

Abstract
The bacterial uptake of steroids and their metabolites remains poorly understood. We investigated two transporters associated with cholate catabolism in Rhodococcus jostii RHA1. Reverse transcriptase quantitative-PCR indicated that an ATP-binding cassette (ABC) transporter and a major facilitator superfamily (MFS) transporter were upregulated 16.7- and 174-fold, respectively, during the exponential phase of growth on cholate compared to growth on pyruvate. Gene knockout analysis established that these transporters are required for the reassimilation of distinct metabolites that accumulate during growth on cholate. The ABC transporter, encoded by camABCD, was essential for uptake of 1β(2′-propanoate)-3aα-H-4α(3″(R)-hydroxy-3″-propanoate)-7aβ-methylhexahydro-5-indanone and a desaturated analog. The MFS transporter, encoded by camM, was essential for uptake of 3,7(R),12(S)-trihydroxy-9-oxo-9,10-seco-23,24-bisnorchola-1,3,5(10)-trien-22-oate. These metabolites differ from cholate metabolites reported to be excreted by proteobacteria in that they retain an isopropanoyl side chain at C-17. The uptake of these metabolites was necessary for maximal growth on cholate: a ΔcamB mutant lacking the permease component of the ABC transporter and a ΔcamM mutant lacking the MFS transporter grew to 74% and 77%, respectively, of the yield of the wild type. This study demonstrates for the first time the requirement for specific transporters for uptake of cholate metabolites and highlights the importance and complexity of transport processes associated with bacterial steroid catabolism.
INTRODUCTION
Steroids are a diverse class of compounds that perform a multitude of functions in eukaryotes (38), such as modulating membrane fluidity (i.e., cholesterol and phytosterols) and hormonal functioning (i.e., testosterone and estradiol) (32). Steroids are released in large quantities through eukaryotic excretion and decomposition, as well as in wastes from agriculture, industry, and sewage treatment (10, 21), and can be quite recalcitrant once in the environment (25). The steroids found in waste can present particular health risks to affected organisms. For example, hormone steroids, found mainly in livestock runoff and sewage effluent, are problematic due to endocrine disruption in humans and in fish and other wildlife (2, 18, 33). Alternatively, certain steroids have therapeutic properties and are routinely prescribed to treat a wide variety of diseases and disorders (16, 19). Ongoing efforts are aimed at discovering new pharmaceutically useful steroids.
Environmental microorganisms have evolved diverse strategies to take up, transform, and degrade steroid compounds. Indeed, microbial steroid degradation is an important part of global carbon cycling (27, 32). These microorganisms could potentially be used for biotechnological purposes. For example, an effective bioremediation method to clean up environments contaminated with steroids could include the application of steroid-degrading bacteria. Additionally, the implementation of microbial biotechnology in industrial steroid production is of interest, as both the cost of manufacturing and the production of toxic waste would be reduced compared to chemical synthesis (8, 11). There are potential clinical applications as well, due to the discovery of a cholesterol degradation gene cluster in Mycobacterium tuberculosis that is implicated in pathogenesis (26, 37). However, the current limited understanding of the genes, enzymes, and pathways involved in microbial steroid transformation hinders its commercial exploitation.
To better understand the pathways involved in microbial steroid catabolism, the transformation of cholesterol, testosterone, and the various bile acids is being investigated. Most of the current knowledge of the catabolism of testosterone and the bile acids is based on the study of Proteobacteria. Nevertheless, the catabolism of bile acids is relatively poorly understood. Bile acids are surface-active steroids that aid in the digestion of dietary fats in animals (15). Considerable quantities of bile acids are released into the environment each day through fecal matter and urine (up to 600 mg per person) (13) and are decomposed by soil and aquatic bacteria (27). Bile acids are lipophilic and can behave as detergents to disrupt the membranes of bacteria, resulting in cell lysis (14). As such, bile acids and their degradation products are both nutrient sources and stress factors for free-living bacteria (3). Microorganisms that degrade bile acids have likely evolved adaptive strategies to reduce their exposure to toxic degradation products while maximizing the nutritional benefit of these substrates. For example, Pseudomonas sp. strain Chol1 exhibited transient extracellular accumulation of intermediates during bile acid degradation, presumably to regulate the intracellular levels of these compounds. During later stages of bile acid metabolism, the extracellular metabolites dissipate, which implies that the intermediates are reassimilated for further conversion (5, 27). However, no uptake system for bile acid intermediates has been described.
Rhodococcus jostii RHA1, a soil actinomycete, has become a model organism for studying microbial steroid catabolism. Its 9.7-Mb genome contains hundreds of steroid catabolism genes arranged in 4 clusters that together permit robust growth on various steroids (23). As noted in our accompanying article, gene expression profiling of RHA1 (24a) demonstrated the upregulation of dozens of genes during growth on cholate, the major constituent of mammalian bile acid, including the genes encoding an ATP-binding cassette (ABC) transporter, camABCD, and a transporter of the major facilitator superfamily (MFS), camM. In this study, functional knockouts of RHA1 were used to elucidate the roles of camABCD and camM in cholate metabolism. Like other microorganisms growing on cholate, RHA1 exhibits the extracellular accumulation and reassimilation of metabolites. Structural analysis of these metabolites allowed us to propose a partial RHA1 cholate degradation pathway, extending what has been described in other microorganisms (5, 6, 27, 36).
MATERIALS AND METHODS
Reagents and chemicals.
Cholate was purchased from Sigma and was at least 98% pure. Restriction enzymes were purchased from New England BioLabs. T4 DNA ligase was purchased from Fermentas.
Cell growth assays.
RHA1 and derivative strains were cultivated at 30°C under aerobic conditions (200 rpm) in M9 mineral medium (9) supplemented with trace elements, vitamin B1 (1), and, unless otherwise indicated, 2.0 mM cholate. Experiments were performed in triplicate unless otherwise indicated. Escherichia coli strains were grown at 37°C (200 rpm) in Luria-Bertani (LB) broth. Bacto agar (1.5% [wt/vol]; Difco) was used for solid medium. Culture growth was measured as the change in optical density at 600 nm (OD600). Protein measurements were performed with a bicinchoninic acid (BCA) protein assay (Thermo Scientific) using bovine serum albumin as the standard. To test for transformation of cholate metabolites, M9 mineral medium was diluted 1-fold with filter-sterilized culture supernatant harvested during the early stationary growth phase of mutant strains that accumulated particular metabolites, and uninoculated medium was used as a blank for OD measurements.
DNA manipulation and construct preparation.
