

Abstract
Mi-2/nucleosome remodeling and deacetylase (NuRD) chromatin remodeling complexes are important regulators of chromatin structure and DNA accessibility. We examined requirements for individual domains of chromodomain helicase DNA-binding protein 4 (CHD4), a core catalytic component of NuRD complexes, as well as the NuRD subunit methyl-binding domain protein 2 (MBD2) and methylated DNA, for NuRD function in the context of tissue-specific transcription. By itself, loss of NuRD activity is not sufficient for transcriptional activation. However, NuRD complexes greatly reduce activation of the B cell-specific mb-1 (Cd79a) gene by the transcription factors EBF1 and Pax5. Using our B cell model system, we determined that the two chromodomains and ATPase/helicase and C-terminal domains (CTD) of CHD4 are all necessary for repression of mb-1 promoters by NuRD. All of these domains except the CTD are required for efficient association of CHD4 with mb-1 promoter chromatin. Loss of MBD2 expression or of DNA methylation impaired association of CHD4 with mb-1 promoter chromatin and enhanced its transcription. We conclude that repressive functions of MBD2-containing NuRD complexes are dependent on cooperative interactions between the major domains of CHD4 with histones and DNA and on binding of methylated DNA by MBD2.
INTRODUCTION
The structure of chromatin controls the accessibility of DNA to many enzyme-mediated processes, including transcription, histone modification, DNA replication and repair. Chromatin remodeling complexes (CRCs) are primary effectors in these processes, controlling access to DNA throughout the genome. The Mi-2 nucleosome remodeling and deacetylase (NuRD) CRC is an important epigenetic regulator in metazoans of many cellular processes, including DNA damage repair, cell cycle progression, and oncogenesis (reviewed in reference 20). Chromodomain helicase DNA-binding protein 4 (CHD4, or Mi-2β) and/or CHD3 (Mi-2α) comprises the catalytic core of NuRD complexes and acts as a scaffold for other factors such as histone deacetylase 1 (HDAC1) and HDAC2, p66α (GATAD2A), p66β (GATAD2B), retinoblastoma-binding protein 4 (RBBP4, or RBAP48) and RBBP7 (RBAP46), metastasis-associated gene proteins 1 to 3 (MTA1-3), and methyl-CpG binding domain proteins 2 and 3 (MBD2 and MBD3) (45, 50, 53, 54). A histone demethylase, lysine-specific demethylase 1 (LSD1, or KRDM1), has also been shown to be a component of the complex in breast cancer cells (46a).
Unique among CRCs with histone deacetylase activity, the NuRD complex can facilitate both closing and opening of chromatin (45, 53). It functions as either a transcriptional corepressor or a coactivator depending upon the developmental context of the gene being regulated. NuRD CRCs function in opposition to other chromatin remodelers such as SWI/SNF and embryonic stem cell BAF (esBAF) at the same promoters (6, 36, 51) and are often localized to areas of transcriptionally active genes and “poised” promoters with bivalent histone tail modifications (37, 38, 49, 52). The dynamic balance between opposing enzymatic activities involved in chromatin remodeling—chromatin opening versus compaction, histone acetylation versus deacetylation, and histone/DNA methylation versus demethylation—determines DNA accessibility to transcription factors and RNA polymerase II (RNAPII) complexes as well as DNA replication and repair enzymes. Indeed, both histone acetyltransferases (HATs) and HDACs have been localized to actively transcribed genes, underscoring the importance of HDACs for dynamic control of actively transcribed genes (47). NuRD complexes act as corepressors of the B cell-specific gene mb-1 (Cd79a) (6), secondary and late primary inflammatory response genes in macrophages (36), fetal γ-globin genes (7), and likely Cd8 genes (10). In contrast, NuRD activity is necessary for activating Cd4 gene expression in double-positive thymocytes (35).
Compositional differences in NuRD complexes help define their regulatory roles. For example, MTA3 is expressed at high levels in NuRD complexes of germinal center B cells, where it interacts with the transcription factor Bcl-6 to repress expression of the plasma cell-specific transcriptional program (5). MBD3-containing complexes regulate expression of 5-hydroxymethylcytosine-marked genes in embryonic stem cells (51) and play a role in colon tumor suppression through recruitment of unphosphorylated c-Jun (2). MBD2-containing NuRD complexes silence globin (7, 39, 40), the Il4 gene (15), and tumor suppressor genes including p14/p16 (25), DAPK1, and KLK10 (29). In addition, it has been shown that MBD2- and MBD3-containing NuRD complexes are biochemically distinct in human epithelial cells (21).
CHD4 is the largest subunit in NuRD complexes, where it performs the ATP-dependent nucleosome mobilization activities of the complex. It contains several highly conserved domains, including two plant homeodomain (PHD) fingers, two chromodomains (CDs), a large and complex SWI2/SNF2-type ATPase/helicase domain, two domains of unknown function (DUFs), and the C-terminal domain (CTD). The PHD domains are zinc fingers that mediate binding to histone tails, preferentially those with unmodified H3K4 and methylated H3K9 (H3K9me) residues (33). CHD4 CDs are unique in their ability to bind DNA and are also necessary for wild-type ATPase activity (3). The X-ray crystallographic structures of the two CHD1 CDs and the ATPase domain indicate that these domains interact and probably function as a unit to bind and mobilize nucleosomes (11). The CHD1 CDs likely regulate the specificity of DNA binding of the ATPase domain, ensuring binding of nucleosomal rather than free DNA. Indeed, both PHD fingers, both chromodomains, and the N-terminal DUF domain interact to regulate the ATPase activity of CHD4 (48). In addition, the CTD of CHD4 (amino acids [aa] 1577 to 1912) interacts with pericentrin A (PCNA) (42). In light of this structural data, major questions that remain unanswered include (i) whether all of the domains of CHD4 are required for performing its various functions within the context of NuRD complexes in their native chromatin environment and (ii) whether the diverse activities of CHD4 are mediated by combinatorial effects of different sets of domains.
We utilized a cell-based assay in which endogenous CHD4 was depleted together with concurrent exogenous CHD4 expression to investigate roles of various domains of CHD4 in functions of NuRD complexes at mb-1 (Cd79a), which is a model B cell-specific gene. Our results underscore the importance of multiple domains of CHD4 for repressive functions of the entire complex. In addition, we show that methylated CpGs are necessary for the binding and function of NuRD complexes at the mb-1 promoter. To investigate the identity of the NuRD complex protein mediating methyl-CpG binding in our cells, we selectively depleted MBD2 and established that MBD2-containing NuRD complexes are responsible for maintaining transcriptional silencing of mb-1 genes.
MATERIALS AND METHODS
Plasmid construction.
Plasmids for expression of early B cell factor 1 and estrogen receptor (EBF1:ER) (Vλ-EBF1:ER-Eλ) and paired box protein 5 (Pax5):ER (MSCV-Pax5:ER-YFP, where MSCV is mouse stem cell virus and YFP is yellow fluorescent protein) fusion proteins, 3′ untranslated region (UTR)-specific Chd4, and luciferase short hairpin RNAs (shRNAs) were reported previously (6, 34). The Mbd2 and Mbd3 shRNA sequences were generated using the Thermo Scientific siDESIGN center (www.thermoscientificbio.com/design-center/). To construct the shRNA expression vectors, PAGE-purified oligonucleotides (Integrated DNA Technologies) included the following: Mbd2, 5′-TGAGATGAGGCGTAAGAATATTCAAGAGATATTCTTACGCCTCATCTCTTTTTTC-3′(sense) and 5′-TCGAGAAAAAAGAGATGAGGCGTAAGAATATCTCTTGAATATTCTTACGCCTCATCTCA-3′ (antisense); Mbd3, 5′-TCCGGAAAGATGTTGATGAATTCAAGAGATTCATCAACATCTTTCCGGTTTTTTC-3′ (sense) and 5′-TCGAGAAAAAACCGGAAAGATGTTGATGAATCTCTTGAATTCATCAACATCTTTCCGGA-3′ (antisense).
Oligonucleotides were phosphorylated, annealed, and ligated into a pQCXIP-based vector (Clontech) modified to contain the coding sequence for cyan fluorescent protein (CFP) and a murine U6 promoter cassette to drive expression of the shRNA (pQCyIPU6).
