

Abstract
HtpX is an integral cytoplasmic membrane metalloprotease well conserved in numerous bacteria. A recent study showed that expression of the Bacillus subtilis htpX gene is under dual negative control by Rok and a novel type of transcriptional regulator, YkrK. Here we report that expression of the B. subtilis htpX gene is strongly heat inducible. Contrary to the previous prediction, ykrK expression has been found to be not subject to autoregulation. We have identified the htpX promoter and the authentic ykrK promoter, which is also distinct from the previously predicted one. We have redefined a conserved inverted repeat sequence to be the YkrK operator, which is somewhat different from the previously proposed one. We provide evidence that YkrK is not a substrate of HtpX and that heat induction of htpX is not YkrK mediated. We have also found that the absence of FtsH or HtpX alone did not impair B. subtilis cell viability on LB agar plates at high temperature, whereas the absence of both FtsH and HtpX caused a severe growth defect under heat stress. This finding supports the notion that FtsH and HtpX may have partially overlapping functions in heat resistance. Finally, we show that htpX expression is subject to transient negative control by sigB under heat stress in a Rok- and YkrK-independent manner. Triple negative control of htpX expression at high temperature by rok, sigB, and ykrK may help cells to prevent uncontrolled and detrimental oversynthesis of the HtpX protease.
INTRODUCTION
When faced with heat stress, bacteria tend to produce misfolded and denatured proteins in the cytoplasm and cell envelopes. These nonnative proteins are potentially toxic to the cells and have to be refolded or degraded. Induction of expression of molecular chaperones and proteases in response to heat stress is a strategy widely used by bacteria to achieve protein quality control against misfolded and denatured proteins.
HtpX is a cytoplasmic membrane-bound Zn2+-dependent metalloprotease well conserved in numerous bacteria (1). Topology studies indicated that the active site of the Escherichia coli HtpX is located on the cytosolic side of the cytoplasmic membrane (26). Biochemical characterization of the E. coli HtpX confirmed its proteolytic activities against membrane and soluble proteins (22). Therefore, HtpX is involved in membrane protein quality control important for growth and survival of the cells, especially under heat stress (1). The E. coli htpX gene is known to be regulated by the CpxR/CpxA two-component system (26). Accumulation of abnormal cytoplasmic membrane proteins can activate this stress response pathway and transduce the signal to htpX.
The htpX gene of Bacillus subtilis encodes an integral membrane metalloprotease of 298 amino acids with a zinc-binding motif, HEXXH (where X represents any residue), at positions 155 to 159. The glutamic acid at position 156 is predicted to be a catalytic residue. HtpX was recently proposed to be one among a group of proteins that was associated with the MreB cytoskeleton of B. subtilis (15). A recent report by Marciniak et al. showed that expression of the B. subtilis htpX gene was under dual negative control by the Rok repressor and YkrK, a novel type of transcriptional regulator (17). However, we have found that some of the propositions and predictions in the report by Marciniak et al. (17) were controversial. In the present study, we have identified the authentic promoters of htpX and ykrK, redefined the YkrK operator, and recharacterized transcriptional regulation of htpX and ykrK. Furthermore, we have found that htpX expression is subject to transient negative control by sigB under heat stress.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The bacterial strains and plasmids used in this study are listed in Table S1 in the supplemental material. The oligonucleotide primers are listed in Table S2 in the supplemental material. Escherichia coli strain DH5α was used as a host for cloning purposes; E. coli strains BL21(DE3) and JM109 were used as hosts for the heterologous production of proteins from pET22b-based and pQE80-based vectors, respectively. E. coli and B. subtilis cells were grown in Luria-Bertani (LB) medium (23). The following antibiotics were used at the indicated concentrations: ampicillin, 100 μg/ml; chloramphenicol, 5 μg/ml; erythromycin, 1 μg/ml; kanamycin, 10 μg/ml.
Construction of plasmids.
To construct plasmids pGS1582, pGS1932, and pGS2010 for the disruption of htpX, ykrK, and ftsH, respectively, DNA fragments were amplified by PCR with primer pairs R935 plus R752, R978 plus T859, and T544 plus T545, respectively, and then cloned individually between the HindIII and BamHI sites of the thermosensitive plasmid pRN5101 (9).
To construct plasmids pGS1829 and pGS1962, which carry transcriptional fusions of the regulatory regions of htpX and ykrK, respectively, with the bgaB gene, DNA fragments were amplified by PCR with primer pairs T586 plus T587 and T901 plus T902, respectively, and then cloned individually between the BamHI and KpnI sites of the integrative promoter probe vector pDL (30).
To construct plasmid pGS1990 for overproduction of His-tagged YkrK in E. coli, a DNA fragment carrying the coding sequence of ykrK was amplified by PCR with primer T944 plus T945 and ligated into BamHI- and HindIII-digested pQE80 (Qiagen).
To construct plasmid pGS1994, which carries a 3-bp mutation (TAT to ATA) in the −10 box of the ykrK σA promoter within the htpX-bgaB fusion, primers T586, T587, T955, and T956 was employed for site-directed mutagenesis. The PCR-amplified DNA fragments were then cleaved with BamHI plus KpnI and cloned into pDL.
To construct plasmid pGS2018, which carries a 3-bp mutation (alteration from AAC to TTG) in the right arm of the inverted repeat within the htpX promoter region, or plasmid pGS2415, which carries a 4-bp mutation (GTTC to CAAG) in the left arm of the inverted repeat, a two-step PCR method (13) using primers T586, T587, T998, and T999 (for pGS2018) or primers T586, T587, A898, and A899 (for pGS2415) was employed for site-directed mutagenesis. The PCR-amplified DNA fragments were then cleaved with BamHI plus KpnI and cloned individually into pDL.
To construct plasmid pGS2197, which carries a 3-bp mutation (AAT to TTA) in the −10 box of the σA promoter of htpX, or plasmid pGS2416, which carries a 3-bp mutation (TAT to ATA) in the −10 box of the σA promoter of ykrK, primers T586, T587, A454, and A455 (for pGS2197) or primers T901, T902, T955, and T956 (for pGS2416) were employed for site-directed mutagenesis. The PCR-amplified DNA fragments were then cleaved with BamHI plus KpnI and cloned individually into pDL.