DNA was amplified, purified, and manipulated according to standard procedures (9). PCRs were performed with Taq Polymerase (Qiagen) or High Fidelity Expand Polymerase (Roche) in a 96-well PTC-200 Peltier Thermal Cycler (MJ Research). Oligonucleotides were from Integrated DNA Technologies. DNA sequencing was performed at the Nucleic Acid Protein Services Unit at the University of British Columbia (UBC). The ΔcamB and ΔcamM gene deletion strains were generated on the basis of previously described methods (35). The camB and camM upstream flanking regions were amplified from fosmid DNA (Table 1) with the following primers: cBupFor, cBupRev, cMupFor, and cMupRev (Table 2). The downstream flanking regions were amplified in a similar fashion using cBdwFor, cBdwRev, cMdwFor, and cMdwRev primers. Triple ligations using T4 DNA ligase, with the cut sites for EcoRI, HindIII, and SphI for camB and EcoRI, HindIII, and XbaI for camM, were performed to clone the amplicons into pK18mobsacB (Table 1) to produce pK18camB and pK18camM, respectively. The gene deletion constructs were transformed by electroporation and propagated in E. coli DH5α. For the complementation assay, primers were designed to include in the amplicons at least 200 bp upstream of the target gene, thereby including the native promoter region. The camB and camM amplicons for the complementation assay were produced from the fosmid DNA, using primers camBpSTf and camBpSTr for camB and primers camMpSTf and camMpSTr for camM.
Table 1.
Strains, fosmids, and plasmids used in this study
| Strain/fosmid/plasmid | Relevant characteristicsa | Source or reference |
|---|---|---|
| Strains | ||
| R. jostii | ||
| RHA1 | Wild type; Nalr | 29 |
| ΔcamB | Derivative of RHA1 with ro04887 truncated | This study |
| ΔcamM | Derivative of RHA1 with ro05792 truncated | This study |
| E. coli | ||
| DH5α | Host for pK18mobsacB and pSET152 | 31 |
| S17-1 | Conjugal mobilization of pK18mobsacB | 31 |
| Fosmids | ||
| RF00110B17 | Source of camABCD; Ampr | 23 |
| RF00110H09 | Source of camM; Ampr | 23 |
| Plasmids | ||
| pK18mobsacB | Kanr | 28 |
| pK18camB | Kanr; pK18mobsacB camB deletion plasmid | This study |
| pK18camM | Kanr; pK18mobsacB camM deletion plasmid | This study |
| pSET152 | Amr; intϕ-C31 | 4 |
| pSTcamB | Amr; intϕ-C31; pSET152 camB complementation plasmid | This study |
| pSTcamM | Amr; intϕ-C31; pSET152 camM complementation plasmid | This study |
Nalr, nalidixic acid resistance; Ampr, ampicillin resistance; Kanr, kanamycin resistance; Amr, apramycin resistance; intϕ-C31, bacteriophage ϕ-C31 attachment site.
Table 2.
Oligonucleotides used in this study
| Primer name | Sequence (5′ to 3′)a | Restriction site |
|---|---|---|
| cBupFor | GCGAAGCTTCGAGATGAAGACGATGTTGTAGCG | EcoRI |
| cBupRev | CGGGAATTCGCGCAGAACAAGATCCTCTCGTTC | HindIII |
| cBdwFor | CGGAAGCTTGCGGTCTTCGTGGTCCTCATC | HindIII |
| cBdwRev | GCGGCATGCACCCACCGAGGAGGCCAGTACC | SphI |
| cMupFor | TCTGAATTCGGTAGTGGACGAGGGTGGCGAC | EcoRI |
| cMupRev | TTATCTAGAGGGATTCGAGAGGATCAGCACG | XbaI |
| cMdwFor | TGTTCTAGACTCCTCATCCGCAACAAGATCGAG | XbaI |
| cMdwRev | TCCAAGCTTCTTTCGACGTGGTCCTCAGCG | HindIII |
| camBColF | GACGACCAGCGCGAGGATGCAG | |
| camBColR | GTCGAAACCCGGAGACGAGGCGTAC | |
| camMColF | ACCAGTAGGGCGCGATCTGATGGAG | |
| camMColR | AGGTGACCCGGCGCGTCAATC | |
| camBpSTf | ATATCTAGAAGACACGTTCGCCGAGTCGGTC | XbaI |
| camBpSTr | AATTCTAGAAGATGTTCGCCAAGACCATCG | XbaI |
| camMpSTf | AAAGAATTCAGACTGAGTGATTCCGTCATGGG | EcoRI |
| camMpSTr | AAATCTAGATGAACACTTGGACTGCCACACCG | XbaI |
| camBpTIPf | CTTGTTACATATGACCATCGTCTGGTCGGGCCTC | NdeI |
| camBpTIPr | ATCAAGCTTTCACACCACCCGTTCCCGCACC | HindIII |
| camBQFor | TCTCGTCCCACACGTTCAAC | |
| camBQRev | CGAAGGTTCCGACCATCATC | |
| camMQFor | CCGTCGCGAAAACATTGG | |
| camMQRev | GGATCGGTCATCACGAACATC |
Restriction enzyme cut sites are underlined.
Gene deletions.
Strains ΔcamB and ΔcamM were generated by the previously described sacB counterselection system (35). Kanamycin-sensitive, sucrose-resistant (Kans/Sucr) colonies were screened by colony PCR using camBColF and camBColR for the camB deletion and camMColF and camMColR for the camM deletion. The regions of deletion in the ΔcamB and ΔcamM strains were sequenced to ensure there were no changes in the regions flanking the deletions.
Gene complementation.
The entire 5-kb, four-gene operon camABCD and the 1.8-kb camM gene were cloned to generate the ΔcamBc and ΔcamMc complementation strains, respectively. Double ligations using T4 DNA ligase with the XbaI cut site for ΔcamBc and the EcoRI and XbaI cut sites for ΔcamMc were performed to clone the amplicons into the pSET152 integrative vector (4) to produce pSTcamB for camB complementation and pSTcamM for camM complementation (Table 1). The complementation constructs were propagated in E. coli DH5α. The ΔcamB and ΔcamM mutants were individually cultivated in LB medium containing 30 μg ml−1 nalidixic acid at 30°C for 48 h (200 rpm). These cultures were then plated on LB agar containing 30 μg ml−1 nalidixic acid and incubated at 30°C for an additional 48 h. The complementation constructs pSTcamB and pSTcamM were transformed by electroporation into E. coli S17-1 donor cells, plated on LB plates containing 50 μg ml−1 apramycin, grown for 24 h at 37°C, and then left at room temperature for an additional 24 h to garner more cell material. Cells from plates of both the donor strain and the RHA1 mutants were suspended in 2.0 ml of LB medium, and 750 μl of each was gently mixed together. The cells were pelleted by centrifugation (9,400 × g for 1 min) and suspended in 1.0 ml of LB medium, and aliquots of 200 μl were spread on nonselective LB plates and incubated overnight at 30°C to facilitate conjugation. Cells were then harvested from plates with confluent growth and suspended in 2.0 ml of LB medium, and aliquots of 100 μl were spread on selective LB agar with 50 μg ml−1 apramycin plus 30 μg ml−1 nalidixic acid and incubated at 30°C for 96 h to facilitate integration in the ϕ-C31 bacteriophage attachment site. Subsequent colonies were replated on selective LB agar with 50 μg ml−1 apramycin plus 30 μg ml−1 nalidixic acid, incubated at 30°C for 48 h, and then screened for the presence of both the truncated and the wild-type target genes using cBupFor and cBdwRev primers for ΔcamBc and cMupFor and cMdwRev primers for ΔcamMc (Table 2). The pSET152 vector cannot exist freely in Streptomyces, and the same was assumed to be true for Rhodococcus (4).