Plasmid DNA encoding human CHD4 was generously provided by G. Blobel (Rockefeller University) and is described in Musselman et al. (34). Upon full sequencing of the construct, an additional mutation was discovered: L1400S. CHD4 was mutated using mutated sense and antisense oligonucleotides (Integrated DNA Technologies) as follows: for the W508E mutation, 5′-GATCCTAATCGAGAAGTGGGGTCAGCCACCATC-3′ and 5′-GACCCCACTTCTCGATTAGGATCTTCTGCACTTTGCC-3′; W547E, 5′-CTTTGTGAAAGAGCAAGGCATGTCTTACTGGCAC-3′ and 5′- CATGCCTTGCTCTTTCACAAAGAACTGCCGCTC-3′; W644E, 5′-GTCCACTACTTGATCAAGGAGCGGGACTTACCTTACGATCAG-3′ and 5′-AGTCCCGCTCCTTGATCAAGTAGTGGACGTGGC-3′; W654E, 5′-CAGGCTTCTGAGGAGAGTGAGGATGTGGAGATCC-3′ and 5′-CTCACTCTCCTCAGAAGCCTGATCGTAAGGTAAGTCC-3′; S851A, 5′-CTGCTGACAGCCTATGAATTGATCACCATTGACATG-3′ and 5′-GATCAATTCATAGGCTGTCAGCAGCACATGGAATTTC-3′; E874Q, 5′-CATCGTGGATCAAGCCCATCGGCTGAAGAAC-3′ and 5′-ATGGGCTTGATCCACGATGAGGCAGGCC-3′; K879A, 5′-CATCGGCTGGCGAACAATCAGTCTAAGTTCTTCCGG-3′ and 5′-CTGATTGTTCGCCAGCCGATGGGCTTCATCC-3′; K987A, 5′-GAAGAAATACTACGCGTACATCCTCACTCGAAATTTTGAAG-3′ and 5′-GAGGATGTACGCGTAGTATTTCTTCTGCATAGGGCTCAG-3′; R1159A, 5′-GCCTTTAGCGCAGCTCACCGGATTGGGCAA-3′ and 5′-CGGTGAGCTGCGCTAAAGGCCTGAATGTCATTATG-3′; and ΔV1853-Q1912, 5′-GCTGGCGGCCGCTCATTTGTGCAGGACTGCATTGG (ΔCTD; antisense only). Outside primers for generation of the two fragments and joining of the two overlapping fragments initially generated by PCR were the following: CD1 mutants, 5′-GTGTCCAGCTCTGAAGGGCAA-3′ and 5′-GTGGCCCTTCTTGTCCACACT-3′; CD2 mutants, 5′-GTCCACTACTTGATCAAGTGGCG-3′ and 5′CTGTCTGTACAGTTTTCCCAAGGC-3′; ATPase mutants, 5′-CAGTCTTCCTGTATTCCCTTTACAAGG-3′ and 5′-GAGTGGAAAGCAAGAAGCAGAACT-3′; R1159, 5′-CAACCATCCATACCTCTTCCCTG-3′ and 5′-TTCTTTGCCACCTGCGTGATGC-3′; and ΔCTD, 5′-CGCAAGAAGGTTCAGGAGTTTG-3′. Overlapping fragments for each of the mutants were gel purified, combined, and PCR amplified using the appropriate outside oligonucleotides listed above. The resulting PCR products were gel purified and ligated into a truncated Bluescript SK+ plasmid (bases 2851 to 579 deleted) containing wild-type CHD4 with three copies of a T7 tag [ΔBSK-(3×)T7-wtCHD4] using the following enzymes: for CD1 mutants, BsaAI and BmgBI; for CD2 mutants, BmgBI and PshAI; for ATPase mutants (all except R1159A), PshAI and ClaI; for the R1159A mutant, ClaI and AgeI; for deletion of bases 1853 to 1912 (Δ1853-1912), SphI and NotI. All PCRs were performed with Pfu DNA polymerase (Agilent Technologies). The resulting mutant CHD4 sequences were cloned into the MSCV-IThy1.1 vector as described previously (34). All plasmids were sequenced to confirm mutations.
Cell lines, transfection, retroviral infection, and flow cytometry.
The μM.2 and μM.2 EBF1:ER stably transfected cell lines were cultured as described previously (27). Generation of retroviruses, infection of cells, induction of ER fusion proteins, staining of cells, and flow cytometry were performed as described previously (6, 26, 34). Data were analyzed using FlowJo, version 7.6.5. When indicated below, cells were treated with 2 μM 5-azacytidine for 48 h.
RNA isolation and qRT-PCR.
RNA isolation and quantitative reverse transcription-PCR (qRT-PCR) were performed on sorted cells as described previously (24). Sorting of transduced cells was performed using a MoFlo XDP (Beckman Coulter) or Synergy (iCyt) cell sorter.
Antibodies.
MBD2-specific (abS0970) rabbit IgG was purchased from Epitomics. CHD4-specific (ab72418) rabbit IgG was purchased from Abcam for use in Western blotting. MBD3-specific (3896) rabbit IgG was purchased from Cell Signaling Technologies, Inc. HDAC2-specific rabbit IgG and T7 epitope-specific goat IgG were used for Western blotting as described previously (34). Rabbit IgG (ab37415), goat T7-specific antibodies (ab9138), rabbit histone H3-specific (ab1791), rabbit histone H3K4 monomethyl-specific (ab8895), rabbit histone H3K4 trimethyl-specific (ab8580), rabbit histone H3K9 acetyl-specific (ab10812), rabbit histone H3K9 trimethyl-specific (ab8898), and mouse monoclonal histone H3K27 trimethyl-specific (ab6002) antibodies were purchased from Abcam for use in chromatin immunoprecipitation (ChIP) assays.
Western blotting.
Nuclear protein extraction and Western blotting of transduced μM.2 cells were performed as described previously (1, 55). Whole-cell extracts were prepared by washing cell pellets twice in ice-cold phosphate-buffered saline (PBS), followed by resuspension in radioimmunoprecipitation assay (RIPA) buffer with protease inhibitors and dithiothreitol (DTT) (25 mM Tris-HCl, pH 7.6, 150 mM NaCl, 1% Nonidet P-40 substitute, 1% sodium deoxycholate, 0.1% SDS, 1× Halt protease inhibitor cocktail [Thermo Scientific], 5 mM DTT). Insolubles were pelleted, and soluble protein extracts were flash frozen in liquid nitrogen.
ChIP assays and chromatin accessibility.
ChIP assays were performed according to the Upstate Biotechnology protocol (16-157) with modifications. For each IP, 2 to 10 million cells were cross-linked with 1% formaldehyde for 10 min with rotation at room temperature (RT). Cross-linking was quenched with 125 mM glycine, and cells were pelleted and washed three times in ice-cold 1× PBS. Cell pellets were resuspended in 100 μl of lysis buffer (10 mM EDTA, 50 mM Tris-HCl, pH 8.1, 1% SDS, 0.5% Empigen BB [Sigma], 1× Halt protease inhibitor cocktail [Thermo Scientific]) per 10 million cells and incubated for 10 min on ice. Chromatin was sonicated to obtain an average DNA length of approximately 200 to 500 bp (30 s on and 1 min off, for six 15-min cycles on high power) using a Bioruptor (Diagenode Inc.). The lysate was cleared of insoluble material by centrifugation and diluted 10-fold in IP buffer (2 mM EDTA, 100 mM NaCl, 20 mM Tris-HCl, pH 8.1, 0.5% Triton X-100, 1× Halt protease inhibitor cocktail [Thermo Scientific]). Ten percent input was removed, and the diluted chromatin was incubated with 5 μg of antibody rotating overnight at 4°C. Immunocomplexes were collected with 2 to 4 h of incubation rotating at 4°C with 15 μl of blocked Pierce protein A/G magnetic beads (Thermo Scientific) per IP and serially washed with 500 μl of buffer A (2 mM EDTA, 20 mM Tris-HCl, pH 8.1, 0.1% SDS, 1% Triton X-100, 150 mM NaCl), buffer B (2 mM EDTA, 20 mM Tris-HCl, pH 8.1, 0.1% SDS, 1% Triton X-100, 500 mM NaCl), and buffer C (1 mM EDTA, 10 mM Tris-HCl, pH 8.1, 1% Nonidet P-40, 1% sodium deoxycholate, 250 mM LiCl) and twice with 1× Tris-EDTA (TE) buffer. Bead-bound complexes were then eluted with 120 μl of elution buffer (EB) (1% SDS, 100 mM NaHCO3) followed by the addition of 1 μg of RNase A to each eluate. Cross-links were reversed by overnight incubation at 65°C. Proteinase K digestion was performed for 1 h at 55°C, and DNA was recovered using QIAquick columns and eluted in 30 μl of EB. qRT-PCR was performed using an Applied Biosystems AB 7300 instrument and the following mb-1 promoter-specific primers: sense, 5′-AGTGGTGATGAACCAGGCCTTG-3′; antisense, 5′-ATATGTGTGGGCTCTGAGTGGAGA-3′.
Chromatin accessibility assays were performed using infected cells as described above. Ligation-mediated PCR (LM-PCR) detection of Sau96I cleavage was performed as described previously (6).
Bisulfite conversion of genomic DNA and pyrosequencing.
Genomic DNA was prepared from sorted cells by resuspending cells in lysis buffer (300 mM NaCl, 50 mM Tris-HCl, pH 8.0, 25 mM EDTA, pH 8.0, 0.2% SDS, 0.2 mg/ml proteinase K), phenol-chloroform extraction, and ethanol precipitation. Precipitated DNA was resuspended in 1× TE buffer, and bisulfite conversion was performed using an EZ DNA Methylation-Gold kit from Zymo research (D5006). Converted mb-1 promoters were amplified by performing PCR with the primers 5′-biotin-TTTTTTTTAGGGATTAGTGGTGATGAA-3′ (forward) and 5′-AAACTTCTAAACCCCCTAACA-3′ (reverse), designed with PyroMark Assay Design software, version 2.0 (Qiagen). Each PCR mixture contained 2 μl of bisulfite-converted DNA, 125 μM concentrations of the deoxynucleoside triphosphates (dNTPs), 0.2 μM each primer, 1× PCR Gold buffer, and 2.4 U of AmpliTaq Gold DNA polymerase (Applied Biosciences) in an 80-μl total volume. Thermocycling conditions were 95° for 5 min with 55 cycles of 95° for 15 s, 60° for 1 min, and 72° for 1 min, followed by a final extension at 72° for 10 min. Pyrosequencing was performed on PCR products using a PyroMark Q96 MD machine (Qiagen) and the following sequencing primers: distal-proximal mb-1 promoter CpGs 1 and 2, 5′-TCCCTTAAATCTCTCTCTAATA-3′; CpGs 3 to 5, 5′-TACCAAATTCCACTCCAAACTCC-3′.
RESULTS
Domain structure of CHD4 and measurement of functional activity in B cells.
A schematic of CHD4 depicts the major domains in this core component of NuRD CRCs (Fig. 1A). In this study, we examined the function of four domains of CHD4: chromodomains 1 and 2 (CD1 and CD2), the ATPase/helicase domain, and the C-terminal domain (CTD). A sequence alignment of CHD4 CDs (Fig. 1B), ATPase/helicase domain (Fig. 1C), and the CTD (Fig. 1D) compared to its paralogs, CHD3 and CHD5, shows remarkable conservation among the domains. Among these domains, the CDs show the most sequence variation (CD1 is 60% identical and CD2 is 62% identical, whereas the ATPase/helicase domain and CTD are 85% and 80% identical, respectively) between the various CHD proteins. CHD3 is widely expressed, especially in T cells (45), while CHD5 is expressed primarily in the nervous system (44).
Fig 1.