To construct plasmid pGS2264 for overproduction of His-tagged HtpX in E. coli, a DNA fragment carrying the coding sequence of htpX was amplified by PCR using primer A569 plus A570 and was then ligated into NdeI- and XhoI-digested pET22b (Novagen). To construct plasmid pGS2305 for overproduction of the E156A mutant of HtpX in E. coli, primers A569, A570, A657, and A658 were employed for site-directed mutagenesis. The PCR-amplified DNA fragment was then ligated into NdeI- and XhoI-digested pET22b.
To construct plasmid pGS2277 for deletion of the chromosomal htpX gene and replacement with the kanamycin resistance gene (kan), a DNA fragment containing a region located upstream of the htpX gene was amplified by PCR using primer T586 plus A591 and digested with BamHI and PstI. After cloning of this DNA fragment into plasmid pDG780 (10), the resulting plasmid was cleaved with BamHI and SalI to obtain an about 1.8-kb DNA fragment containing the kan gene. Another DNA fragment containing a region located downstream of the htpX gene was also amplified by PCR using primer A592 plus T682 and digested with SalI and EcoRI. These two DNA fragments were then ligated together into BamHI- and EcoRI-digested plasmid pMAD (2).
Construction of disruption mutants of B. subtilis.
Disruption of the chromosomal ftsH, htpX, or ykrK gene by integration of pGS2010, pGS1582, or pGS1932, respectively, through a Campbell-like single-crossover recombination was performed as described previously (9). The integration plasmid was introduced into B. subtilis cells by the protoplast method (5). The correctness of integrants was verified by PCR.
Construction of deletion and double mutants of B. subtilis.
Construction of the htpX deletion mutant BM1889, in which the htpX gene was replaced with the kanamycin resistance gene kan via a two-step gene replacement event, was performed according to the method described previously (2). The correct replacement of the chromosomal htpX sequence was verified by PCR. To construct the ftsH htpX double mutant BM1892, the pRN5101-derived plasmid pGS2010 was introduced into the htpX deletion mutant BM1889. Chromosomal integration of pGS2010 through a single-crossover recombination was carried out as described above. The correct integration was verified by PCR.
Construction of strains with a bgaB fusion integrated at the amyE locus.
Competent cells of B. subtilis were prepared and transformed by pDL-derived integrative plasmids as described elsewhere (7). Transformants were selected by growth on chloramphenicol-containing LB agar plates. Correct integrants were screened by a method described elsewhere (20).
RNA isolation and primer extension analysis.
Total RNA was isolated according to the method of Zuber and Losick (32). B. subtilis cells were grown in 50 ml of LB medium to an optical density at 600 nm (OD600) of 0.6. After harvesting the cells by centrifugation, cell pellets were resuspended in 5 ml of protoplasting buffer (15 mM Tris-HCl [pH 8.0], 0.45 M sucrose, and 8 mM EDTA). Lysozyme (50 mg) was then added to the mixture, and the mixture was incubated at room temperature for 15 min. After centrifugation at 4,000 × g, the resulting protoplast pellet was resuspended in 4 ml of cold lysing buffer (10 mM Tris-HCl [pH 8.0], 10 mM NaCl, and 1.5% SDS) and kept on ice for 5 min. After that, 4 ml of saturated NaCl solution was added and the mixture was kept on ice for another 5 min. After centrifugation at 15,000 × g, the supernatant was extracted with an equal volume of phenol-chloroform. The aqueous phase was then mixed with an equal volume of 8 M LiCl, and the mixture was incubated at −20°C for at least 2 h. After centrifugation at 15,000 × g, the RNA in the pellet was resuspended in diethyl pyrocarbonate-treated H2O and stored at −70°C for later use. Primer extension was performed according to a standard method (3). For each extension reaction, 25 μg of total RNA was used. Primers A275 and A073 were labeled with [γ-32P]ATP by T4 DNA kinase and used to initiate reverse transcription by avian myeloblastosis virus reverse transcriptase for the determination of the transcriptional initiation sites of htpX and ykrK, respectively.
Northern blot analysis.
Total RNA was isolated as described above and was run by electrophoresis on a denaturing agarose gel containing 2% formaldehyde. Northern blotting was performed using the standard method (23). A DNA fragment corresponding to the internal sequence of htpX was amplified by PCR (R935 and R936 as primers), internally labeled with the Klenow enzyme plus [α-32P]dCTP, and used as the probe. Bands were visualized by using a Molecular Dynamics PhosphorImager. The phosphorimage was analyzed with ImageQuant software.
Overproduction and purification of the His-tagged YkrK protein.
Overproduction of the His-tagged YkrK protein in E. coli JM109 cells bearing plasmid pGS1990 and purification by affinity chromatography on a nickel-nitrilotriacetic acid (Ni-NTA) agarose column were carried out exactly as described previously (14).
EMSAs.
Electrophoretic mobility shift assays (EMSAs) for examining the interaction of YkrK with DNA were carried out according to a method described elsewhere (3). A DNA fragment corresponding to positions −77 to + 97 (relative to the transcriptional initiation site of htpX) and containing either the wild-type inverted repeat in the htpX promoter region or a mutation in either arm of the inverted repeat was amplified by PCR using primers A024 and A025. After digestion by BamHI, this DNA fragment was end labeled with the Klenow enzyme plus [α-32P]dCTP and used as the probe. Various amounts of purified His-tagged YkrK were then mixed with approximately 1 nM 32P-labeled DNA probe in the binding solution (final volume, 30 μl) containing 40 mM Tris-HCl (pH 7.4), 50 mM KCl, 1 mM EDTA, 1 mM dithiothreitol (DTT), 0.01% bovine serum albumin (BSA), and 0.1 μg of poly(dI-dC) · poly(dI-dC). After incubation at room temperature for 20 min, the mixtures were run on a 6% native polyacrylamide gel. Bands were visualized by using a Molecular Dynamics PhosphorImager. The phosphorimage was analyzed with ImageQuant software.