RNA extraction, cDNA synthesis, and reverse transcriptase quantitative PCR (RT-QPCR).
Samples (10 ml) were taken when cultures reached approximately 35%, 65%, and 100% of their maximal OD600 values, corresponding to exponential, late exponential, and stationary phases, respectively. To each sample, 100 ml of stop solution (95% reagent alcohol-5% acid phenol [vol/vol], pH 5) was added. The samples were then centrifuged at 4,000 × g and 4°C for 10 min. Cells were suspended in 1.0 ml supernatant plus 2.0 ml of RNA Protect (Qiagen), incubated for 5 min at room temperature, and then centrifuged at 18,000 × g for 2 min. The pellets were frozen on dry ice and stored at −80°C. RNA extraction and purification were performed according to previously described methods (12).
cDNA was synthesized using the commercially available ThermoScript RT-PCR System protocol (Invitrogen), with the following modifications: 5 μg RNA was incubated with 50 ng of random hexamers plus 10 mM deoxynucleoside triphosphate (dNTP) mixture. The mixture was brought to 12 μl with diethyl pyrocarbonate (DEPC)-treated water, incubated at 65°C for 5 min, and then cooled on ice for 10 min. The following mixture was prepared and added to each reaction mixture: 5× cDNA synthesis buffer (Invitrogen), 0.1 M dithiothreitol (DTT), 10 units RNaseOUT (Invitrogen), 15 units ThermoScript RT brought to 10 μl with DEPC-treated water. The samples were incubated at 25°C for 10 min, 50°C for 50 min, and then 85°C for 5 min. Two units of RNase H was added, and the samples were incubated at 37°C for 20 min. Fresh cDNA samples were used immediately for RT-QPCR analysis.
RT-QPCR measured the expression of targeted genes compared to that of an internal-standard gene, ro01702, encoding DNA polymerase IV (12). The TaqMan probes and primers used in this study (Table 2) were designed with Primer Express Software v2.0 from Applied Biosystems. Each QPCR mixture contained 1.0 μl of the 20 μl of cDNA produced, 200 nM TaqMan probe, and 10 μl 2× TaqMan universal PCR master mix from Applied Biosystems, in a total volume of 20 μl. Duplicate singleplex reactions for the housekeeping gene and the target gene were performed in a Stratagene Mx3000P real-time PCR system for 2 min at 50°C and 10 min at 95°C, followed by 40 cycles of 15 s at 95°C and 1 min at 60°C. The quantitative PCR cycle threshold (CT) results were analyzed using the comparative CT (ΔΔCT) method. Student's one-sample t test was used to evaluate whether the gene targets were differentially regulated. The expression ratios were calculated with the formula E−ΔΔCT, where E is the average amplification efficiency derived from the standard curves for the target and housekeeping genes (20).
GC-MS.
For gas chromatography-mass spectrometry (GC-MS) analysis, 1.0-ml samples were periodically collected and cells were removed by centrifugation. The supernatant was then acidified with 10% H2SO4 (50:1 ratio [vol/vol]), extracted twice at room temperature for 30 min with an equal volume of ethyl acetate, dried under nitrogen gas, and dissolved in 200 μl pyridine. 5α-Cholestane (0.25 mM) was used as an internal standard. The extracts were derivatized at room temperature with 200 μl bis(trimethylsilyl)trifluoroacetamide/trimethylchlorosilane and analyzed with an Agilent 6890 series gas chromatograph equipped with an HP-5ms 30-m by 0.25-mm capillary column (Hewlett-Packard) and an HP 5973 mass-selective detector (22). The gas chromatography conditions were as follows: the injector temperature was set at 280°C, the transfer line temperature was 290°C, and the flow rate was 1 ml min−1 with helium. The temperature program of the oven was 104°C for 2 min, increased to 290°C at a rate of 15°C per min, and was then held at 290°C for 15 min. The mass spectrometer conditions were set to electron emission scanning at 40 to 800 m/z at 1.97 scans per second. Because authentic standards were not available for the metabolites detected, their relative accumulation was calculated on the basis of the total ion current signal, with samples normalized on the basis of the internal standard. Very approximate concentrations of the metabolites were estimated by assuming the total ion current signal was proportional to the amounts of carbon in cholate and the metabolites.
SPE and HPLC.
Two cholate metabolites, 1β(2′-propanoate)-3aα-H-4α(3″(R)-hydroxy-3″-propanoate)-7aβ-methylhexahydro-5-indanone (HHIDP) and 3,7(R),12(S)-trihydroxy-9-oxo-9,10-seco-23,24-bisnorchola-1,3,5(10)-trien-22-oate (THSBNC), were produced by the ΔcamB and ΔcamM mutants, respectively, and purified by solid-phase extraction (SPE) and high-performance liquid chromatography (HPLC). Briefly, 100-ml cultures were grown on cholate for 5 days. Cells were harvested by centrifugation (16,000 × g for 5 min), and the supernatant was stored at 4°C. A 100-ml sample of the supernatant (pH adjusted to 7) was deposited on a Strata-XL-A 100-μm polymeric strong-anion 2-g/20-ml column (Phenomenex) conditioned with acetonitrile and equilibrated with water. The metabolites were eluted with 20 ml of 95% acetonitrile-5% formic acid. Samples were concentrated under nitrogen flow. Compounds were chromatographically purified using a Waters 2695 Separations HPLC module (Milford, MA) equipped with a Waters 2996 photodiode array detector and a Luna 3-μm PFP(2) 50- by 4.6-mm column (Phenomenex) using a solvent system of 0.2% aqueous formic acid and 0.2% formic acid in methanol for THSBNC purification or water and methanol for purification of HHIDP. The column was equilibrated with 40% methanol containing 0.2% formic acid, and 500 μl of the concentrated SPE containing THSBNC was injected. The eluate was monitored at 280 nm. THSBNC was eluted using a gradient of 40 to 62% methanol containing 0.2% formic acid over 13 min at 1 ml min−1. Under these conditions, the retention time for THSBNC was 7.5 min. Recovered samples were dried under nitrogen, dissolved in 300 μl methanol, and deposited a second time on the same column, equilibrated as described above. THSBNC was eluted using a biphasic gradient consisting of 40 to 48.5% methanol containing 0.2% formic acid at 1 ml min−1 over 5 min, followed by 48.5 to 56.5% methanol containing 0.2% formic acid at 0.5 ml min−1 over 10 min. Under these conditions, the retention time of THSBNC was 10.0 min. For purification of HHIDP, 400 μl of the concentrated SPE was injected onto the column, which was equilibrated with 25% methanol in water. The eluate was monitored at 230 nm. HHIDP was eluted using a gradient of 25 to 60% methanol over 15 min at 0.8 ml min−1. Under these conditions, the retention time for HHIDP was 8.5 min. The recovered sample was dried under nitrogen. The purity of THSBNC and HHIDP was assessed by GC-MS under the conditions described above.
NMR spectroscopy.