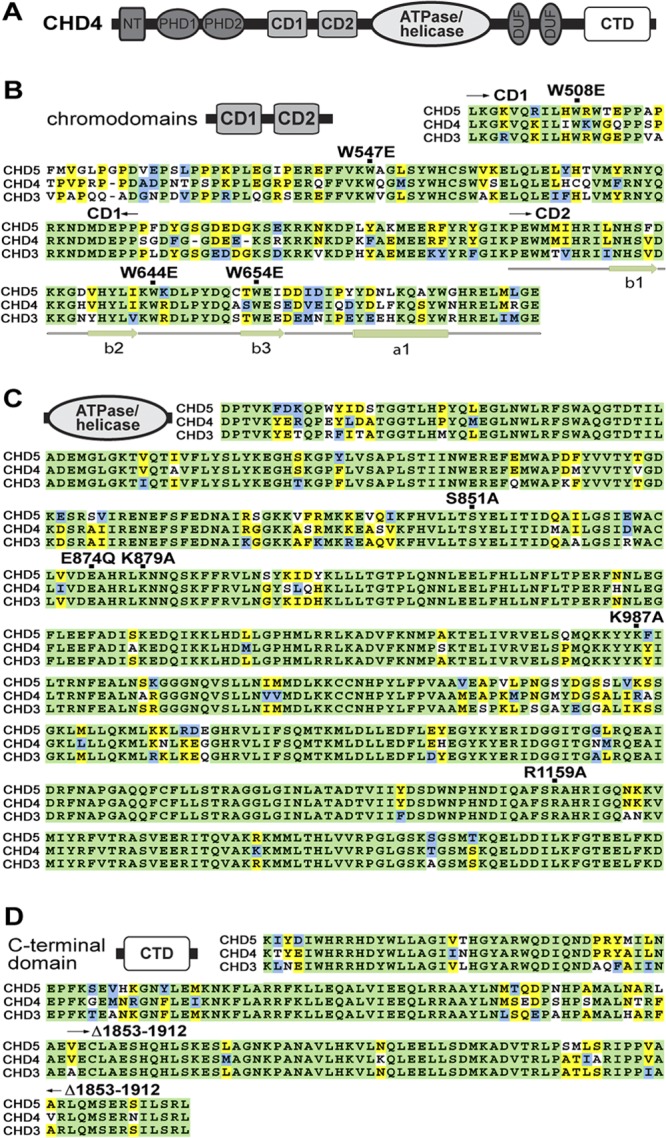
Functional domains are highly conserved between CHD proteins. (A) Schematic diagram of CHD4 domain organization. CHD4 contains two chromodomains (CD1 and CD2), a large, complex ATPase/helicase domain, and the C-terminal domain (CTD). Other domains include two PHD fingers and two domains of unknown function. (B to D) Alignment of CDs, ATPase/helicase domains, and CTD for human CHD3, CHD4, and CHD5 proteins. Highly conserved residues are shaded green, while moderately and weakly conserved residues are shown in blue and yellow, respectively.
We previously utilized a plasmacytoma cell-based system enabling depletion and reconstitution of CHD4 to examine functions of the CHD4 PHD domains in vivo (34). Here, we employed our system to explore functions of the other major CHD4 domains as well as roles of binding to methylated DNA by NuRD complexes. We employed 3′ UTR-specific shRNA to significantly deplete endogenous CHD4 from μM.2 murine plasmacytoma cells (Fig. 2E, second lane) (72% knockdown). These cells express all components of the B cell receptor except Igα (a transmembrane protein encoded by mb-1 [Cd79a]) (13). Exogenous expression of the B cell-specific transcription factors early B cell factor 1 (EBF1) and paired box protein 5 (Pax5) is required to efficiently activate mb-1 transcription in these cells, resulting in the display of membrane-bound IgM (mIgM) on the cell surface (Fig. 2A, second panel compared to first panel). Detection of mIgM by flow cytometry provides an indirect measure of mb-1 gene activation. Knockdown of endogenous CHD4 potently increases detection of mIgM in response to these transcriptional activators (Fig. 2A, third panel) by relieving chromatin compaction and facilitating DNA demethylation of mb-1 (6). Accordingly, CHD4 depletion resulted in a 513-fold increase in mb-1 transcripts over control cells expressing firefly luciferase (Luc)-specific shRNA (Fig. 2D, compare second and third bars).
Fig 2.
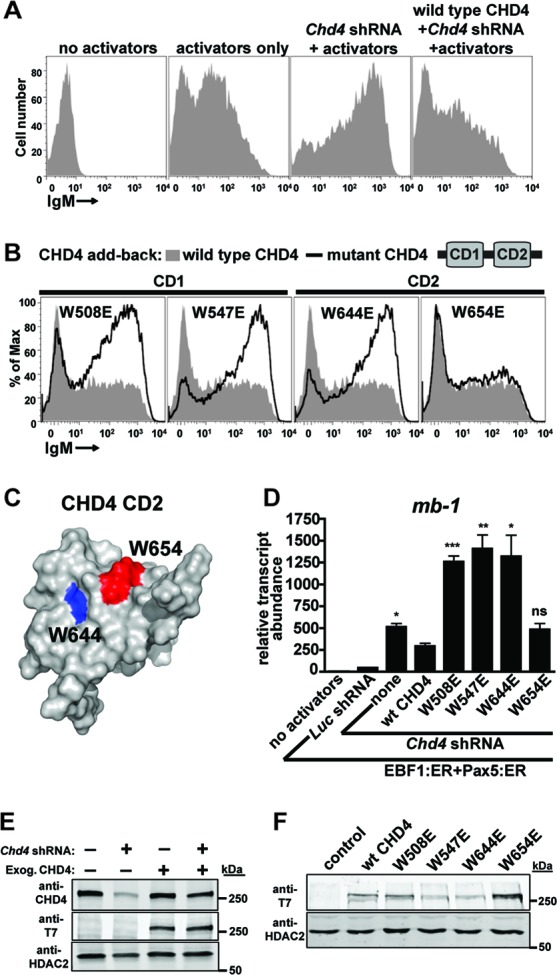
Both chromodomains of CHD4 are necessary for the transcriptional repressive function of NuRD complexes. (A) Flow cytometric analyses demonstrate increased display of membrane-bound IgM (mIgM) in response to activating transcription factors (EBF1:ER and Pax5:ER) and depletion of endogenous CHD4. In the absence of transcriptional activators, μM.2 plasmacytoma cells do not express plasma membrane-bound IgM (mIgM; first panel). When activated by 4-hydroxytamoxifen (4-OHT), the B cell-specific transcription factors EBF1 (as EBF1:ER) and Pax5 (as Pax5:ER) synergistically activate endogenous mb-1 (Cd79a) genes (encoding the Igα component of mIgM). mb-1 expression enables the assembly of mIgM on the B cell surface (second panel). Depletion of endogenous CHD4 by 3′ UTR-specific shRNA potently increases the display of mIgM in response to EBF1 and Pax5 activators (third panel). Exogenous expression of 3×T7-wt CHD4 with concurrent depletion of endogenous CHD4 restores moderate mIgM expression (fourth panel). Empty retroviruses were used as controls. (B) Flow cytometry demonstrates the effects of mutations in chromodomains 1 or 2 (CD1 or CD2) of CHD4 on mIgM expression in μM.2 cells compared to wild-type CHD4. All panels contain the transcriptional activators EBF1:ER and Pax5:ER in addition to Chd4 shRNA. All conditions included 4-OHT. Empty retroviruses and luciferase (Luc) shRNA were used as controls. (C) W654 is more solvent exposed than W644. Structure of CHD4 CD2 (Protein Data Bank [PDB] code 2ee1) with mutated residue W644 shown in blue and W654 shown in red. (D) Relative abundance of mb-1 transcripts from sorted cells in panel B. Asterisks indicate statistical significance relative to wild-type CHD4: *, P < 0.01; **, P < 0.001; ***, P < 0.0001. Data are representative of three independent experiments. (E to F) Western blots showing CHD4 depletion and/or expression of wild-type and mutant CHD4 were analyzed by blotting with anti-CHD4 (total CHD4) or anti-T7 epitope (exogenous CHD4) antibodies. HDAC2 served as a nuclear protein loading control. (E) Whole-cell extracts were prepared from sorted cells expressing the indicated shRNA and/or protein. (F) Nuclear extracts were prepared from μM.2 cells sorted for exogenous CHD4 expression and subjected to Western blotting with antibodies as shown. All data are representative of three independent experiments.
Exogenous expression of wild-type CHD4 with concurrent depletion of endogenous CHD4 attenuates mIgM display significantly (Fig. 2A, compare third and fourth panels). qRT-PCR analysis following CHD4 reconstitution detected the partial restoration of repression by NuRD. Reconstitution with wild-type CHD4 reduced mb-1 transcript abundance by 43% compared to depletion of endogenous CHD4 alone (Fig. 2D, compare third and fourth bars). However, mb-1 transcripts were 6.6-fold more abundant in cells with wild-type CHD4 reconstitution than in cells without CHD4 depletion, containing EBF1:ER and Pax5:ER transcriptional activators alone (Fig. 2D, compare second and fourth bars). Western analysis demonstrated that total CHD4 levels in the reconstituted cells (including expression of both endogenous and exogenous CHD4) comprised only 62% of endogenous CHD4 levels (Fig. 2E, compare the first and fourth lanes), explaining why transcriptional repression was not completely restored by CHD4 reconstitution.
Both CHD4 chromodomains are necessary for repression of mb-1 promoters by NuRD complexes.
CD1 (W508E and W547E) and CD2 (W644E and W654E) point mutants were designed to disrupt the chromodomains by changing highly conserved hydrophobic tryptophan residues to charged glutamic acid. Indeed, both CD1 point mutants and one CD2 mutant were completely defective for transcriptional repression, as indicated by high levels of mIgM expression compared to cells with wild-type CHD4 reconstitution (Fig. 2B, black line versus shaded area). Similar to the W508 and W547 residues of CD1, the W644 residue of CD2 comprises the hydrophobic core of the domain and is likely required for structural stability. In contrast, W654 is much more solvent exposed than W644; therefore, mutation of this residue is less likely to disrupt the structure and function of CD2 (Fig. 2C). qRT-PCR analysis of mb-1 transcript abundance revealed a substantial dominant negative effect of defective CD mutants of CHD4, with transcripts 2.4- to 2.7-fold more abundant in defective mutants than cells with endogenous CHD4 knockdown alone (Fig. 2D, compare none with W508E, W547E, and W644E). Presumably, this occurs due to incorporation of mutant CHD4 into complexes containing residual endogenous CHD4 and/or sequestration of other NuRD complex components. Western blot analysis of nuclear extracts confirmed expression of wild-type and CD mutant CHD4 proteins in the nucleus (Fig. 2F). The W508E mutant was expressed at a level similar to that of endogenous CHD4, while W547E and W644E mutants were expressed at 36% and 38% of wild-type levels, respectively. The W654E mutant with wild-type activity was overexpressed slightly compared to wild-type CHD4 (1.6-fold higher than the wild type). Interestingly, total CHD4 expression was approximately equivalent regardless of exogenous expression of T7 epitope-tagged CHD4 (Fig. 2E, first and third lanes). This indicates that cells regulate the expression of CHD4 tightly, which is logical given its diverse roles in cellular processes.