DNase I footprinting analysis.
The same DNA fragment as that used in EMSA was end labeled with 32P and used as the probe. In 30 μl of reaction mixtures, various amounts of purified His-tagged YkrK were mixed with approximately 10 ng of the DNA probe in the binding solution [40 mM Tris-HCl (pH 7.4), 1 mM DTT, 50 mM KCl, 0.01% BSA, 1 μg of poly(dI-dC) · poly(dI-dC), and 5 mM CaCl2], and the mixture was incubated for 15 min at room temperature. DNase I (200 ng) was then added, and incubation was continued at room temperature for 2 min. Reactions were terminated by the addition of EDTA to a final concentration of 10 mM. After extraction with phenol-chloroform, the DNA in the supernatant was precipitated with ethanol, resuspended in 10 μl of the loading solution (80% deionized formamide, 0.02% bromophenol blue, and 0.02% xylene cyanol), and denatured at 95°C for 3 min. Samples were then electrophoresed on an 8% polyacrylamide gel containing 8 M urea and analyzed by phosphorimaging.
Partial purification of His-tagged HtpX and the E156A mutant.
Partial purification of His-tagged HtpX and the E156A mutant was carried out according to a method described elsewhere (27), but with some modifications. E. coli cells carrying plasmid pGS2264 (HtpX) or pGS2305 (E156A) were grown at 37°C in 1 liter of LB medium to an OD600 of 0.5. IPTG (isopropyl-β-d-thiogalactopyranoside) was then added at a final concentration of 0.4 mM and cells were allowed to continue to grow for 2 h. After centrifugation, cell pellets were washed three times with TE buffer (50 mM Tris-HCl [pH 8.0] and 10 mM EDTA) and then resuspended in TE buffer supplemented with 1% Triton X-100. A French press was used to break the cells. After centrifugation at 2,500 × g, membrane materials in the supernatants were collected by ultracentrifugation at 115,000 × g. The resulting pellets were resuspended in a solution containing 50 mM 3-(cyclohexylamino)-1-propane sulfonic acid (CAPS; pH 11.0), 0.6% N-lauroylsarcosine, and 1% Triton X-100. After incubation at room temperature for 30 min, samples were clarified by ultracentrifugation. The supernatants were dialyzed extensively against buffer A (50 mM Tris-HCl [pH 8.0], 100 mM NaCl, 5% glycerol, and 1% Triton X-100) and then incubated at 4°C for 30 min with Ni-NTA agarose, which was preequilibrated with buffer A. After centrifugation of the mixture, the HtpX- or E156A-bound Ni-NTA agarose was washed three times with buffer A and subjected to proteolytic activity assays.
In vitro assays for proteolytic activities of HtpX and E156A.
Assays for proteolytic activities of partially purified His-tagged HtpX and E156A were performed as described elsewhere (22), with slight modifications. The relative concentrations of His-tagged HtpX and E156A were estimated by Western blotting with antibody against His tag as the probe. A roughly equal amount of HtpX and E156A was separately incubated with 3 mM casein or 1.5 mM purified YkrK at 37°C for 6 h in 50 μl of reaction buffer containing 50 mM Tris-HCl (pH 8.0), 100 mM NaCl, 5% glycerol, 0.1% Triton X-100, and 5 mM zinc acetate. The reaction mixtures were then mixed with a quarter volume of 5× SDS sample buffer, and the mixture was incubated at 37°C for 5 min and subjected to SDS–13% PAGE.
Assay of heat survival.
Heat survival rates were determined as described previously (28). Different strains were cultured at 37°C to an OD600 of 1.5. After serial dilutions, cells were plated on LB agar and incubated at 37 and 55°C separately for 20 h. Colonies were then counted and calculated as the numbers of CFU.
β-Galactosidase activity assay.
The activity of the thermostable β-galactosidase BgaB was measured as described previously (24), but with some modifications (29). A portion (at least 2 ml) of the bacterial culture was pelleted and resuspended in an equivalent volume of chilled BgaB buffer (25 mM potassium phosphate [pH 6.4], 50 mM KCl, and 1 mM MgSO4). The absorbance of resuspended cells was measured at 600 nm. For cell permeabilization, 50 μl of 0.1% SDS and 100 μl of chloroform were added to 1 ml of resuspended cells. Permeabilization was achieved by vortexing. The reaction was then initiated by adding 0.2 ml of o-nitrophenyl-β-d-galactopyranoside (ONPG) at a concentration of 4 mg per ml. After incubation at 55°C for 30 min, 0.5 ml of 1 M Na2CO3 was added to stop the reaction. The reaction mixture was centrifuged to remove cell debris and chloroform. The absorbance of the supernatant was recorded at 420 nm and 550 nm. BgaB activity is given in Miller units (19).
Other methods.
The genomic DNA of B. subtilis was isolated by the method described previously (21). Western blotting was carried out by a standard method (23). Protein concentrations were determined by the bicinchoninic acid protein assay method according to the instructions of the assay kit manufacturer (Pierce Biotechnology, Inc.) with bovine serum albumin as the standard.
RESULTS AND DISCUSSION
Heat induction of htpX and ykrK.