Nuclear magnetic resonance (NMR) measurements were carried out at the UBC Chemistry High-Resolution NMR Analytical Services and Research Support Facility, with approximately 1 mg of HPLC-purified THSBNC or HHIDP dissolved in CD2Cl2 or deuterated methanol, respectively. NMR spectra were recorded at 300 K on a Bruker Avance 600-MHz spectrometer equipped with a TCI-Z 5-mm 13C, 15N/1H inverse-detect cryoprobe with a pulsed-field gradient z-gradient coil and selective pulse capability. The proton one-dimensional spectrum was recorded with a spectral width of 12 ppm and 32,000 complex points. Homonuclear two-dimensional correlated spectroscopy (COSY) was recorded with 1,000 complex points in the detected dimension and 256 complex points in the indirect dimension. All spectra were processed with SpinWorks 3.1.8.1 software.
RESULTS
Growth of RHA1 on cholate.
RHA1 was grown on 2.0 mM cholate as the sole carbon and energy source. After a lag phase of about 24 h, RHA1 achieved a doubling time of 3.4 ± 0.3 h (Fig. 1). After approximately 52 h, RHA1 reached a maximal OD600 of 1.76 ± 0.02 and a final growth yield of 21.6 ± 0.2 mg of protein per g of cholate. Cultures with 5.0 mM or more cholate failed to grow.
Fig 1.
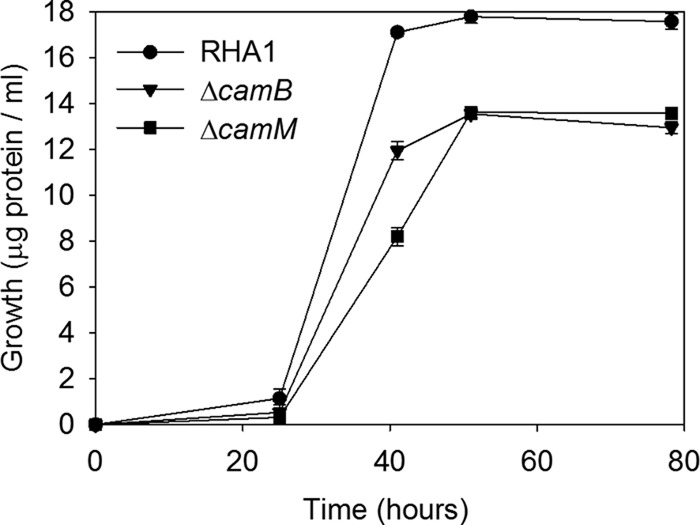
Growth of RHA1 and derivative strains on 2.0 mM cholate. The error bars show standard deviations (n = 3).
Metabolite accumulation.
Using GC-MS, we observed that three main metabolites accumulated in the culture supernatant during the growth of RHA1 on cholate. The first major metabolite was identified as 3,7(R),12(S)-trihydroxy-9-oxo-9,10-seco-23,24-bisnorchola-1,3,5(10)-trien-22-oate (THSBNC) (Fig. 2A) based on the following characterization. GC-MS of the trimethylsilyl derivative: Retention time (Rt) = 20.08 min; MS (70 eV, electron impact ionization [EI]), m/z: 678 (4.7%), 588 (30.7%), 367 (14.3%), 339 (27.7%), 294 (11.4%), 279 (31.3%), 251 (13.1%), 193 (13.7%), 73 (100%). 1H NMR (600 MHz, CD2Cl2): δ = 7.01 (d, J = 8.2 Hz, 1H, 1), δ = 6.65 (d, J = 2.6 Hz, 1H, 4), δ = 6.59 (dd, 2J = 5.5 Hz, 3J = 2.7 Hz, 1H, 2), δ = 4.18 (m, 1H, 7), δ = 3.81 (m, 1H, 12), δ = 2.98 (dd, 2J = 9.1 Hz, 3J = 4.8 Hz, 1H, 11β), δ = 2.84 (dd, 2J = 11.3 Hz, 3J = 3.8 Hz, 1H, 6β), δ = 2.80 (dd, 2J = 8.8 Hz, 3J = 5.1 Hz, 1H, 11α), δ = 2.48 (t, J = 4.8 Hz, 1H, 20), δ = 2.36 (dd, 2J = 12.4 Hz, 3J = 2.7 Hz, 1H, 6α), δ = 2.30 (m, 1H, 17), δ = 2.24 (s, 3H, 19), δ = 1.85 (m, 1H, 14), δ = 1.58 to 1.65 (m, 2H, 15β, 16β), δ = 1.32 to 1.38 (m, 2H, 15α, 16α), δ = 1.27 (d, J = 6.0 Hz, 3H, 21), δ = 0.96 (s, 3H, 18). The proton at C-8 could not be assigned. We propose that the hydroxyl group, originating from the C-7 hydroxyl of cholate, remains in the (R) orientation in THSBNC and subsequent metabolites. However, we did not confirm this experimentally.
Fig 2.

Structures of major cholate metabolites. (A) THSBNC. (B) HHIDP.
The second major cholate metabolite was identified as 1β(2′-propanoate)-3aα-H-4α(3″(R)-hydroxy-3″-propanoate)-7aβ-methylhexahydro-5-indanone (HHIDP) (Fig. 2B) based on the following characterization. GC-MS of the trimethylsilyl derivative: Rt = 15.09 min. MS (70 eV, EI); m/z: 528 (5.8%), 513 (26.5%), 438 (45.7%), 381 (23.8%), 348 (45.5%), 233 (36.7%), 217 (24.7%), 147 (32.5%), 73 (100%). 1H NMR (600 MHz, MeOD): δ = 4.17 (qd, 2J = 4.4 Hz, 3J = 1.9 Hz, 1H, 3″), δ = 2.78 (dd, 2J = 11.6 Hz, 3J = 8.9 Hz, 1H, 2″β), δ = 2.61 (dd, 2J = 13.8 Hz, 3J = 4.5 Hz, 1H, 2″α), δ = 2.51 (m, 1H, 6β), δ = 2.48 (dd, 2J = 7.6 Hz, 3J = 3.6 Hz, 1H, 4), δ = 2.42 (td, 2J = 6.8 Hz, 3J = 3.6 Hz, 1H, 2′), δ = 2.31 (qd, 2J = 7.4 Hz, 3J = 2.9 Hz, 1H, 6β), δ = 2.31 (m, 1H, 6α), δ = 2.14 (m, 1H, 3a), δ = 2.08 (m, 1H, 7β), δ = 1.94 (m, 1H, 7α), δ = 1.89 (m, 1H, 2β), δ = 1.78 (t, 1H, J = 12.2 Hz, 1), δ = 1.70 (t, 1H, J = 9.7 Hz, 3β), δ = 1.54 (m, 1H, 2α), δ = 1.43 (qd, 2J = 6.3 Hz, 3J = 5.4 Hz, 3α), δ = 1.22 (d, 3H, J = 6.9 Hz, 3′), δ = 0.97 (s, 3H, 8).