The CHD4 ATPase/helicase and C-terminal domains are necessary for NuRD complex function.
The SWI2/SNF2 ATPase/helicase domain of CHD4 is large (465 residues) and complex. It consists of several subdomains and many secondary structural motifs. We designed and tested five ATPase/helicase domain mutants: S851A, E874Q, K879A, K987A, and R1159A. These mutants were designed based on alignment of CHD4 with the structure of the Sulfolobus solfataricus SWI2/SNF2 ATPase core in complex with DNA (4). S851, K879, and K987 are putative DNA contacts, while mutations corresponding to CHD4 E874Q and R1159A were expected to be defective for ATP hydrolysis. In particular, CHD4 E874 is part of the active-site DEXX box motif. Flow cytometry measuring mIgM display as an indicator of repression by CHD4 at the mb-1 locus revealed that E874Q, K879A, and R1159A were completely defective for repressive activity while S851A and K987A were partially defective (Fig. 3A). qRT-PCR analysis confirmed these results, demonstrating significant increases in mb-1 transcript abundance upon expression of mutant CHD4 proteins (Fig. 3C, K879A, K987A, and R1159A). In addition, all defective mutants displayed various degrees of dominant negative effects similar to results seen with the CD mutants; mb-1 transcript abundance was significantly (∼3-fold) higher in cells expressing defective CHD4 mutants than in cells with endogenous CHD4 depletion alone. Western blot analysis confirmed expression of all CHD4 mutants in the nucleus. Cells expressed S851A and K987A mutants at similar levels compared to wild-type CHD4. E874Q, K879A, and R1159A were expressed at 35%, 61%, and 49% of wild-type levels, respectively (Fig. 3D).
Fig 3.
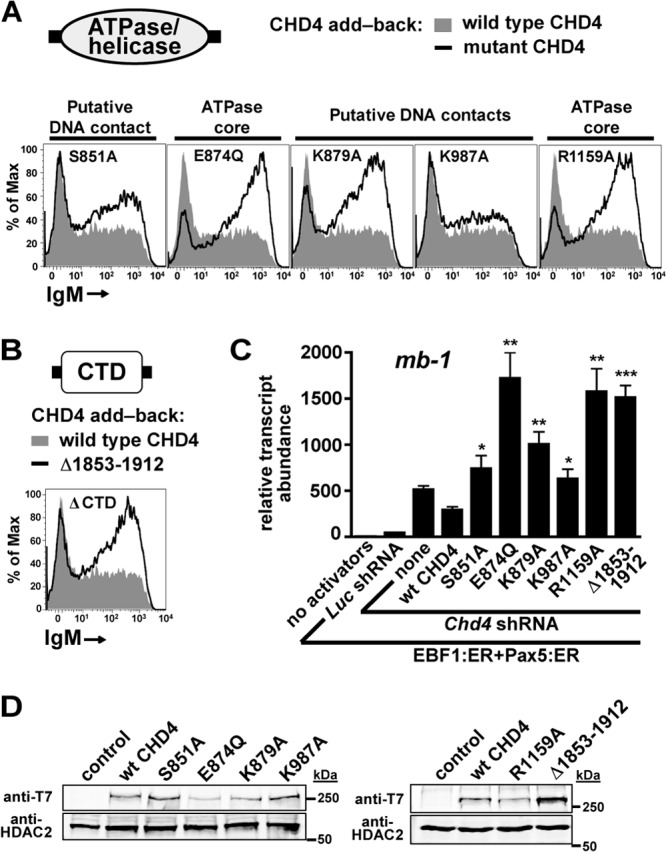
The ATPase domain and C terminus of CHD4 are necessary for the transcriptional repressive function of NuRD complexes. (A and B) Flow cytometry demonstrates the effects of mutations in the ATPase/helicase domain or C-terminal domain (CTD) of CHD4 on mIgM expression in μM.2 cells compared to wild-type CHD4. All panels contain the transcriptional activators EBF1:ER and Pax5:ER in addition to Chd4 shRNA and activation by 4-OHT. (C) Relative abundance of mb-1 transcripts from sorted cells in (A to B). Empty retroviruses and luciferase shRNA were used as controls. Asterisks indicate statistical significance relative to wild-type CHD4: *, P < 0.01; **, P < 0.001; ***, P < 0.0001. (D) Western blots showing expression of ATPase/helicase and CTD mutants were analyzed by blotting with anti-T7 epitope antibodies. Nuclear extracts were prepared from μM.2 cells sorted for exogenous CHD4 expression. HDAC2 served as a nuclear protein loading control. All data shown represent three independent experiments.
The C-terminal domain of CHD4 is required for NuRD complex function but not association with chromatin.
We wished to investigate the function of the CHD4 CTD due to its predicted alpha-helical domain structure (unpublished observations), which is characteristic of many cofactor-transcription factor interactions, as well as its published interaction with pericentrin A (42). A single C-terminal truncation, Δ1853–1912, was designed to delete the C-terminal end of the predicted domain. The high level of mIgM expression as measured by flow cytometry (Fig. 3B) showed that this mutant was completely defective for repression. qRT-PCR confirmed this result and identified a potent dominant negative effect associated with expression of the CTD mutant (Fig. 3C). Western analysis detected expression of the CTD mutant at a level 2.2-fold higher than that of the wild-type CHD4 (Fig. 3D).
ChIP analysis of mb-1 promoter occupancy of exogenous CHD4 [anti-T7 ChIP, (3×)T7-wt CHD4] revealed that all tested mutants exhibited significantly reduced occupancy compared to wild-type CHD4, with the exceptions of the CD1 and CTD mutants (Fig. 4A). Of note, we observed a basal level of CHD4 promoter occupancy likely reflecting the many components and multifunctional nature of the NuRD complex. Since the CHD4 CTD mutant was defective for mb-1 repression but displayed wild-type occupancy of the promoter, it is likely that the CTD domain is either required for the association of other NuRD complex members with CHD4 or the recruitment of corepressors such as Polycomb repressor complex 2 (PRC2). ChIP analysis of histone modifications on the mb-1 promoter (Fig. 4B) in the presence and absence of CHD4 depletion lends indirect support to the corepressor recruitment hypothesis, as trimethylation of histone H3K27 (H3K27me3, a repressive mark maintained by Ezh2, a component of PRC2) markedly decreases upon CHD4 depletion (3.2-fold reduction over transcriptional activators alone). This finding parallels the detection of decreased H3K27me3 at the promoters of rRNA genes upon CHD4 depletion (49). CHD4 depletion was associated with reduced histone H3 occupancy (a 2-fold reduction) and increased H3K9 acetylation (2.5-fold increase; a mark associated with actively transcribed genes). EBF1:ER and Pax5:ER transcriptional activators were associated with significantly increased H3K4 monomethylation (2-fold) and combined with CHD4 depletion significantly increased H3K4 trimethylation (1.9-fold). We also investigated effects of the transcriptional activators and CHD4 depletion on H3K9 trimethylation but did not find any significant variation under these conditions (unpublished observations).
Fig 4.
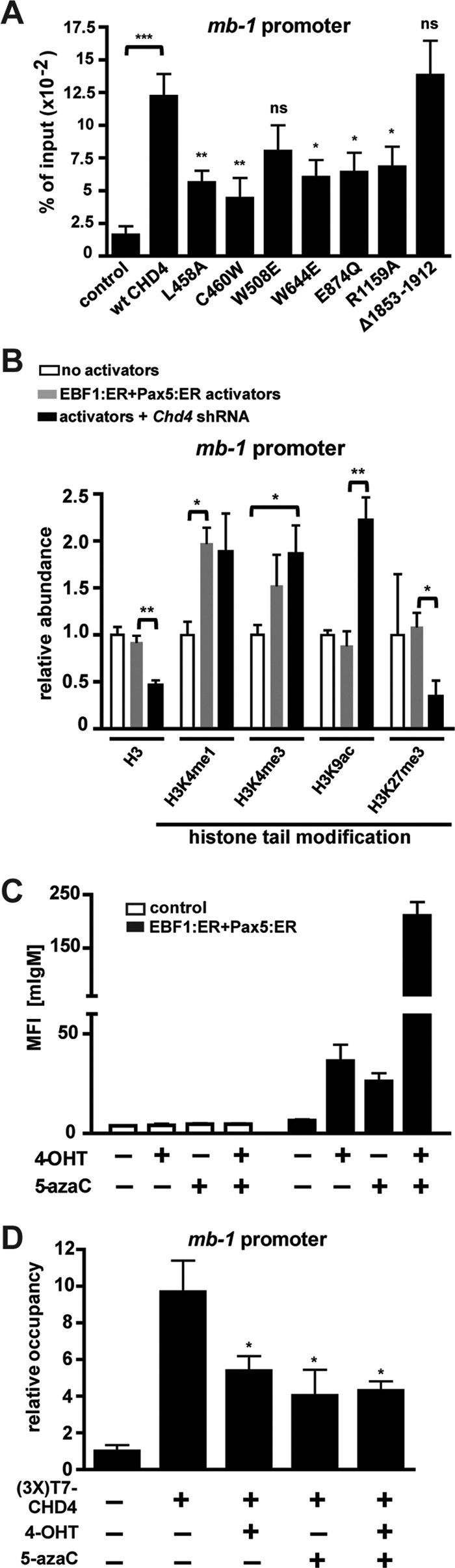
Intact CHD4 domains and methylated promoter DNA are required for occupancy of mb-1 promoters by CHD4. (A) ChIP assays using anti-T7 epitope antibodies measure promoter occupancy of exogenous (3×)T7-CHD4 (wild type or mutant) expressed in sorted μM.2 cells. RT-qPCR amplification of a 122-bp region of the mb-1 promoter was performed. Empty retroviruses were used as controls. IP values are shown as percentages of input, with background IgG IP values subtracted. Asterisks indicate statistical significance relative to wild-type CHD4: *, P < 0.01; **, P < 0.001; ***, P < 0.0001. Data shown represent five independent experiments. (B) CHD4 depletion results in activating histone modifications and histone H3 depletion from the mb-1 promoter. ChIP showing the relative abundance of the indicated histone H3 posttranslational modifications at the mb-1 promoter in the absence and presence of CHD4 depletion and/or transcriptional activators. Data were normalized to the histone H3 occupancy of each sample as well as IPs of control cells (empty retrovirus and Luc shRNA), with background IgG IP signals subtracted. Asterisks indicate statistical significance: *, P < 0.01; **, P < 0.001. Data shown represent three independent experiments. (C) Mean fluorescence intensities (MFI) of mIgM measured by flow cytometry of μM.2 cells in the absence or presence of EBF1:ER, Pax5:ER, and 5-azacytidine (5-azaC). 4-OHT was included where noted. Three independent experiments are shown. (D) ChIP assays using anti-T7 epitope antibodies detected occupancy of mb-1 promoters by exogenous (3×)T7-CHD4 in the presence or absence of 5-azaC and/or 4-OHT treatment in sorted μM.2 cells containing EBF1:ER and Pax5:ER. IP values are shown normalized to control (empty retrovirus), with background IgG IP values subtracted. Asterisks indicate statistical significance relative to untreated cells expressing (3×)T7-CHD4: *, P < 0.01. Data shown represent five independent experiments.
