB. 2007. Shortcomings in methodology complicate measurements of serum retinol binding protein (RBP4) in insulin-resistant human subjects. Diabetologia 50:814–823 [DOI] [PubMed] [Google Scholar]
- 32. Graham TE, et al. 2006. Retinol-binding protein 4 and insulin resistance in lean, obese, and diabetic subjects. N. Engl. J. Med. 354:2552–2563 [DOI] [PubMed] [Google Scholar]
- 33. Haider DG, et al. 2007. Serum retinol-binding protein 4 is reduced after weight loss in morbidly obese subjects. J. Clin. Endocrinol. Metab. 92:1168–1171 [DOI] [PubMed] [Google Scholar]
- 34. Ingelsson E, et al. 2009. Circulating retinol-binding protein 4, cardiovascular risk factors and prevalent cardiovascular disease in elderly. Atherosclerosis 206:239–244 [DOI] [PubMed] [Google Scholar]
- 35. Jackson CJ, Nguyen M. 1997. Human microvascular endothelial cells differ from macrovascular endothelial cells in their expression of matrix metalloproteinases. Int. J. Biochem. Cell Biol. 29:1167–1177 [DOI] [PubMed] [Google Scholar]
- 36. Kannel WB, McGee DL. 1979. Diabetes and cardiovascular disease. The Framingham study. JAMA 241:2035–2038 [DOI] [PubMed] [Google Scholar]
- 37. Kannel WB, McGee DL. 1979. Diabetes and glucose tolerance as risk factors for cardiovascular disease: the Framingham study. Diabetes Care 2:120–126 [DOI] [PubMed] [Google Scholar]
- 38. Kawaguchi R, et al. 2007. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science 315:820–825 [DOI] [PubMed] [Google Scholar]
- 39. Kern TS. 2007. Contributions of inflammatory processes to the development of the early stages of diabetic retinopathy. Exp. Diabetes Res. 2007:95103 doi:10.2337/db11-1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Reference deleted.
- 41. Kowalska I, et al. 2008. Serum retinol binding protein 4 is related to insulin resistance and nonoxidative glucose metabolism in lean and obese women with normal glucose tolerance. J. Clin. Endocrinol. Metab. 93:2786–2789 [DOI] [PubMed] [Google Scholar]
- 42. Reference deleted.
- 43. Li ZZ, Lu XZ, Liu JB, Chen L. 2010. Serum retinol-binding protein 4 levels in patients with diabetic retinopathy. J. Int. Med. Res. 38:95–99 [DOI] [PubMed] [Google Scholar]
- 44. Libermann TA, Baltimore D. 1990. Activation of interleukin-6 gene expression through the NF-kappa B transcription factor. Mol. Cell. Biol. 10:2327–2334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lim S, et al. 2008. Insulin-sensitizing effects of exercise on adiponectin and retinol-binding protein-4 concentrations in young and middle-aged women. J. Clin. Endocrinol. Metab. 93:2263–2268 [DOI] [PubMed] [Google Scholar]
- 46. Lu Z, et al. 2012. Toll-like receptor 4 activation in microvascular endothelial cells triggers a robust inflammatory response and cross talk with mononuclear cells via interleukin-6. Arterioscler. Thromb. Vasc. Biol. 32:1696–1706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lumeng CN, Deyoung SM, Bodzin JL, Saltiel AR. 2007. Increased inflammatory properties of adipose tissue macrophages recruited during diet-induced obesity. Diabetes 56:16–23 [DOI] [PubMed] [Google Scholar]
- 48. Lumeng CN, Deyoung SM, Saltiel AR. 2007. Macrophages block insulin action in adipocytes by altering expression of signaling and glucose transport proteins. Am. J. Physiol. Endocrinol. Metab. 292:E166–E174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Masuyama H, Inoue S, Hiramatsu Y. 2011. Retinol-binding protein 4 and insulin resistance in preeclampsia. Endocr. J. 58:47–53 [DOI] [PubMed] [Google Scholar]