A third cholate metabolite appeared to be a desaturated analog of HHIDP and was tentatively identified as 1-ylidene(2′-propanoate)-3aα-H-4α(3″(R)-hydroxy-3″-propanoate)-7aβ-methylhexahydro-5-indanone (YHHIDP). GC-MS of the trimethylsilyl derivative: Rt = 15.34 min. MS (70 eV, EI); m/z: 526 (2.3%), 511 (11.0%), 436 (15.3%), 379 (4.2%), 346 (63.6%), 233 (7.6%), 217 (9.3%), 147 (16.5%), 73 (100%). Based on the GC-MS molecular ion and fragmentation pattern, as well as the proposed catabolic pathway for cholate, we hypothesize that the structure of YHHIDP is identical to that of HHIDP, with the addition of a double bond in the side chain between C-1 and C-2′. However, YHHIDP could not be isolated in sufficient amounts for NMR analysis, and thus, the location of the double bond remains speculative. In this study, all subsequent references to HHIDP imply both the saturated and desaturated compounds.
As approximated by their GC peak areas, both HHIDP and THSBNC reached their maximum levels of accumulation 35 to 40 h after inoculation, during the mid-log phase of RHA1 growth (Fig. 3 and 4). By late stationary phase, these compounds were no longer present in the supernatant, indicating their complete reassimilation. Other unidentified metabolites were also found to transiently accumulate, but in much smaller amounts.
Fig 3.
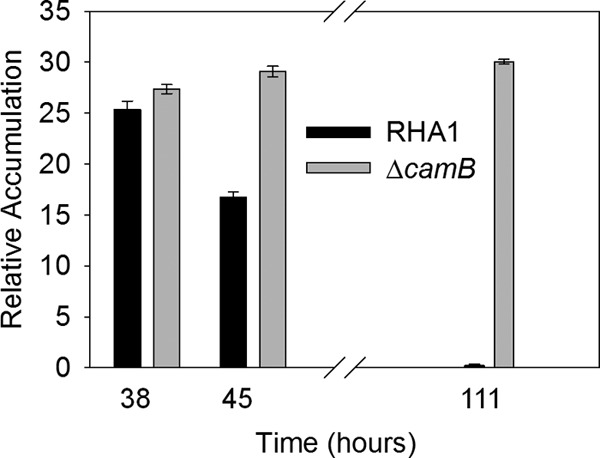
Accumulation of HHIDP by RHA1 and the ΔcamB mutant during growth on 2.0 mM cholate. The desaturated analog accounted for approximately 25% of the total amount of HHIDP detected. The error bars show standard deviations (n = 3).
Fig 4.
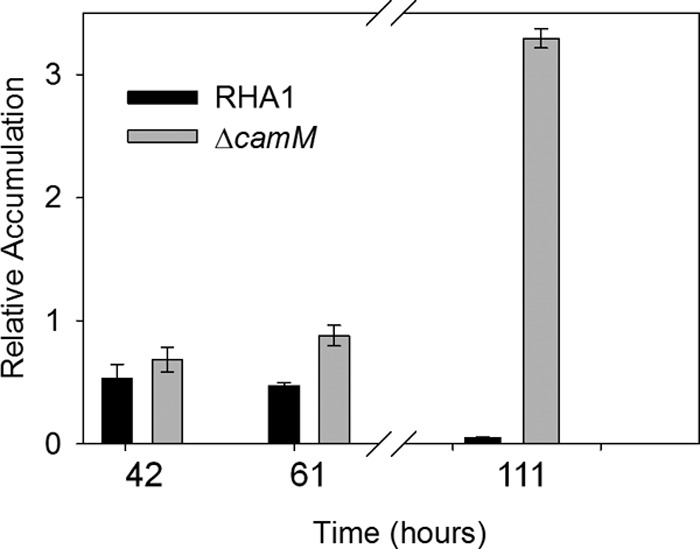
Accumulation of THSBNC by RHA1 and the ΔcamM mutant during growth on 2.0 mM cholate. The error bars show standard deviations (n = 3).
Upregulation of transporters during growth on cholate.
A transcriptomic analysis revealed that many genes were upregulated during growth of RHA1 on cholate compared to growth on pyruvate (24a). Among these were genes encoding two transporters: a four-gene operon encoding an ABC transporter, CamABCD (ro04888 to ro04885), and a gene encoding an MFS transporter, CamM (ro05792). Cam signifies cholate metabolite transporter. Examination of the camABCD operon revealed that CamC represents a fusion of an ATPase domain with a permease domain. Thus, we hypothesize the subunit organization shown in Fig. 5. The transcriptomic data revealed that during mid-log phase of RHA1 growth on cholate, compared to pyruvate, the gene encoding the permease component of the ABC transporter (camB) exhibited 7.4-fold upregulation, and the MFS transporter gene (camM) showed 26.6-fold upregulation.
Fig 5.

Proposed cholate catabolic pathway of RHA1. The structures in green are proposed based on the Chol1 cholate pathway (5), the RHA1 cholesterol pathway (36), and the current findings; they have not yet been identified in the RHA1 cholate pathway. The inset shows the hypothesized subunit organization of the CamABCD transporter. The mechanism of excretion of THSBNC and HHIDP is still unknown.
To verify the transcriptomic data, RT-QPCR was performed on camB and camM. The former was maximally expressed during exponential phase on cholate, while camM was maximally expressed during late exponential phase (Table 3). During stationary phase, neither camB nor camM was significantly upregulated in cholate- versus pyruvate-grown cells. These data suggest that these transporters are important for cholate catabolism by RHA1.
Table 3.
Fold change in gene expression of RHA1 grown on cholate versus pyruvate, based on RT-QPCR
| Gene | Fold change at growth stage: |
||
|---|---|---|---|
| Exponential | Late exponential | Stationary | |
| camB | 16.7 ± 0.4 | 8.9 ± 0.2 | 0.01 ± 0.01 |
| camM | 39.0 ± 0.2 | 174.1 ± 0.7 | 0.06 ± 0.02 |
The camB mutant.
The CamABCD ABC transporter was inactivated by deleting the camB gene to generate a ΔcamB mutant. The mutant was grown on cholate under the same conditions as the wild-type strain. Like wild-type RHA1, the ΔcamB mutant exhibited an initial lag phase of 24 h, followed by exponential growth in which the strain achieved a doubling time of 3.7 ± 0.3 h (Fig. 1). The ΔcamB mutant reached a maximum OD600 of 1.47 ± 0.01 after 41 h incubation. The final growth yield of the ΔcamB mutant was 15.9 ± 0.2 mg of protein per g of cholate, 74% of that of the wild type. Thus, the growth kinetics of the mutant were similar to those of the wild type, but the final biomass was lower.
Extraction of extracellular metabolites at various stages during growth on cholate revealed that the mutant transiently accumulated the same metabolites as the wild type. However, unlike wild-type RHA1, ΔcamB was unable to reassimilate HHIDP (Fig. 3). Maximal accumulation of HHIDP roughly corresponded to the point of maximum growth of ΔcamB (Fig. 1 and 3). The molar quantity of HHIDP accumulated by ΔcamB was approximately equivalent to 25% of the original cholate added, consistent with its reduced growth yield.