Methylation of promoter CpGs is necessary for transcriptional repression and CHD4 occupancy.
CpG methylation has long been associated with transcriptional repression. In order to dissect the individual roles played by methylated DNA and the NuRD complex, we used 5-azacytidine, a DNA methyltransferase inhibitor, to globally demethylate CpGs in plasmacytoma cells. However, since proliferation of the cells decreased after incubation with 5-azacytidine, mb-1 promoter CpGs were demethylated only by an average of 30% (data not shown). Despite modest demethylation of promoter CpGs, 5-azacytidine facilitated a potent transcription factor-dependent increase in mIgM display (Fig. 4C), which is very similar to results seen upon CHD4 depletion (6). ChIP of exogenous CHD4 with and without 5-azacytidine treatment revealed that modest CpG demethylation significantly decreased CHD4 occupancy of the mb-1 promoter (Fig. 4D). In addition, expression of the transcription factors EBF1 and Pax5 in the absence of CpG demethylation produced a similar decrease in occupancy, suggesting that transcription factor binding to target sequences in promoters is sufficient to displace repressive CRCs in vivo. Expression of the transcriptional activators in combination with 5-azacytidine did not significantly decrease occupancy further. These experiments demonstrate that CpG demethylation and transcription factor (EBF1 and Pax5) DNA binding both reduce promoter occupancy by CHD4 and, correspondingly, NuRD complex-mediated transcriptional repression. Together with earlier data which demonstrated that mb-1 promoters are demethylated upon CHD4 depletion (6), these results indicate that the NuRD complex is anchored by methyl-CpGs in the promoter and suggest that its occupancy prevents CpG demethylation.
MBD2 is necessary for NuRD complex occupancy and function at the mb-1 promoter.
MBD2 and MBD3 are members of the NuRD complex that contain single methyl-CpG binding domains (MBDs). MBD3 binds hydroxymethylated CpGs preferentially to methylated CpGs (12, 41, 51). However, MBD2 displays strong methyl-CpG binding activity that may aid in localizing NuRD complexes to inactive chromatin. Because we found that methylation of promoter CpGs correlates positively with CHD4 occupancy of the mb-1 promoter, we decided to investigate whether MBD2 plays an important role in tethering the NuRD complex to repressed promoters including the mb-1 promoter. We designed shRNA specific for Mbd2 and expressed it in μM.2 cells with or without the transcriptional activators EBF1:ER and Pax5:ER. MBD2 depletion facilitated a potent increase in mIgM display on the cell surface but only in response to the transcriptional activators (Fig. 5A). This response was similar to that observed with CHD4 depletion, indicating that MBD2 is necessary for the transcriptional repressive function of the NuRD complex at the mb-1 promoter. qRT-PCR analysis of mb-1 transcripts confirmed this result (Fig. 5B). MBD2 depletion increased mb-1 transcript abundance by almost 3-fold without transcriptional activators and by 12-fold over that obtained in the presence of EBF1:ER and Pax5:ER. Notably, MBD2 depletion combined with transcriptional activators increased mb-1 transcript abundance by >100-fold over control cells without activators. qRT-PCR analysis indicated that Mbd2 shRNA depleted Mbd2 transcripts by 74% (Fig. 5C). Western blot analysis did not detect MBD2 in cells expressing Mbd2 shRNA (Fig. 5D). However, anti-MBD2 signal strength was poor in these blots, and this likely reflects the lower limit of detection for the antibody under these conditions. We next performed ChIPs of exogenous CHD4 with or without MBD2 depletion. This experiment demonstrated that MBD2 is necessary for wild-type occupancy of the NuRD complex at the mb-1 promoter and likely accomplishes this through interaction with methylated CpGs (Fig. 5E). This result is almost identical to results we obtained from promoter demethylation experiments using 5-azacytidine.
Fig 5.
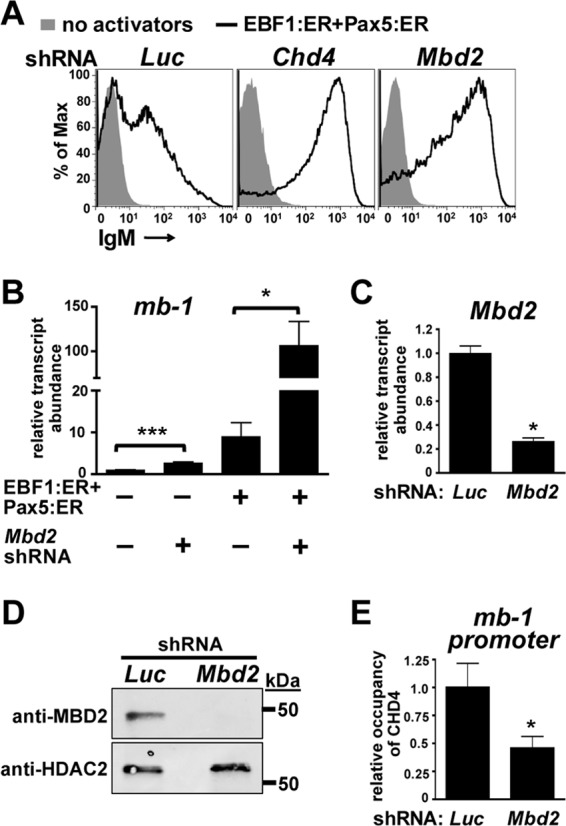
MBD2 is necessary for promoter binding and repressive function of NuRD complexes. (A) Flow cytometric analysis measures mIgM upon expression of Mbd2 shRNAs in the presence and absence of transcriptional activators EBF1:ER and Pax5:ER. Luc and Chd4 shRNA were used as negative and positive controls, respectively. (B) Relative abundance of mb-1 transcripts from sorted cells in panel A. Asterisks indicate statistical significance: *, P < 0.01; ***, P < 0.0001. Empty retroviruses and luciferase (Luc) shRNA were used as controls. Data shown represent four independent experiments. (C) Relative abundance of Mbd2 transcripts from sorted cells in the presence or absence of Mbd2 shRNA expression. Luc shRNA was used as a negative control. (D) Western blots showing MBD2 depletion were analyzed by blotting with anti-MBD2 antibodies. Whole-cell extracts were prepared from sorted cells. HDAC2 served as a nuclear protein loading control. (E) ChIP assays using anti-T7 epitope antibodies measure mb-1 promoter occupancy of exogenous (3×)T7-CHD4 with or without expression of Mbd2 shRNA in sorted μM.2 cells. IP values are shown normalized to control (Luc shRNA), with background IgG IP values subtracted. The asterisk indicates statistical significance relative to the control: *, P < 0.01. Data shown represent six independent experiments.
We next investigated the methylation status of five individual CpGs in the mb-1 promoter with and without MBD2 depletion using sodium bisulfite conversion of genomic DNA followed by pyrosequencing (Fig. 6A). Our results indicate that promoter proximal CpGs 1 and 2 are significantly demethylated upon MBD2 depletion (10.5 and 12.5% loss in methylation, respectively) but only in the presence of transcriptional activators. This result indicates that significant transcriptional activation is necessary for promoter demethylation as NuRD complex dissociation from the promoter is not sufficient for demethylation to occur. We obtained similar results upon CHD4 depletion (6). These results led us to ask whether promoter accessibility correlates with CpG methylation status and NuRD complex occupancy. To answer this question, we performed a chromatin accessibility assay, which measures protection of genomic DNA from Sau96I restriction endonuclease digestion in intact nuclei (Fig. 6B). Nuclei were obtained from cells expressing Luc (control), Chd4, Mbd2, and Mbd3 shRNAs with or without transcriptional activators and with or without 5-azacytidine treatment and incubated with a limiting concentration of Sau96I enzyme (test). Signals obtained from completely cleaved DNA are shown (control). mb-1 promoters in μM.2 cells were relatively inaccessible to Sau96I digestion in the absence of transcriptional activators regardless of shRNA expression or DNA demethylation. In the presence of the transcriptional activators EBF1:ER and Pax5:ER, CHD4 depletion resulted in a 6.4-fold increase in accessibility, while MBD2 depletion and 5-azacytidine treatment resulted in modest 2-fold and 1.5-fold increases in accessibility, respectively. Expression of Mbd3 shRNA did not affect accessibility in our assay. Although Mbd3 shRNA depleted Mbd3 transcripts by 86% as measured by RT-qPCR (Fig. 6C), MBD3 protein was not significantly depleted upon Western blot analyses 2 to 5 days after shRNA expression (Fig. 6D). This may reflect a high stability of MBD3 in the plasmacytoma cells. We observed a 3-fold increase in the transcript abundance of other NuRD complex components, Chd4 and Mbd3, upon MBD2 depletion, which likely represents an attempt by cells to compensate for the lost function of MBD2-containing NuRD complexes (Fig. 6E). These data confirm that the observed effects of MBD2 depletion were not due to off-target effects of CHD4 or MBD3 depletion and underscore the importance of this complex in maintaining cellular homeostasis. Collectively, these data demonstrate that significant increases in promoter accessibility and demethylation occur only upon full transcriptional activation of the promoter, as a result of efficient transcription factor binding and recruitment of the RNA polymerase II (RNAPII) complex. They also indicate that NuRD complex dissociation from the promoter is necessary, but not sufficient, for these events to occur.