The htpX and ykrK genes of B. subtilis are adjacent and divergently transcribed (Fig. 1). Such genetic organization is also present at least in B. amyloliquefaciens, B. licheniformis, and B. pumilus (see Fig. S1 in the supplemental material). Marciniak et al. recently reported that environmental osmotic stress and overproduction of membrane proteins could strongly induce expression of the htpX-gfp fusion in B. subtilis (17). When heat induction of htpX was investigated by Marciniak et al., gfp was also used as the reporter gene and the green fluorescent protein (GFP) fluorescence of the htpX-gfp fusion was monitored by flow cytometry (17). It was found that heat stress (42°C or 50°C for 60 min relative to 37°C for 60 min) resulted in only a relatively mild increase in expression of htpX-gfp (see Fig. 2D of reference 17). Technically speaking, heat stress at 42°C seemed to be not severe enough for B. subtilis (which usually requires at least 50°C for heat stress). However, heat stress at 50°C might cause a significant loss of fluorescence intensity for GFP, since it was reported that both GFP and GFP fusion protein were thermally sensitive and that the fluorescence intensity of GFP decreased linearly with increasing temperature from 15°C to 80°C (31). These could explain why only a mild effect of heat stress on expression of htpX-gfp was observed. In this study, we used the thermostable β-galactosidase BgaB as a reporter (30). A DNA fragment containing the regulatory region of htpX or ykrK was transcriptionally fused to bgaB and integrated separately into the chromosome by a double crossover at the amyE locus of B. subtilis cells. It was found that not only expression of htpX-bgaB was strongly induced by heat stress at 51°C but also expression of ykrK-bgaB was moderately heat inducible (Fig. 2A and B). We also used Northern blot analysis to confirm the strong induction of htpX transcription by heat stress at 51°C (see Fig. S2 in the supplemental material). Therefore, at least htpX should be a member of the heat shock stimulon of B. subtilis.
Fig 1.

Nucleotide sequence of the B. subtilis ykrK-htpX intergenic region. Nucleotide positions are numbered relative to the transcriptional initiation site of htpX. An inverted repeat that serves as the YkrK operator is indicated by an inverted pair of solid arrows above the sequence. The positions of mutagenesis within the inverted repeat (AAC to TTG in the right arm and GTTC to CAAG in the left arm, as described in the legend to Fig. 2C and D, respectively) are underlined with dotted lines. The −35 and −10 boxes of the σA promoter of ykrK are underlined, whereas those of htpX are overlined. The position of mutagenesis within the −10 box of the σA promoter of ykrK (TAT to ATA, as described in the legend to Fig. 2A) is overlined with a dashed line. The translational start sequences of ykrK and htpX are denoted in boldface. SD, Shine-Dalgarno sequence.
Fig 2.

(A) Effect of heat stress, disruption of ykrK, or mutation in the −10 box of the σA promoter of ykrK (TAT to ATA) on expression of the ykrK promoter region-bgaB fusion. The transcriptional fusion of the regulatory region of ykrK to bgaB was integrated at the amyE locus. B. subtilis cells were grown at 37°C in LB medium to an OD600 of 0.3, and then the culture was split into two portions. One portion was maintained at 37°C (circles), and the other was shifted to 51°C (squares) at time zero. The cells were then grown for various times as indicated. Solid lines and solid symbols, no disruption (BM1506); solid lines and open symbols, disruption of ykrK (BM1537); dotted lines and solid symbols, mutation in −10 box (BM2026). (B) Effect of heat stress or ykrK disruption on expression of the htpX promoter region-bgaB fusion. Solid symbols, no disruption (BM1302); open symbols, disruption of ykrK (BM1482); circles, 37°C; squares, 51°C. (C) Effect of mutation in the right arm of the inverted repeat (AAC to TTG) on expression of the htpX promoter region-bgaB fusion. Solid lines and solid symbols, no mutation (BM1302); solid lines and open symbols, with mutation (BM1579); dotted lines and solid symbols, with mutation and in ykrK mutant (BM2098); circles, 37°C; squares, 51°C. (D) Effect of mutation in the left arm of the inverted repeat (GTTC to CAAG) on expression of the htpX promoter region-bgaB fusion. Solid lines and solid symbols, no mutation (BM1302); solid lines and open symbols, with mutation (BM2024); dotted lines and solid symbols, with mutation and in ykrK mutant (BM2025); circles, 37°C; squares, 51°C. (E) Effect of htpX disruption on expression of the htpX promoter region-bgaB fusion. Solid symbols, no disruption (BM1302); open symbols, disruption of htpX (BM1495); circles, 37°C; squares, 51°C. The values shown in panels A to E are means from two independent experiments. Individual values did not differ by more than 15% from the means. (F) Effect of deletion of sigB on expression of the htpX promoter region-bgaB fusion. B. subtilis cells were grown at 37°C in LB medium to an OD600 of 0.3, and then the culture was split into two portions. One portion was maintained at 37°C for another 15 min and the other was shifted to 51°C for 15 min. Stippled bars, no deletion (BM1302); solid bars, deletion of sigB (BM1501). Each value is the mean ± SD of at least three determinations.
ykrK expression is not autoregulated by YkrK.
Marciniak et al. claimed to identify the sequence 5′-TGAWCTTA-3′ (where W represents A or T) as the YkrK binding motif (17). This motif is present within the ykrK-htpX intergenic region at three locations: downstream of the predicted σA promoter of htpX, overlapping with the −35 box of the predicted σA promoter of ykrK, and, with more deviation from the consensus, between these two predicted promoters (see Fig. 6B of reference 17). Since one of the three proposed YkrK binding sites happened to overlap the −35 box of the ykrK σA promoter, they predicted that ykrK expression might be subject to autoregulation (17). However, we found in this study that disruption of the ykrK gene had no effect on expression of ykrK-bgaB (Fig. 2A), whereas the same mutation led to a marked derepression of htpX-bgaB (Fig. 2B). This finding casts some doubt on the correctness of the previously predicted ykrK promoter.
Fig 6.

(A) Western blot analysis of HtpX and the active-site mutant E156A. His-tagged HtpX and E156A were partially purified from crude extracts of E. coli BL21(DE3) cells carrying the HtpX-overproducing plasmid pGS2264 and E156A-overproducing plasmid pGS2305, respectively, by Ni-NTA affinity column. An equal amount of partially purified HtpX (lane 1) and E156A (lane 2) was separately loaded onto an SDS–13% polyacrylamide gel and subjected to Western blotting with antibody against the His tag as the probe. Lane M, prestained protein markers with molecular masses shown on the left. (B) SDS-PAGE analysis of proteolysis by HtpX and E156A. An equal amount of partially purified HtpX and E156A was separately incubated with casein (0.5 mM) or YkrK (0.25 mM) in the presence of 5 mM Zn+2 at 37°C for 0 h (lanes 1, 3, and 5) or 6 h (lanes 2, 4, and 6). The reaction mixtures were then loaded onto an SDS–13% polyacrylamide gel. Bands were visualized with Coomassie brilliant blue staining. Lane M, protein markers with molecular masses shown on the left.