RHA1 and ΔcamB were precultured on cholate and then washed and harvested during mid-log phase. Twenty percent of these cells were added to medium with HHIDP as the major organic substrate. The medium was obtained by mixing equal volumes of cell-free supernatant from ΔcamB cultures with fresh medium lacking cholate. Wild-type RHA1 grew slowly on this medium, reaching stationary phase after 13 days (Fig. 6). In contrast, ΔcamB exhibited no growth on the medium, reaffirming that a functional CamABCD transporter is required for assimilation and catabolism of HHIDP (Fig. 5).
Fig 6.
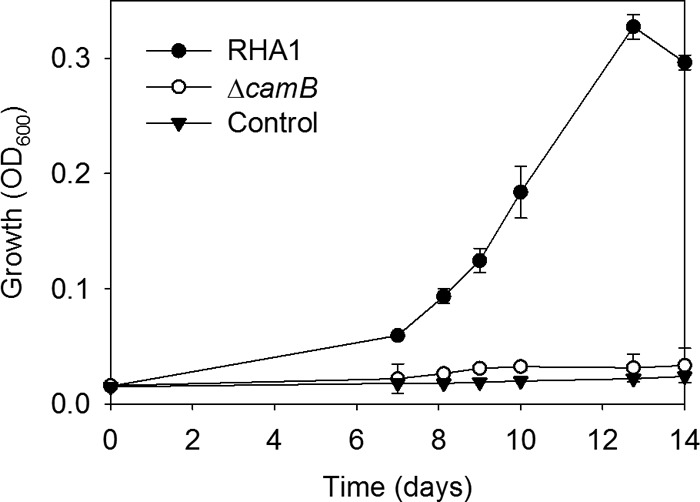
Growth of RHA1, the ΔcamB mutant, and an uninoculated control on HHIDP as the major organic substrate. The error bars show standard deviations (n = 3).
The ΔcamBc complemented strain was grown on cholate under the same conditions as the ΔcamB mutant and the wild type. Compared to these strains, the ΔcamBc strain grew more slowly yet reached 98% of the final protein yield of the wild type. In addition, the ΔcamBc strain produced the same metabolites as wild-type RHA1 and also reassimilated these metabolites, so that none were detected in the supernatants of stationary-phase cultures. Thus, the cloned version of camABCD essentially restored the wild-type cell yield on cholate and restored the capacity of ΔcamB to take up extracellular HHIDP.
The camM mutant.
The camM MFS transporter was inactivated via gene deletion to generate a ΔcamM mutant. The mutant grew on cholate with a doubling time of 3.9 ± 0.4 h and reached final cell density after 50 h (Fig. 1). For the ΔcamM mutant, the OD600 could not be measured, since compounds accumulating in the medium interfered with optical density measurements. The final growth yield of the ΔcamM mutant was 16.7 ± 0.3 mg of protein per g of cholate, 77% of that of the wild type. Like the ΔcamB mutant, the ΔcamM mutant exhibited growth kinetics similar to those of the wild type but achieved a lower growth yield.
The ΔcamM strain transiently accumulated the same metabolites as the wild type. However, the ΔcamM mutant accumulated THSBNC to a higher level than the wild type and was unable to reassimilate the compound (Fig. 4). Indeed, THSBNC remained in the culture supernatant throughout late stationary phase. The molar quantity of THSBNC accumulated by the ΔcamM mutant was approximately equivalent to 25% of the original cholate added, consistent with its reduced growth yield. Pure THSBNC did not absorb at OD600 and so was not responsible for the absorbance observed in the culture.
RHA1 and the ΔcamM mutant were precultured on cholate and then washed and harvested during mid-log phase. Twenty percent of these cells were added to medium with THSBNC as the major organic substrate. The medium was obtained as described above but using ΔcamM supernatant. Wild-type cells grew, reaching stationary phase after 8 days. In contrast, the ΔcamM mutant exhibited no growth on this metabolite (Fig. 7), confirming that CamM is required for assimilation and catabolism of THSBNC (Fig. 5).
Fig 7.
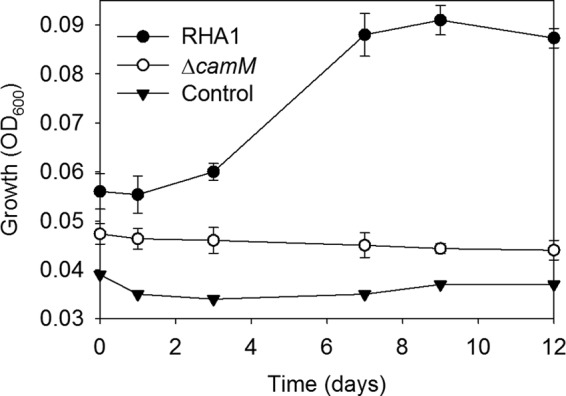
Growth of RHA1, the ΔcamM mutant, and an uninoculated control on THSBNC as the major organic substrate. The error bars show standard deviations (n = 3).
The ΔcamMc complemented strain was grown on cholate under the same conditions as the above-mentioned strains. Compared to the ΔcamM mutant and the wild type, the ΔcamMc strain grew more slowly yet reached 89% of the final protein yield of the wild type. In addition, the ΔcamMc strain produced and reassimilated the same metabolites as wild-type RHA1, so that none were detected in the supernatants of stationary-phase cultures. Thus, the cloned version of camM sufficiently restored the wild-type cell yield on cholate and restored the capacity of the ΔcamM mutant to take up extracellular THSBNC.
Growth on cholate conjugates.
RHA1 and the ΔcamB and ΔcamM mutants were grown on each of two conjugated bile acids, glycocholate and taurocholate. On 2.0 mM taurocholate, there was no change in growth kinetics, final growth yield, or metabolite accumulation compared to growth on cholate for all three strains (data not shown). However, on 2.0 mM glycocholate, only the wild type and the ΔcamB mutant exhibited the same growth phenotypes as cholate-grown cells. In contrast, the ΔcamM mutant grew significantly more slowly on glycocholate than on cholate, taking about 5 days to reach stationary phase instead of 2 days. The final growth yield on glycocholate was 17.0 ± 0.3 μg ml−1, which is similar to that of the wild type. Finally, there were no accumulated metabolites in the supernatant of stationary-phase ΔcamM strain cultures grown on glycocholate (data not shown).
DISCUSSION
The results of this study support the cholate catabolism pathway summarized in Fig. 3. The transient accumulation of extracellular metabolites observed during the growth of RHA1 on cholate is consistent with what has been reported for bile acids in a variety of proteobacteria (5, 11, 27). Moreover, the structure of the steroid nucleus of the RHA1 metabolite THSBNC is similar to that of the metabolites excreted by other cholate-degrading organisms (5, 17, 27). However, metabolites identified in previous studies have a ketone group at position C-17, whereas THSBNC harbors an isopropanoyl group instead. The natural occurrence of this compound has not been previously reported. Additionally, this is the first report of HHIDP as a cholate metabolite. Since HHIDP has an isopropanoyl side chain at C-1 (equivalent to C-17 in cholate), it appears that in RHA1, complete side chain degradation may occur after A-ring oxidation and B-ring opening. This is consistent with previous reports regarding actinobacteria summarized by Hayakawa (13). In contrast, during growth on cholate, the proteobacterial strain Chol1 forms 7,12-dihydroxy-1,4-androstadien-3,17-dione, or DHADD (5), the structure of which suggests that degradation of the C-17 side chain reaches completion prior to aromatization of ring A. Thus, a major difference between cholate catabolism by Actinobacteria and Proteobacteria appears to be the order of side chain degradation relative to A- and B-ring degradation.