Fig 6.
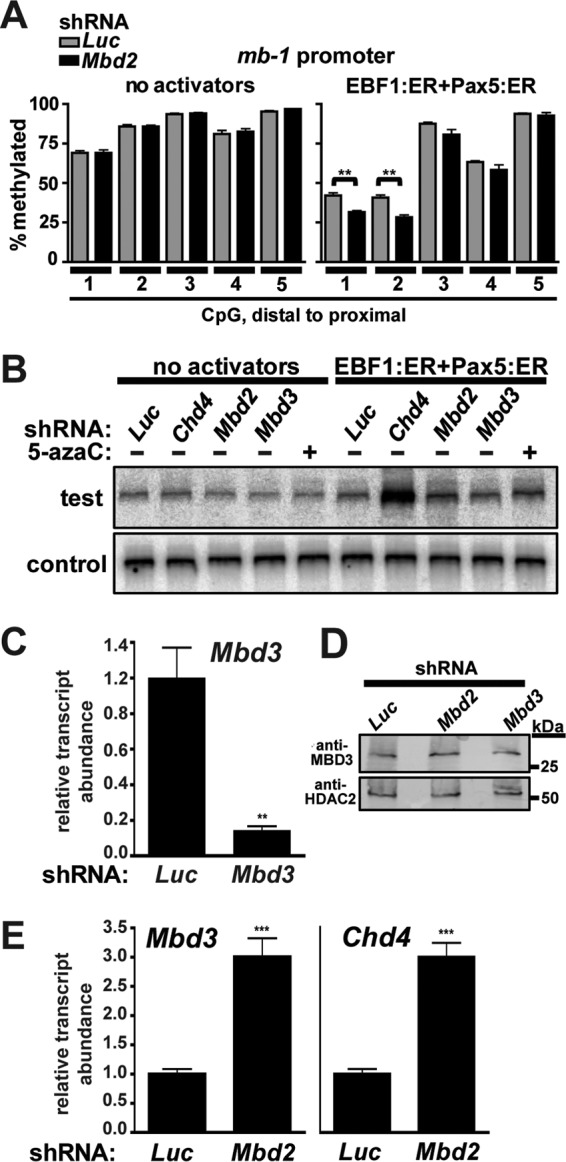
CpG methylation status and accessibility of the mb-1 promoter correlate with promoter occupancy by MBD2-NuRD complexes. (A) Depletion of MBD2 facilitates modest demethylation of CpGs 1 and 2 at the mb-1 promoter. Bars represent percent methylation of individual CpGs in the mb-1 promoter in the absence and presence of MBD2 depletion and/or transcriptional activators. CpG 1 is the most distal from the transcriptional start site, while CpG 5 is the most proximal. The asterisks indicate statistical significance: **, P < 0.001. Data shown represent four independent experiments. (B) Accessibility of mb-1 promoter chromatin in the absence or presence of transcriptional activators EBF1:ER, Pax5:ER, shRNA, and 5-azacytidine. Relative cleavage by Sau96I (test) was detected using LM-PCR. Total cleavable DNA was measured following digestion of samples to completion (control). Empty retroviruses and Luc shRNA were used as negative controls. (C) Relative abundance of Mbd3 transcripts from sorted cells in the presence or absence of Mbd3 shRNA expression. (D) Western blots showing MBD3 depletion were analyzed by blotting with anti-MBD3 antibodies. Whole-cell extracts were prepared from sorted cells 96 h after infection. HDAC2 served as a nuclear protein loading control. (E) Relative abundance of Mbd3 and Chd4 transcripts from sorted cells in the presence or absence of Mbd2 shRNA expression. The asterisks indicate statistical significance: ***, P < 0.0001.
DISCUSSION
In our studies, we examined the regulation of a model B cell-specific gene to obtain insights into the various mechanisms responsible for the transcriptional repressive functions of NuRD complexes. Release of NuRD-mediated repression is necessary for full activation of mb-1 genes by EBF1 and Pax5. We show that point mutations in the major domains of CHD4 abrogate NuRD activity and that expression of these mutants exerts a dominant negative effect that inhibits the function of remaining NuRD complex components. These results reveal an exquisitely fine-tuned mechanism of chromatin remodeling in which the role of each domain in the central catalytic scaffold is crucial for function. Two studies recently used biophysical techniques to demonstrate that the PHD and chromodomains of CHD4 interact with the ATPase domain to regulate chromatin remodeling activity (32, 48). These data support our findings that all of these domains are necessary for the transcriptional repressive function of CHD4 and, indeed, the entire NuRD complex. If any one of the major domains of CHD4 is nonfunctional, the activity of the entire NuRD complex is adversely affected as its association with nucleosomal DNA is compromised. Thus, histone tail modifications such as acetylation and methylation are also affected by mutation or depletion of CHD4. Since MBD2 binding to methylated CpGs is necessary for stable chromatin occupancy of NuRD complexes at the mb-1 promoter, this subunit is required for the other activities of the complex as well. Our findings underscore the requirement for the presence and function of different NuRD components for performance of the important activities of the complex.
Although it seems puzzling at first why the ATPase/helicase domain mutants exhibit reduced promoter occupancy in ChIP experiments, these data likely reflect the increased dissociation of the NuRD complex from nucleosomal DNA in the absence of ATP-driven DNA translocation. While most CHD4 mutants tested exhibited decreased promoter occupancy, the data indicate a residual level of promoter association that is nevertheless nonfunctional. This result is consistent with the increased dissociation hypothesis, reflecting a basal level of occupancy due to other constituents of the NuRD complex and other domains of CHD4 itself. Comparable results obtained upon promoter demethylation and MBD2 depletion also support this conclusion.
Similar to the chromodomains and ATPase/helicase domain, the C terminus of CHD4 is necessary for NuRD function. However, unlike mutations of the other domains, the CTD mutant exhibits wild-type promoter occupancy. This result suggests that the CTD is involved in the recruitment of other proteins, such as corepressors necessary for NuRD function, or that it may be involved in anchoring another subunit of the NuRD complex necessary for enzymatic activity but not chromatin localization. Potential candidates for recruited repressors include Polycomb group (PcG) complexes such as PRC2 (37), which di- and trimethylates histone H3K27 through its Ezh2 methyltransferase subunit, or other repressive chromatin remodelers such as Sin3a/HDAC complexes. Future analysis of the subunit composition of CHD4 CTD mutant NuRD complexes compared to wild-type complexes and association of the CHD4 CTD with other nuclear proteins will shed light on this important question.
Our study has also established the importance of methylated DNA in the recruitment and repressive function of NuRD complexes. We show that methylated CpGs contribute to wild-type CHD4 occupancy and transcriptional repression of mb-1 promoters. Methyl-CpGs in the mb-1 promoter recruit the NuRD complex through its MBD2 subunit. DNA methylation has long been associated with compact chromatin. NuRD is one of many enzymatic complexes present in the cell that likely establishes and maintains chromatin compaction associated with DNA methylation. It has been shown in epithelial cells that different subsets of NuRD complexes contain either MBD2 or MBD3 (21) which likely perform different functions in the cell such as preferential binding of methyl- versus hydroxymethyl-CpGs, (51), respectively, and participating in the recruitment of specific transcription factors (2). MBD3 is important for the selective targeting of NuRD complexes to genes in embryonic stem cells, where it anchors NuRD to regions enriched in 5-hydroxymethyl-CpGs (16, 17, 51). The amino acid sequence of MBD3 is very similar to MBD2, with the important distinctions that a large portion of the N-terminal sequence of MBD2 is missing from MBD3 and that the MBD in MBD3 does not preferentially bind methyl-CpGs in vitro (12, 41). It is important to note, however, that MBD2 and MBD3 are found in other repressive CRCs such as MeCp1 and Sin3A complexes and that MBD2 interacts with DNA methyltransferase 1 (DNMT1). Therefore, our result showing transcription factor-dependent activation of mb-1 transcription upon MBD2 depletion may be partly due to inhibition of the activities of other complexes and enzymes. Despite this possibility, enabling NuRD complex activity is a major part of MBD2 function at the mb-1 promoter because of the correlations we observed between MBD2 depletion, reduced CHD4 occupancy, CHD4 depletion, and activation of mb-1 transcription.
Other factors including sequence-specific DNA-binding proteins likely participate in recruitment of NuRD complexes to the mb-1 promoter. Investigators have demonstrated that NuRD complexes suppress transcriptional networks associated with pluripotency and support lineage commitment, particularly in embryonic stem cells (37, 38, 52). In more differentiated cells, the lymphocyte-specific transcription factor Ikaros (Ikzf1) is closely associated with NuRD complexes, directing both the distribution and activity of these complexes in thymocytes and T cells (18, 43, 52). Significantly, subnuclear localization of NuRD is altered by Ikaros. Although NuRD complexes associated with genes controlling transcription, cell cycle, DNA damage, apoptosis, and catabolic processes whether or not Ikaros was present, NuRD complexes containing Ikaros specifically associated with genes involved in lymphocyte differentiation. In the absence of Ikaros, NuRD complex occupancy of proproliferation genes was substantially increased. This redistribution led to dysregulated proliferation and a preleukemic gene expression signature, including expression of progenitor cell markers. In addition to Ikaros, GATA1 is another lineage-specific transcription factor that promotes a specific program of differentiation through recruitment of NuRD to developmentally appropriate sets of genes. GATA1 activates transcription of adult globin genes in erythroid cells of mice and humans by interacting with the zinc finger cofactor FOG1 to recruit the NuRD complex (8, 9, 22, 30, 31), which is mediated through its RbAp48 (RBBP4) subunit (7, 30). These data underscore the complex roles of NuRD in both transcriptional repression and activation; the NuRD complex may be directed by different transcriptional regulators to sets of target genes that vary depending on the cell type and developmental context. Together, these studies indicate that the NuRD complex plays an important role in promoting cellular differentiation versus pluripotency/self-renewal in diverse cell types.