Identification of the authentic ykrK promoter.
The putative −35 and −10 boxes of the previously predicted σA promoter of ykrK are TTGAAC and TATATC, respectively (see Fig. 6B of reference 17). The spacing between the −35 and −10 boxes is only 13 nucleotides rather than the usual 16 to 18 nucleotides. To our knowledge, such short spacing has not been observed in other known σA-dependent promoters of B. subtilis. In this study, sequence analysis of the ykrK-htpX intergenic region revealed a potential σA promoter of ykrK (Fig. 1; see Fig. S1 in the supplemental material). Its −35 and −10 boxes, with a spacing of 17 nucleotides, are TTGAAA and TATGAT, respectively. To determine whether this potential σA promoter is active, we first used site-directed mutagenesis to alter its −10 box (TATGAT to ATAGAT) in the ykrK-bgaB fusion. We found that this mutation totally abolished ykrK-bgaB expression under normal conditions and heat stress (Fig. 2A). This result suggests that this promoter is the only authentic promoter of ykrK within the ykrK-htpX intergenic region under the assay conditions used. This result also revealed that the previously predicted σA promoter of ykrK was not functional under the assay conditions used, since it was present in the mutated DNA fragment and exhibited no promoter activity. We next used primer extension analysis to map the transcriptional start site of ykrK. It was found that one major extension product was obtained (Fig. 3A). The deduced transcriptional start site is at an appropriate distance from the σA promoter identified in this report (Fig. 1 and 3A), thus confirming it to be the authentic promoter of ykrK.
Fig 3.
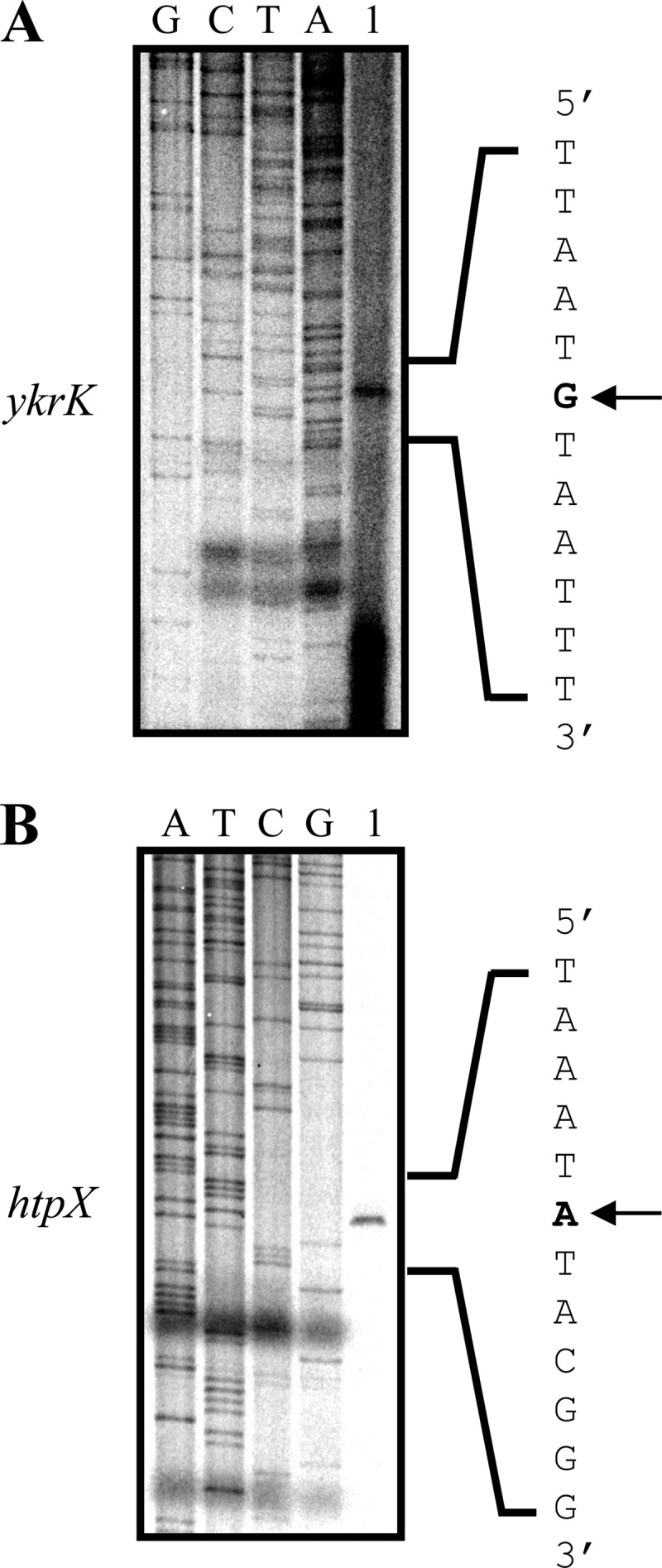
Primer extension analyses of the transcriptional initiation sites of ykrK (A) and htpX (B). Total RNA was isolated from B. subtilis cells grown in LB medium to an OD600 of 0.5. Lanes 1, primer extension products. Dideoxy sequencing ladders obtained with the same primer used for primer extension analysis are shown in lanes G, C, T, and A. The sequences shown are complementary to those read from the ladders. The arrows indicate the transcriptional initiation sites.
Identification of htpX promoter.