Despite the longer side chain of RHA1 cholate metabolites compared to analogous proteobacterial metabolites, our results are otherwise consistent with a cholate catabolic pathway in RHA1 very similar to that proposed for proteobacteria (13). Specifically, the formation of the secosteroid THSBNC suggests that aromatization of ring A and opening of ring B happen in similar fashions in RHA1 (Fig. 3) and in proteobacteria, like Comamonas testosteroni TA441 and Pseudomonas sp. Chol1 (5, 17). In addition, the occurrence of HHIDP also agrees with the more general microbial secosteroid pathway, in which the removal of ring A and the partial degradation of ring B result in formation of 3aα-H-4α(3′-propanoate)-7aβ-methylhexahydro-1,5-indanedione (HIP) before the unknown final steps leading to CO2 (30, 34).
A frequently stated hypothesis is that the metabolites that accumulate during microbial cholate degradation represent precursors to rate-limiting steps in the pathway that occur at high cholate concentrations. At these “bottlenecks,” the excess metabolites are excreted into the extracellular environment until conditions permit the cell to reassimilate those metabolites and further degrade them. The lower final growth yields of the ΔcamB and ΔcamM mutants, each approximately 75% that of the wild type, indicate the extent to which reassimilated HHIDP and THSBNC, respectively, contribute to growth. This process of excretion and reassimilation could be an adaptive strategy employed by diverse bacteria that would maintain low intracellular levels of these metabolites, which are potentially toxic to the cell (5), while maximizing the nutritional benefits of cholate. Additionally, the rapid conversion of cholate to metabolites could be an adaptive strategy to lower the cholate concentration and reduce its toxicity. The toxicity of cholate and its degradation intermediates is indicated by the inability of RHA1 to grow in cultures with 5 mM or greater cholate.
We found additional support for the “bottleneck” hypothesis in the growth phenotype of the ΔcamM mutant on glycocholate. The mutant took longer to reach stationary phase than the wild type yet reached the same final growth yield without accumulation of THSBNC. We hypothesize that the initial steps in the degradation of glycocholate are slower than for cholate, due to the altered conformation of the side chain. This would result in a reduced accumulation of THSBNC, thus eliminating the requirement for its excretion. This lack of metabolite accumulation during growth on glycocholate was observed only with the ΔcamM mutant and not with the ΔcamB mutant. Indeed, the same patterns of metabolite accumulation were observed in stationary phase of the ΔcamB mutant grown on either of the conjugated bile acids or cholate. This could be because the ΔcamB mutant is required for transport of metabolites formed later in the catabolic pathway, at which point the rates of metabolite generation and efflux may be equal for all bile acid substrates.
This study contributes to the general knowledge of both the ABC and MFS superfamilies by identifying new members of each superfamily with defined transport functions. Since these transporters have novel substrates, they may contribute to understanding the relationship between protein structure and substrate specificity. Prior to this study, the possibility of transporters involved in cholate metabolite uptake was only postulated (5); we have now provided the evidence for and identification of such transporters. Further studies on the structures of the CamABCD and CamM systems, as well as investigations of their specificities, will contribute to the elucidation of these transporters.
Efflux transporters for the excretion of the aforementioned metabolites likely exist. It is possible that these transporters have broad specificity and perform a general function in the removal of potentially toxic compounds from the bacterial cell. Such transporters would likely be constitutively expressed, in contrast to camABCD and camM, which were the only RHA1 transporters significantly upregulated during growth on cholate (24a). Notably, we did not detect upregulation of a transporter involved in cholate uptake. The alpha orientation of all the hydroxyl groups on cholate creates distinct hydrophobic and hydrophilic parts of the molecule, which allows it to behave as a detergent (27). It is commonly thought that this property of cholate allows it to enter bacterial cells efficiently via passive diffusion (7), and our results suggest that this may be the mechanism by which RHA1 acquires cholate. Such a passive mechanism is in contrast to an active transporter for cholesterol described in RHA1 and other actinobacteria (24).
The cholate pathway is less well understood than the cholesterol and testosterone pathways, and metabolite reassimilation appears to be a unique facet of the first pathway. Indeed, understanding this aspect of cholate catabolism may have implications in a biotechnological context. For example, production of toxic steroid metabolites or metabolite reassimilation could potentially be rate limiting. Alternatively, excretion of desired products could increase the efficiency of a manufacturing process. The phenomenon of metabolite accumulation and reassimilation could thus impact the application of steroid-transforming bacteria in the manufacture of steroid-related therapeutics or bioremediation of steroid-contaminated environments.
ACKNOWLEDGMENTS
We thank Jie Liu for technical assistance with the construction of the mutant strains and Gordon Stewart for technical assistance with GC-MS, as well as Justin LeBlanc for technical advice and training for RT-QPCR. We thank Julian Davies and Karen Lu for the pSET152 vector. We thank Martin Tanner for advice on NMR analysis.
This research was supported by operating grants from the Natural Sciences and Engineering Research Council (NSERC) to W.W.M. and the Canadian Institutes for Health Research (CIHR) to L.D.E. I.C. is the recipient of a postdoctoral fellowship from the Fonds de Recherche en Santé du Québec and the Michael Smith Foundation for Health Research.