Model of NuRD transcriptional repression.
We have established that CHD4 is necessary for association with chromatin and transcriptional repressive function of the entire NuRD complex (6). Our data indicate that the structures of both PHD domains (34), both chromodomains, both lobes of the ATPase domain, and the C-terminal domain (CTD) of CHD4 must be intact to maintain the repressive function of the complex. In our model, the PHD, chromo-, and ATPase domains interact to bind nucleosomal DNA (Fig. 7A), while the interaction between these domains stabilizes a structural unit that performs ATP hydrolysis to maintain a condensed chromatin state. This model is supported by the structure of the chromo- and ATPase domains in CHD1 (11) and a recently published study employing small-angle X-ray scattering to generate a low-resolution structure of the PHD, chromo-, ATPase, and DUF1 domains of CHD4 (48). In contrast, the CHD4 CTD binds corepressor molecules or complexes necessary for NuRD repressive function, but it is not necessary for association of the complex with chromatin.
Fig 7.
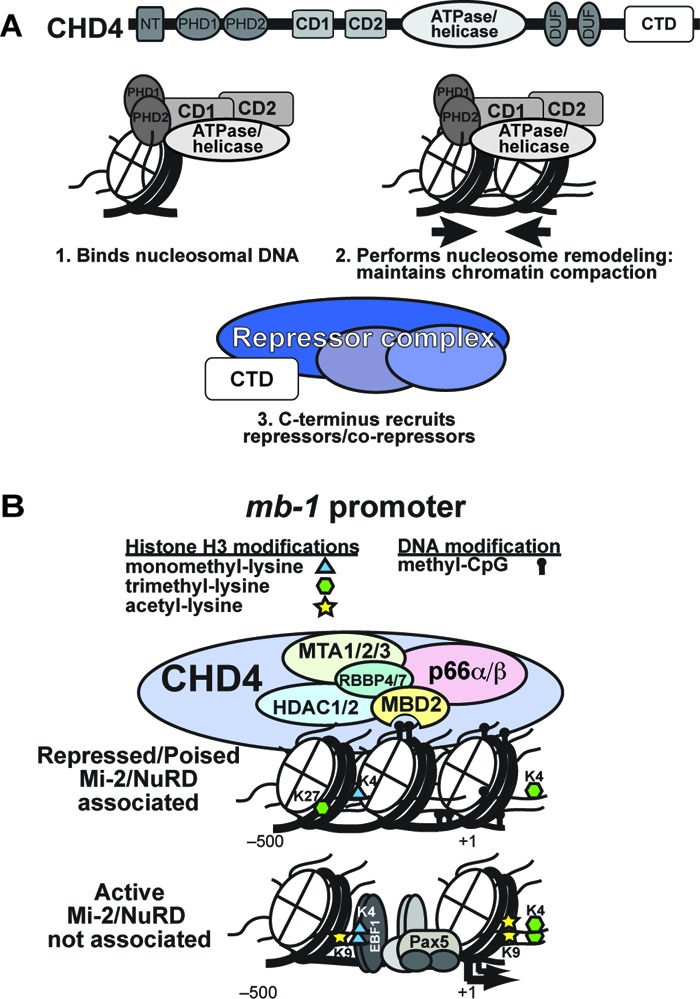
Multiple domains of CHD4, MBD2, and DNA methylation contribute synergistically to the regulation of transcription by NuRD chromatin remodeling complexes. (A) CHD4 is the core subunit of the NuRD complex. Its various domains perform functions central to activities of the complex. The PHD fingers (PHD1 and PHD2), chromodomains (CD1 and CD2), and ATPase/helicase domains interact with each other to bind nucleosomal DNA and histone H3 tails (28, 34, 48). In addition, the close interactions of CD1 and CD2 with the ATPase/helicase domain facilitate chromatin remodeling driven by ATP hydrolysis, which opposes the actions of activating chromatin remodeling complexes (CRCs) by maintaining the chromatin in a more condensed state. The C-terminal domain (CTD) is likely involved in recruitment of corepressor complexes. (B) The mb-1 promoter serves as a model for NuRD CRC function on repressed and poised lineage-specific genes. Binding of methylated CpGs and nucleosomal DNA as well as the remodeling activity of CHD4 is necessary for stable association of the NuRD complex with chromatin. Through concerted actions of its various subunits, the NuRD CRC maintains the repressed state by regulating nucleosome density and histone posttranslational modifications (particularly trimethylated H3K27 and acetylated H3K9) and by preventing promoter binding by transcription factors that recruit activating CRCs. The NuRD CRC and corepressor complexes act in a delicate balance with activating transcription factors, CRCs, and HATs to regulate transcriptional programs. LSD1 (KRDM1; not shown) is expressed in plasma cells and may be an additional component of NuRD complexes in B cells.
Our model for transcriptional regulation of the mb-1 promoter focuses on maintenance of repression by the NuRD complex and events leading to activation (Fig. 7B). The concerted action of all tested domains of CHD4 is necessary for performance of NuRD activities. Methylated CpGs are required to anchor the complex to the promoter, likely through binding of MBD2 to promoter proximal CpGs. In turn, NuRD promoter occupancy prevents binding of activating transcription factors, which if unregulated would lead to recruitment of the RNAPII complex, transcriptional activation, and demethylation of promoter DNA. During the first steps of transcriptional regulation, DNA methyltransferase 1 (DNMT1) methylates newly synthesized promoter DNA, which is then bound by the NuRD complex. Repressive histone modifications such as histone H3 trimethyl lysine 27, unacetylated H3K9, and unmethylated H3K4 also help recruit NuRD CRCs. NuRD complexes then mobilize and compact nucleosomes, maintain histone tail deacetylation and H3K4 demethylation, and likely recruit corepressors to preserve transcriptional silencing. Expression of transcriptional activators in the appropriate developmental context leads to promoter binding and eviction of the NuRD complex, followed by DNA demethylation and transcriptional activation mediated by the remodeling activities of activating CRCs such as SWI/SNF. NuRD-mediated repression of the rho-globin gene in primary erythroid cells is also dependent on MBD2, supporting our model and providing evidence that the significance of our findings is not limited to the mb-1 promoter (19).
Recent studies have indicated that the NuRD complex plays a vital role in maintaining a dynamic balance between the functions of activating CRCs and histone acetyltransferases to attenuate various transcriptional programs (reviewed in reference 14). Previously, we documented opposing attenuating and activating activities of different CRCs (6). However, it is becoming increasingly clear that NuRD itself performs opposing functions based on core complex composition, other associated factors, and chromatin configuration which all interact to regulate developmentally appropriate transcriptional programs that differ between cell types. The current research highlights the delicate interplay between CRCs, transcription factors, and DNA- and chromatin-modifying enzymes that forms the basis of cellular identity and function. A major question concerns how NuRD complexes are targeted to specific genes during B cell development. In this regard, recent studies revealed a potential role of Ikaros, which directs NuRD binding during thymocyte development (52). Genome-wide studies have underscored the important roles played by a network of transcription factors, including EBF1, in orchestrating the balance between transcriptional activation and repression during B cell development (23, 46). Our data suggest the importance of NuRD-mediated repression in this network, which provides a barrier that must be overcome by EBF1. Despite existing studies of NuRD function in different developmental frameworks, much work remains to be done in investigating functions of the complex at specific sites and in specific cellular milieus.
In summary, our studies and those of other investigators (49) that have focused on defining mechanisms of CRC regulation of a single gene or set of related genes complement broader genome-wide factor occupancy and gene expression studies. Together with global regulatory studies, they will build a more instructive view of the connection between the many roles played by CRCs in transcriptional regulation with the details of how these functions are accomplished and themselves regulated.
ACKNOWLEDGMENTS
We thank C. Musselman for helpful discussions, C. Hennessy and D. Schwartz for invaluable assistance with pyrosequencing, G. Blobel for the CHD4 cDNA plasmid, and K. Lukin for assistance with preparation of figures.
This research was supported by NIH grants R01 AI054661 and AI081878 to J.H. and GM096863 and CA113472 to T.G.K. J.R. was supported by a generous grant from the Rocky Mountain Chapter of the Arthritis Foundation and NIH postdoctoral training grant T32 AI07405. C.D. is supported by the Cancer Research Institute Predoctoral Emphasis Pathway in Tumor Immunology Fellowship.
We declare that we do not have any conflicts of interest.