We also used primer extension analysis to map the transcriptional start site of htpX. The result showed that one major extension product was obtained (Fig. 3B). The deduced transcriptional start site is at an appropriate distance from the previously predicted σA promoter of htpX (Fig. 1 and 3B; see Fig. 6B of reference 17). Mutation in the −10 box of this σA promoter (TATAAT to TATTTA) totally abolished expression of htpX-bgaB under normal conditions and heat stress (data not shown), demonstrating it to be the only promoter of htpX within the ykrK-htpX intergenic region under the assay conditions used. Although the transcripts of the divergently transcribed htpX and ykrK genes are not overlapped, their promoters are partially overlapped (Fig. 1). This kind of promoter arrangement is conserved in some other Bacillus species (see Fig. S1 in the supplemental material). It is conceivable that the binding of one RNA polymerase molecule to either promoter may possibly affect the binding of another RNA polymerase molecule to the opposing promoter. Therefore, transcription from these two promoters may be mutually influenced. A preliminary experiment showed that mutation in the −10 box of the σA promoter of ykrK significantly enhanced htpX-bgaB expression under normal conditions and heat stress (see Fig. S3 in the supplemental material). This result suggests that active transcription of the ykrK promoter can negatively affect htpX transcription.
Redefining the YkrK operator.
One of the three YkrK binding sites previously proposed according to the previously defined YkrK binding motif is located downstream of the authentic ykrK promoter (see Fig. 6B of reference 17). Thus, YkrK binding to this proposed operator was supposed to repress ykrK transcription by a roadblock mechanism (4, 6). However, our data clearly indicate that expression of ykrK-bgaB is not autoregulated by YkrK (Fig. 2A). This result casts some doubt on the correctness of the proposed operator. Sequence alignments of the htpX promoter regions of some Bacillus species revealed a conserved inverted repeat sequence, 5′-GTTC-N4-GAAC-3′, located downstream of the authentic σA promoter of htpX (see Fig. S1 in the supplemental material). The previously defined YkrK binding motif 5′-TGAACTTA-3′ encompasses only the right half of this conserved inverted repeat. To investigate the role of this inverted repeat in htpX expression, we mutated either half of the inverted repeat in the regulatory region of the htpX-bgaB fusion. It was found that base substitutions in either half of the inverted repeat caused a marked derepression of htpX under normal conditions and heat stress (Fig. 2C and D), thus demonstrating the importance of this inverted repeat in htpX expression. When the expression of the htpX-bgaB fusion bearing the aforementioned mutation in either half of the inverted repeat was examined in the ykrK null mutant, no further derepression of bgaB expression was observed under normal conditions and heat stress (dotted lines in Fig. 2C and D). These results suggest that this inverted repeat might mediate negative regulation of htpX expression by YkrK.
To further establish that the inverted repeat is required for YkrK binding in vitro, we constructed plasmid pGS1990, which was able to overproduce His-tagged YkrK in E. coli, and then purified His-tagged YkrK by affinity chromatography on an Ni-NTA agarose column to about 90% homogeneity. A 32P-labeled DNA fragment containing the wild-type inverted repeat and spanning from positions −77 to +97 (relative to the transcriptional initiation site of htpX) was used as the probe in an EMSA. The same 32P-labeled DNA fragment, but containing a mutation in either half of the inverted repeat, was used as a control probe (Fig. 4). The results showed that purified His-tagged YkrK could retard the DNA fragment containing the wild-type inverted repeat but could not retard the two control probes under the assay conditions used (Fig. 4). These results indicate that the inverted repeat is required for YkrK binding.
Fig 4.

EMSAs of the interaction of YkrK with various DNA probes. A 32P-labeled DNA fragment containing the wild-type inverted repeat in the htpX promoter region and spanning from positions −77 to +97 (relative to the transcriptional initiation site of htpX) was used as the probe (probe I) in lanes 1 to 5. The same 32P-labeled DNA fragment but containing a mutation in the left arm of the inverted repeat (GTTC to CAAG) was used as a control probe (probe II) in lanes 6 to 10. A 32P-labeled DNA fragment containing a mutation in the right arm of the inverted repeat (AAC to TTG) was used as a second control probe (probe III) in lanes 11 to 15. About 1 nM 32P-labeled DNA probe was used in each reaction mixture (final volume, 30 μl). Lanes 1, 6, and 11, DNA probe alone; lanes 2 to 5, 7 to 10, and 12 to 15, DNA probe plus increasing amounts of purified His-tagged YkrK (3, 6, 12, and 24 ng for each set of four lanes, respectively).
We also used DNase I footprinting analysis to determine the location of the YkrK binding site in the htpX promoter region. The same DNA fragment as that used in the EMSA was used as the probe. When the upper strand of the DNA probe was end labeled with 32P, YkrK could protect the sequences from positions −11 to +24 against digestion by DNase I (Fig. 5A and C). This protected region contains the inverted repeat (from positions +7 to +18) mentioned above. When the lower strand of the DNA probe was end labeled with 32P, YkrK was found to protect the segment from positions −3 to +26 (Fig. 5B and C), which also contains the inverted repeat. Taken together, the results from both in vivo and in vitro analyses support the notion that the inverted repeat is the YkrK operator. It is possible that the flanking or the spacer sequence of the inverted repeat may also contribute to the binding of YkrK to DNA.
Fig 5.

DNase I footprinting analysis of YkrK binding to the htpX promoter region. (A) A BamHI-HindIII DNA fragment containing the htpX promoter region and spanning from positions −77 to +97 was labeled with 32P at the HindIII site of the upper strand and used as the probe for incubation with or without purified His-tagged YkrK. Lanes 1 and 6, no YkrK; lanes 2 to 5, increasing amounts of YkrK (200, 400, 600, and 800 ng, respectively). The numbers on the left indicate the positions of bases relative to the transcriptional initiation site of htpX. The protected regions are indicated by brackets. (B) The same BamHI-HindIII DNA fragment used for panel A was labeled with 32P at the BamHI site of the lower strand and used as the probe for incubation with or without YkrK. Lanes 1 and 6, no YkrK; lanes 2 to 5, increasing amounts of YkrK (200, 400, 600, and 800 ng, respectively). (C) Sequences of the protected regions.
Computer search for potential target genes of YkrK.