- 50. Mills JP, Furr HC, Tanumihardjo SA. 2008. Retinol to retinol-binding protein (RBP) is low in obese adults due to elevated apo-RBP. Exp. Biol. Med. (Maywood) 233:1255–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Moiseyev G, et al. 2006. RPE65 is an iron(II)-dependent isomerohydrolase in the retinoid visual cycle. J. Biol. Chem. 281:2835–2840 [DOI] [PubMed] [Google Scholar]
- 52. Mostafaie N, et al. 2011. Circulating retinol-binding protein 4 and metabolic syndrome in the elderly. Wien. Med. Wochenschr. 161:505–510 [DOI] [PubMed] [Google Scholar]
- 53. Norseen J, et al. 2012. Retinol-binding protein 4 inhibits insulin signaling in adipocytes by inducing proinflammatory cytokines in macrophages through a c-Jun N-terminal kinase- and toll-like receptor 4-dependent and retinol-independent mechanism. Mol. Cell. Biol. 32:2010–2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Qi Q, et al. 2007. Elevated retinol-binding protein 4 levels are associated with metabolic syndrome in Chinese people. J. Clin. Endocrinol. Metab. 92:4827–4834 [DOI] [PubMed] [Google Scholar]
- 55. Ross R. 1999. Atherosclerosis—an inflammatory disease. N. Engl. J. Med. 340:115–126 [DOI] [PubMed] [Google Scholar]
- 56. Rovin BH, Dickerson JA, Tan LC, Hebert CA. 1995. Activation of nuclear factor-kappa B correlates with MCP-1 expression by human mesangial cells. Kidney Int. 48:1263–1271 [DOI] [PubMed] [Google Scholar]
- 57. Sasaki M, Otani T, Kawakami M, Ishikawa SE. 2010. Elevation of plasma retinol-binding protein 4 and reduction of plasma adiponectin in subjects with cerebral infarction. Metabolism 59:527–532 [DOI] [PubMed] [Google Scholar]
- 58. Solini A, Santini E, Madec S, Rossi C, Muscelli E. 2009. Retinol-binding protein-4 in women with untreated essential hypertension. Am. J. Hypertens. 22:1001–1006 [DOI] [PubMed] [Google Scholar]
- 59. Stratton IM, et al. 2000. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ 321:405–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Takebayashi K, Suetsugu M, Wakabayashi S, Aso Y, Inukai T. 2007. Retinol binding protein-4 levels and clinical features of type 2 diabetes patients. J. Clin. Endocrinol. Metab. 92:2712–2719 [DOI] [PubMed] [Google Scholar]
- 61. Ushio-Fukai M. 2006. Redox signaling in angiogenesis: role of NADPH oxidase. Cardiovasc. Res. 71:226–235 [DOI] [PubMed] [Google Scholar]
- 62. van Hecke MV, et al. 2005. Inflammation and endothelial dysfunction are associated with retinopathy: the Hoorn Study. Diabetologia 48:1300–1306 [DOI] [PubMed] [Google Scholar]
- 63. Viemann D, et al. 2006. TNF induces distinct gene expression programs in microvascular and macrovascular human endothelial cells. J. Leukoc. Biol. 80:174–185 [DOI] [PubMed] [Google Scholar]
- 64. Violi F, Basili S, Nigro C, Pignatelli P. 2009. Role of NADPH oxidase in atherosclerosis. Future Cardiol. 5:83–92 [DOI] [PubMed] [Google Scholar]
- 65. Xie Y, Lashuel HA, Miroy GJ, Dikler S, Kelly JW. 1998. Recombinant human retinol-binding protein refolding, native disulfide formation, and characterization. Protein Expr. Purif. 14:31–37 [DOI] [PubMed] [Google Scholar]
- 66. Xu H, et al. 2003. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Invest. 112:1821–1830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Reference deleted.
- 68. Yang Q, et al. 2005. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature 436:356–362 [DOI] [PubMed] [Google Scholar]
- 69. Yao-Borengasser A, et al. 2007. Retinol binding protein 4 expression in humans: relationship to insulin resistance, inflammation, and response to pioglitazone. J. Clin. Endocrinol. Metab. 92:2590–2597 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.