Footnotes
Published ahead of print 15 October 2012
REFERENCES
- 1. Agalioti T, et al. 2000. Ordered recruitment of chromatin modifying and general transcription factors to the IFN-β promoter. Cell 103:667–678 [DOI] [PubMed] [Google Scholar]
- 2. Aguilera C, et al. 2011. c-Jun N-terminal phosphorylation antagonises recruitment of the Mbd3/NuRD repressor complex. Nature 469:231–235 [DOI] [PubMed] [Google Scholar]
- 3. Bouazoune K, et al. 2002. The dMi-2 chromodomains are DNA binding modules important for ATP-dependent nucleosome mobilization. EMBO J. 21:2430–2440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Durr H, Korner C, Muller M, Hickmann V, Hopfner KP. 2005. X-ray structures of the Sulfolobus solfataricus SWI2/SNF2 ATPase core and its complex with DNA. Cell 121:363–373 [DOI] [PubMed] [Google Scholar]
- 5. Fujita N, et al. 2004. MTA3 and the Mi-2/NuRD complex regulate cell fate during B lymphocyte differentiation. Cell 119:75–86 [DOI] [PubMed] [Google Scholar]
- 6. Gao H, et al. 2009. Opposing effects of SWI/SNF and Mi-2/NuRD chromatin remodeling complexes on epigenetic reprogramming by EBF and Pax5. Proc. Natl. Acad. Sci. U. S. A. 106:11258–11263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gnanapragasam MN, et al. 2011. p66α-MBD2 coiled-coil interaction and recruitment of Mi-2 are critical for globin gene silencing by the MBD2-NuRD complex. Proc. Natl. Acad. Sci. U. S. A. 108:7487–7492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gregory GD, et al. 2010. FOG1 requires NuRD to promote hematopoiesis and maintain lineage fidelity within the megakaryocytic-erythroid compartment. Blood 115:2156–2166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Harju-Baker S, Costa FC, Fedosyuk H, Neades R, Peterson KR. 2008. Silencing of Aγ-globin gene expression during adult definitive erythropoiesis mediated by GATA-1-FOG-1-Mi2 complex binding at the −566 GATA site. Mol. Cell Biol. 28:3101–3113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Harker N, et al. 2011. Pre-TCR signaling and CD8 gene bivalent chromatin resolution during thymocyte development. J. Immunol. 186:6368–6377 [DOI] [PubMed] [Google Scholar]
- 11. Hauk G, McKnight JN, Nodelman IM, Bowman GD. 2010. The chromodomains of the Chd1 chromatin remodeler regulate DNA access to the ATPase motor. Mol. Cell 39:711–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hendrich B, Tweedie S. 2003. The methyl-CpG binding domain and the evolving role of DNA methylation in animals. Trends Genet. 19:269–277 [DOI] [PubMed] [Google Scholar]
- 13. Hombach J, Tsubata T, Leclerq L, Stappert H, Reth M. 1990. Molecular components of the B-cell antigen receptor complex of the IgM class. Nature 343:760–762 [DOI] [PubMed] [Google Scholar]
- 14. Hu G, Wade PA. 2012. NuRD and pluripotency: a complex balancing act. Cell Stem Cell 10:497–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hutchins AS, et al. 2002. Gene silencing quantitatively controls the function of a developmental trans-activator. Mol. Cell 10:81–91 [DOI] [PubMed] [Google Scholar]
- 16. Kaji K, et al. 2006. The NuRD component Mbd3 is required for pluripotency of embryonic stem cells. Nat. Cell Biol. 8:285–292 [DOI] [PubMed] [Google Scholar]
- 17. Kaji K, Nichols J, Hendrich B. 2007. Mbd3, a component of the NuRD co-repressor complex, is required for development of pluripotent cells. Development 134:1123–1132 [DOI] [PubMed] [Google Scholar]
- 18. Kim J, et al. 1999. Ikaros DNA-binding proteins direct formation of chromatin remodeling complexes in lymphocytes. Immunity 10:345–355 [DOI] [PubMed] [Google Scholar]
- 19. Kransdorf EP, et al. 2006. MBD2 is a critical component of a methyl cytosine-binding protein complex isolated from primary erythroid cells. Blood 108:2836–2845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lai AY, Wade PA. 2011. Cancer biology and NuRD: a multifaceted chromatin remodelling complex. Nat. Rev. Cancer 11:588–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Le Guezennec X, et al. 2006. MBD2/NuRD and MBD3/NuRD, two distinct complexes with different biochemical and functional properties. Mol. Cell Biol. 26:843–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lejon S, et al. 2011. Insights into association of the NuRD complex with FOG-1 from the crystal structure of an RbAp48.FOG-1 complex. J. Biol. Chem. 286:1196–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lin YC, et al. 2010. A global network of transcription factors, involving E2A, EBF1 and Foxo1, that orchestrates B cell fate. Nat. Immunol. 11:635–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lukin K, et al. 2010. Compound haploinsufficiencies of Ebf1 and Runx1 genes impede B cell lineage progression. Proc. Natl. Acad. Sci. U. S. A. 107:7869–7874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Magdinier F, Wolffe AP. 2001. Selective association of the methyl-CpG binding protein MBD2 with the silent p14/p16 locus in human neoplasia. Proc. Natl. Acad. Sci. U. S. A. 98:4990–4995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Maier H, Colbert J, Fitzsimmons D, Clark DR, Hagman J. 2003. Activation of the early B-cell-specific mb-1 (Ig-α) gene by Pax-5 is dependent on an unmethylated Ets binding site. Mol. Cell Biol. 23:1946–1960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Maier H, et al. 2004. Early B cell factor cooperates with Runx1 and mediates epigenetic changes associated with mb-1 transcription. Nat. Immunol. 5:1069–1077 [DOI] [PubMed] [Google Scholar]
- 28. Mansfield RE, et al. 2011. Plant homeodomain (PHD) fingers of CHD4 are histone H3-binding modules with preference for unmodified H3K4 and methylated H3K9. J. Biol. Chem. 286:11779–11791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mian OY, et al. 2011. Methyl-binding domain protein 2-dependent proliferation and survival of breast cancer cells. Mol. Cancer Res. 9:1152–1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Miccio A, Blobel GA. 2010. Role of the GATA-1/FOG-1/NuRD pathway in the expression of human beta-like globin genes. Mol. Cell Biol. 30:3460–3470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Miccio A, et al. 2010. NuRD mediates activating and repressive functions of GATA-1 and FOG-1 during blood development. EMBO J. 29:442–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Morra R, Lee BM, Shaw H, Tuma R, Mancini EJ. 2012. Concerted action of the PHD, chromo and motor domains regulates the human chromatin remodelling ATPase CHD4. FEBS Lett. 586:2513–2521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Musselman CA, et al. 2009. Binding of the CHD4 PHD2 finger to histone H3 is modulated by covalent modifications. Biochem. J. 423:179–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Musselman CA, et al. 2012. Bivalent recognition of nucleosomes by the tandem PHD fingers of the CHD4 ATPase is required for CHD4-mediated repression. Proc. Natl. Acad. Sci. U. S. A. 109:787–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Naito T, Gómez-Del Arco P, Williams CJ, Georgopoulos K. 2007. Antagonistic interactions between Ikaros and the chromatin remodeler Mi-2β determine silencer activity and Cd4 gene expression. Immunity 27:723–734 [DOI] [PubMed] [Google Scholar]
- 36. Ramirez-Carrozzi VR, et al. 2006. Selective and antagonistic properties of SWI/SNF and Mi-2β nucleosome remodeling complexes during an inflammatory response. Genes Dev. 20:282–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Reynolds N, et al. 2012. NuRD suppresses pluripotency gene expression to promote transcriptional heterogeneity and lineage commitment. Cell Stem Cell 10:583–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Reynolds N, et al. 2012. NuRD-mediated deacetylation of H3K27 facilitates recruitment of polycomb repressive complex 2 to direct gene repression. EMBO J. 31:593–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rupon JW, Wang SZ, Gaensler K, Lloyd J, Ginder GD. 2006. Methyl binding domain protein 2 mediates γ-globin gene silencing in adult human βYAC transgenic mice. Proc. Natl. Acad. Sci. U. S. A. 103:6617–6622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rupon JW, Wang SZ, Gnanapragasam M, Labropoulos S, Ginder GD. 2011. MBD2 contributes to developmental silencing of the human epsilon-globin gene. Blood Cells Mol. Dis. 46:212–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Saito M, Ishikawa F. 2002. The mCpG-binding domain of human MBD3 does not bind to mCpG but interacts with NuRD/Mi2 components HDAC1 and MTA2. J. Biol. Chem. 277:35434–35439 [DOI] [PubMed] [Google Scholar]
- 42. Sillibourne JE, Delaval B, Redick S, Sinha M, Doxsey SJ. 2007. Chromatin remodeling proteins interact with pericentrin to regulate centrosome integrity. Mol. Biol. Cell 18:3667–3680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sridharan R, Smale ST. 2007. Predominant interaction of both Ikaros and Helios with the NuRD complex in immature thymocytes. J. Biol. Chem. 282:30227–30238 [DOI] [PubMed] [Google Scholar]
- 44. Thompson PM, Gotoh T, Kok M, White PS, Brodeur GM. 2003. CHD5, a new member of the chromodomain gene family, is preferentially expressed in the nervous system. Oncogene 22:1002–1011 [DOI] [PubMed] [Google Scholar]
- 45. Tong JK, Hassig CA, Schnitzler GR, Kingston RE, Schreiber SL. 1998. Chromatin deacetylation by an ATP-dependent nucleosome remodelling complex. Nature 395:917–921 [DOI] [PubMed] [Google Scholar]
- 46. Treiber T, et al. 2010. Early B cell factor 1 regulates B cell gene networks by activation, repression, and transcription-independent poising of chromatin. Immunity 32:714–725 [DOI] [PubMed] [Google Scholar]
- 46a. Wang Y, et al. 2009. LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell 138:660–672 [DOI] [PubMed] [Google Scholar]
- 47. Wang Z, et al. 2009. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 138:1019–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Watson AA, et al. 2012. The PHD and chromo domains regulate the ATPase activity of the human chromatin remodeler CHD4. J. Mol. Biol. 422:3–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xie W, et al. 2012. The chromatin remodeling complex NuRD establishes the poised state of rRNA genes characterized by bivalent histone modifications and altered nucleosome positions. Proc. Natl. Acad. Sci. U. S. A. 109:8161–8166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xue Y, et al. 1998. NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol. Cell 2:851–861 [DOI] [PubMed] [Google Scholar]
- 51. Yildirim O, et al. 2011. Mbd3/NURD complex regulates expression of 5-hydroxymethylcytosine marked genes in embryonic stem cells. Cell 147:1498–1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang J, et al. 2012. Harnessing of the nucleosome-remodeling-deacetylase complex controls lymphocyte development and prevents leukemogenesis. Nat. Immunol. 13:86–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang Y, LeRoy G, Seelig HP, Lane WS, Reinberg D. 1998. The dermatomyositis-specific autoantigen Mi2 is a component of a complex containing histone deacetylase and nucleosome remodeling activities. Cell 95:279–289 [DOI] [PubMed] [Google Scholar]
- 54. Zhang Y, et al. 1999. Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev. 13:1924–1935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhao F, McCarrick-Walmsley R, Akerblad P, Sigvardsson M, Kadesch T. 2003. Inhibition of p300/CBP by early B-cell factor. Mol. Cell. Biol. 23:3837–3846 [DOI] [PMC free article] [PubMed] [Google Scholar]