Since the adjacent downstream sequence TTA of the inverted repeat is also conserved in some other Bacillus species (see Fig. S1 in the supplemental material), we therefore used the search pattern GTTC-N4-GAACTTA (where N represents any base), allowing one mismatch in GAACTTA, to search the SubtiList database (http://genolist.pasteur.fr/SubtiList/) for other possible target genes of YkrK. The search region was set to be the regulatory region and within 300 bp upstream of a predicted gene. This search identified yrkA (encoding a hypothetical protein) and yvdA (possible carbonic anhydrase) as potential target genes. Only one potential YkrK binding site was found to be located 204 bp upstream of the −35 box of a putative σA promoter of yvdA, and thus, it seems unlikely for YkrK to act as a transcriptional repressor for yvdA expression. Since a potential YkrK binding site was found to be located in the promoter spacer region of a putative σA promoter of yrkA, we then tested whether the ykrK gene could regulate yrkA expression. A PCR-amplified DNA fragment containing the regulatory region of yrkA was fused to bgaB and integrated at the amyE locus of wild-type B. subtilis cells and the ykrK mutant. It was found that no significant difference in expression of the yrkA-bgaB fusion was observed between the wild type and the ykrK mutant (data not shown), suggesting that yrkA is not a YkrK target gene. Together, these analyses imply that YkrK is either a gene-specific (local) regulator or a regulator for, at most, very few genes.
Heat induction of htpX is not YkrK mediated.
Although transcription of htpX was strongly induced by heat stress, the mechanism underlying heat induction of htpX remained unknown. Figure 2B shows that disruption of ykrK increased the absolute levels of expression from the htpX-bgaB fusion under both normal conditions and heat stress but did not significantly alter the induction factor of htpX-bgaB by heat stress. This result implied that heat induction of htpX might not be YkrK mediated. To test this hypothesis, we examined the thermostability of YkrK in vitro. After being preheated at 51°C for 30 min or not, YkrK was immediately incubated with the specific DNA probe in the binding solution at 4°C (to prevent possible renaturation of YkrK) for 20 min. The reaction mixtures were then loaded onto a native polyacrylamide gel for EMSA. It was found that no difference in the DNA-binding ability of YkrK was observed regardless of preheating of YkrK at 51°C or not (data not shown). We also investigated the effect of high temperature on the formation of a YkrK-DNA complex in vitro. YkrK was incubated with the DNA probe in the binding solution at 37°C or 51°C for 20 min and then subjected to an EMSA. It was found that no difference in the formation of the YkrK-DNA complex was observed regardless of incubation at 37°C or 51°C (data not shown). Collectively, these results are consistent with the idea that YkrK is not a thermosensor and that heat induction of htpX is not YkrK mediated. The precise mechanism for heat induction of htpX remains to be clarified.
htpX expression is not autoregulated by HtpX.
HtpX is known to be involved in degradation of misfolded membrane proteins (1). Expression of the B. subtilis htpX gene has recently been shown to be induced by overproduction of membrane proteins (17). One might speculate that inactivation of htpX might increase the production of misfolded membrane proteins under heat stress and, therefore, that accumulated misfolded proteins would induce transcription from the htpX promoter. To test this hypothesis, we constructed an htpX disruption mutant and integrated the htpX-bgaB fusion at its amyE locus. The result showed that expression of the htpX-bgaB fusion in the wild type (BM1302) was not significantly different from that in the htpX mutant (BM1495) under both normal conditions and heat stress (Fig. 2E), suggesting that htpX expression is not autoregulated by HtpX. This is somewhat unexpected. A possible explanation is described below.
YkrK is not a substrate of HtpX.
If YkrK were a substrate of HtpX, inactivation of htpX would enhance the stability and level of the YkrK repressor and, therefore, reduce the level of htpX-bgaB expression under both normal conditions and heat stress. However, this was not the case (Fig. 2E), implying that YkrK might not be a substrate of HtpX.
To further establish that YkrK is not a substrate of HtpX, we partially purified His-tagged HtpX and His-tagged active-site mutant E156A of HtpX by affinity chromatography on an Ni-NTA agarose column (Fig. 6A). Partially purified HtpX or E156A was then incubated separately with casein or purified YkrK for 6 h. It was found that YkrK was not degraded by HtpX but casein was degraded under the same assay conditions (Fig. 6B). In a control experiment, casein was not degraded by E156A, suggesting that casein degradation was caused by a functional HtpX protease. Together, these results support the notion that YkrK is not a substrate of HtpX.
The htpX gene is required for heat survival of B. subtilis in the absence of ftsH.
FtsH is an ATP-dependent membrane protease with cytosolic and membrane proteins as the substrates and is conserved in numerous bacteria (16). Both FtsH and HtpX are involved in membrane protein quality control (1). It had been proposed that FtsH and HtpX of E. coli might have some overlapping or complementary proteolytic functions (26). In this study, we constructed an ftsH mutant (BM1574), an htpX mutant (BM1889), and an ftsH htpX double mutant (BM1892) of B. subtilis for heat survival assays. It was found that inactivation of either ftsH or htpX alone had no significant effect on the heat survival of cells grown at 55°C on LB agar plates, whereas inactivation of both ftsH and htpX caused a severe defect of growth at 55°C on LB agar plates (Table 1). This result is somewhat similar to that observed in E. coli cells grown on L-agar plates (26) and supports the notion that FtsH and HtpX have partially overlapping functions in heat resistance. This mutually compensatory function of FtsH and HtpX may probably explain why inactivation of htpX alone failed to enhance htpX-bgaB expression.
Table 1.
Heat survival ratea
| Strain | No. of CFU after incubation at: |
Survival rate (%) | |
|---|---|---|---|
| 37°C | 55°C | ||
| 168 | 3.2 × 108 | 2.8 × 108 | 87.5 |
| BM1889 (HtpX−) | 3.5 × 108 | 3.0 × 108 | 85.7 |
| BM1574 (FtsH−) | 2.3 × 108 | 1.9 × 108 | 82.6 |
| BM1892 (FtsH− HtpX−) | 9.3 × 107 | 6.5 × 105 | 0.7 |
The genotypes of the strains are listed in Table S1 in the supplemental material. The relevant phenotypes of the strains are given within parentheses. The titers of various strains after incubation at 37 and 55°C on LB agar plates are given as the numbers of CFU. Heat survival assays were performed at least three times with each strain. Representative results are shown.