Footnotes
Published ahead of print 28 September 2012
REFERENCES
- 1. Bauchop T, Elsden SR. 1960. The growth of micro-organisms in relation to their energy supply. J. Gen. Microbiol. 23:457–469 [DOI] [PubMed] [Google Scholar]
- 2. Belfroid AC, et al. 1999. Analysis and occurrence of estrogenic hormones and their glucuronides in surface water and waste water in The Netherlands. Sci. Total Environ. 225:101–108 [DOI] [PubMed] [Google Scholar]
- 3. Bernstein H, Bernstein C, Payne CM, Dvorak K. 2009. Bile acids as endogenous etiologic agents in gastrointestinal cancer. World J. Gastroenterol. 15:3329–3340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bierman M, et al. 1992. Plasmid cloning vectors for the conjugal transfer of DNA from Escherichia coli to Streptomyces spp. Gene 116:43–49 [DOI] [PubMed] [Google Scholar]
- 5. Birkenmaier A, et al. 2007. Biochemical and genetic investigation of initial reactions in aerobic degradation of the bile acid cholate in Pseudomonas sp. strain Chol1. J. Bacteriol. 189:7165–7173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bortolini O, Medici A, Poli S. 1997. Biotransformations on steroid nucleus of bile acids. Steroids 62:564–577 [DOI] [PubMed] [Google Scholar]
- 7. Cabral DJ, Small DM, Lilly HS, Hamilton JA. 1987. Transbilayer movement of bile acids in model membranes. Biochemistry 26:1801–1804 [DOI] [PubMed] [Google Scholar]
- 8. Donova MV. 2007. Transformation of steroids by actinobacteria: a review. Appl. Biochem. Microbiol. 43:1–14 [PubMed] [Google Scholar]
- 9. Elder RT. 1983. Cloning techniques review of Molecular cloning: a laboratory manual. Bioscience 33:721–722 [Google Scholar]
- 10. Elhmmali MM, Roberts DJ, Evershed RP. 1997. Bile acids as a new class of sewage pollution indicator. Environ. Sci. Technol. 31:3663–3668 [Google Scholar]
- 11. Fernandes P, Cruz A, Angelova B, Pinheiro HM, Cabral JMS. 2003. Microbial conversion of steroid compounds: recent developments. Enzyme Microbiol. Technol. 32:688–705 [Google Scholar]
- 12. Goncalves ER, et al. 2006. Transcriptomic assessment of isozymes in the biphenyl pathway of Rhodococcus sp. strain RHA1. Appl. Environ. Microbiol. 72:6183–6193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hayakawa S. 1982. Microbial transformation of bile acids: a unified scheme for bile acid degradation, and hydroxylation of bile acids. Z Allg. Mikrobiol. 22:309–326 [DOI] [PubMed] [Google Scholar]
- 14. Helenius A, Simons K. 1975. Solubilization of membranes by detergents. Biochim. Biophys. Acta 415:29–79 [DOI] [PubMed] [Google Scholar]
- 15. Hofmann AF, Mysels KJ. 1988. Bile salts as biological surfactants. Colloids Surfaces 30:145–173 [Google Scholar]
- 16. Hogg JA. 1992. Steroids, the steroid community, and Upjohn in perspective: a profile of innovation. Steroids 57:593–616 [DOI] [PubMed] [Google Scholar]
- 17. Horinouchi M, Kurita T, Hayashi T, Kudo T. 2010. Steroid degradation genes in Comamonas testosteroni TA441: isolation of genes encoding a Delta 4(5)-isomerase and 3 alpha- and 3 beta-dehydrogenases and evidence for a 100 kb steroid degradation gene hot spot. J. Steroid Biochem. Mol. Biol. 122:253–263 [DOI] [PubMed] [Google Scholar]
- 18. Jobling S, Nolan M, Tyler CR, Brighty G, Sumpter JP. 1998. Widespread sexual disruption in wild fish. Environ. Sci. Technol. 32:2498–2506 [Google Scholar]
- 19. Johnston JO. 1998. Aromatase inhibitors. Crit. Rev. Biochem. Mol. Biol. 33:375–405 [PubMed] [Google Scholar]
- 20. LeBlanc JC, Goncalves ER, Mohn WW. 2008. Global response to desiccation stress in the soil actinomycete Rhodococcus jostii RHA1. Appl. Environ. Microbiol. 74:2627–2636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lorenzen A, Chapman R, Hendel JG, Topp E. 2005. Persistence and pathways of testosterone dissipation in agricultural soil. J. Environ. Qual. 34:854–860 [DOI] [PubMed] [Google Scholar]
- 22. Mathieu JM, et al. 2010. 7-Ketocholesterol catabolism by Rhodococcus jostii RHA1. Appl. Environ. Microbiol. 76:352–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McLeod MP, et al. 2006. The complete genome of Rhodococcus sp. RHA1 provides insights into a catabolic powerhouse. Proc. Natl. Acad. Sci. U. S. A. 103:15582–15587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mohn WW, et al. 2008. The actinobacterial mce4 locus encodes a steroid transporter. J. Biol. Chem. 283:35368–35374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24a. Mohn WW, et al. 2012. Gene cluster encoding cholate catabolism in Rhodococcus spp. 194:6712–6719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Moldowan JM, Dahl J, Mccaffrey MA, Smith WJ, Fetzer JC. 1995. Application of biological marker technology to bioremediation of refinery by-products. Energ. Fuel 9:155–162 [Google Scholar]
- 26. Pandey AK, Sassetti CM. 2008. Mycobacterial persistence requires the utilization of host cholesterol. Proc. Natl. Acad. Sci. U. S. A. 105:4376–4380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Philipp B. 2011. Bacterial degradation of bile salts. Appl. Microbiol. Biotechnol. 89:903–915 [DOI] [PubMed] [Google Scholar]
- 28. Schafer A, et al. 1994. Small mobilizable multipurpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145:69–73 [DOI] [PubMed] [Google Scholar]
- 29. Seto M, et al. 1995. A novel transformation of polychlorinated-biphenyls by Rhodococcus sp. strain RHA1. Appl. Environ. Microbiol. 61:3353–3358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sih CJ, Wang KC. 1963. Mechanisms of steroid oxidation by microorganisms. II. Isolation and characterization of 3a-alpha-H-4-alpha-[3′-propionic acid]-7a-beta-methyl-hexahydro-1,5-indanedione. J. Am. Chem. Soc. 85:2135–2137 [Google Scholar]
- 31. Simon R, Priefer U, Puhler A. 1983. A broad host range mobilization system for Invivo genetic engineering: transposon mutagenesis in Gram negative bacteria. BioTechnol. 1:784–791 [Google Scholar]
- 32. Streck G. 2009. Chemical and biological analysis of estrogenic, progestagenic and androgenic steroids in the environment. Trends Anal. Chem. 28:635–652 [Google Scholar]
- 33. Thomas KV, et al. 2002. An assessment of in vitro androgenic activity and the identification of environmental androgens in United Kingdom estuaries. Environ. Toxicol. Chem. 21:1456–1461 [PubMed] [Google Scholar]
- 34. Thomas ST, VanderVen BC, Sherman DR, Russell DG, Sampson NS. 2011. Pathway profiling in Mycobacterium tuberculosis: elucidation of cholesterol-derived catabolite and enzymes that catalyze its metabolism. J. Biol. Chem. 286:43668–43678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. van der Geize R, Hessels GI, van Gerwen R, van der Meijden P, Dijkhuizen L. 2001. Unmarked gene deletion mutagenesis of kstD, encoding 3-ketosteroid Delta1-dehydrogenase, in Rhodococcus erythropolis SQ1 using sacB as counter-selectable marker. FEMS Microbiol. Lett. 205:197–202 [DOI] [PubMed] [Google Scholar]
- 36. van der Geize R, et al. 2007. A gene cluster encoding cholesterol catabolism in a soil actinomycete provides insight into Mycobacterium tuberculosis survival in macrophages. Proc. Natl. Acad. Sci. U. S. A. 104:1947–1952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yam KC, et al. 2009Studies of a ring-cleaving dioxygenase illuminate the role of cholesterol metabolism in the pathogenesis of Mycobacterium tuberculosis. PLoS Pathog. 5:e1000344 doi:10.1371/journal.ppat.1000344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ying GG, Kookana RS, Ru YJ. 2002. Occurrence and fate of hormone steroids in the environment. Environ. Int. 28:545–551 [DOI] [PubMed] [Google Scholar]