It should be mentioned that the ftsH single mutant of B. subtilis was previously reported to display a heat-sensitive phenotype since it failed to grow in liquid Spizizen minimal medium at 50°C for at least 10 h (8). In this study, we found that the ftsH single mutant could grow, albeit more slowly, to the same cell density as the wild-type cells after growth at 51°C in liquid LB medium for 9 h (data not shown). The ftsH single mutant could also grow at 55°C on LB agar plates, as described above.
htpX expression is subject to transient negative control by sigB under heat stress.
SigB, the general stress sigma factor of B. subtilis, can be activated by energy depletion or by exposure to environmental stress (11). Under heat stress, rapidly and transiently activated SigB can associate with core RNA polymerase to positively regulate a large group of genes belonging to class II heat shock genes of the heat shock stimulon of B. subtilis (12, 25). To examine whether sigB was involved in heat induction of htpX, we integrated the htpX-bgaB fusion at the amyE locus of the sigB deletion mutant 1A780 (see Table S1 in the supplemental material). The absence of SigB activity in the sigB deletion mutant was verified by the lack of expression of gsiB, a gene known to be SigB regulated (18), in this mutant (data not shown). It was found, unexpectedly, that the level of htpX-bgaB expression was about 2-fold higher in the sigB mutant (BM1501) than in the wild type (BM1302) after heat treatment of cells at 51°C for 15 min (Fig. 2F). No significant difference in the level of htpX-bgaB expression was observed between the wild type and the sigB mutant grown at 37°C (Fig. 2F). These results indicate that under heat stress htpX expression is subject to transient negative control by sigB. Since no typical σB-dependent promoter sequence is present in the regulatory region of htpX and deletion of sigB did not transiently affect expression of rok or ykrK at 51°C (data not shown), sigB probably indirectly controls htpX expression in a Rok- and YkrK-independent manner. Triple negative control of htpX expression at high temperature by rok, sigB, and ykrK may help cells to prevent uncontrolled and detrimental oversynthesis of the HtpX protease.
ACKNOWLEDGMENTS
This research was supported by grant NSC 97-2311-B-010-003-MY3 from the National Science Council and a grant, Aim for the Top University Plan, from the Ministry of Education of the Republic of China.
Footnotes
Published ahead of print 5 October 2012
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1. Akiyama Y. 2009. Quality control of cytoplasmic membrane proteins in Escherichia coli. J. Biochem. 146:449–454 [DOI] [PubMed] [Google Scholar]
- 2. Arnaud M, Chastanet A, Debarbouille M. 2004. New vector for efficient allelic replacement in naturally nontransformable, low-GC-content, gram-positive bacteria. Appl. Environ. Microbiol. 70:6887–6891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ausubel FM, et al. 1995. Current protocols in molecular biology. John Wiley & Sons Inc, New York, NY [Google Scholar]
- 4. Belitsky BR, Sonenshein AL. 2011. Roadblock repression of transcription by Bacillus subtilis CodY. J. Mol. Biol. 411:729–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chang S, Cohen SN. 1979. High frequency transformation of Bacillus subtilis protoplasts by plasmid DNA. Mol. Gen. Genet. 168:111–115 [DOI] [PubMed] [Google Scholar]
- 6. Choi SK, Saier MH., Jr 2005. Regulation of sigL expression by the catabolite control protein CcpA involves a roadblock mechanism in Bacillus subtilis: potential connection between carbon and nitrogen metabolism. J. Bacteriol. 187:6856–6861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Contente S, Dubnau D. 1979. Characterization of plasmid transformation in Bacillus subtilis: kinetic properties and the effect of DNA conformation. Mol. Gen. Genet. 167:251–258 [DOI] [PubMed] [Google Scholar]
- 8. Deuerling E, Mogk A, Richter C, Purucker M, Schumann W. 1997. The ftsH gene of Bacillus subtilis is involved in major cellular processes such as sporulation, stress adaptation and secretion. Mol. Microbiol. 23:921–933 [DOI] [PubMed] [Google Scholar]
- 9. Fedhila S, Msadek T, Nel P, Lereclus D. 2002. Distinct clpP genes control specific adaptive responses in Bacillus thuringiensis. J. Bacteriol. 184:5554–5562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guerout-Fleury AM, Shazand K, Frandsen N, Stragier P. 1995. Antibiotic-resistance cassettes for Bacillus subtilis. Gene 167:335–336 [DOI] [PubMed] [Google Scholar]
- 11. Hecker M, Pane-Farre J, Volker U. 2007. SigB-dependent general stress response in Bacillus subtilis and related gram-positive bacteria. Annu. Rev. Microbiol. 61:215–236 [DOI] [PubMed] [Google Scholar]
- 12. Helmann JD, et al. 2001. Global transcriptional response of Bacillus subtilis to heat shock. J. Bacteriol. 183:7318–7328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Higuchi R, Krummel B, Saiki RK. 1988. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res. 16:7351–7367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huang SC, Lin TH, Shaw GC. 2011. PrcR, a PucR-type transcriptional activator, is essential for proline utilization and mediates proline-responsive expression of the proline utilization operon putBCP in Bacillus subtilis. Microbiology 157:3370–3377 [DOI] [PubMed] [Google Scholar]
- 15. Kawai Y, et al. 2011. A widespread family of bacterial cell wall assembly proteins. EMBO J. 30:4931–4941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Langklotz S, Baumann U, Narberhaus F. 2012. Structure and function of the bacterial AAA protease FtsH. Biochim. Biophys. Acta 1823:40–48 [DOI] [PubMed] [Google Scholar]
- 17. Marciniak BC, Trip H, Fusetti F, Kuipers OP. 2012. Regulation of ykrL (htpX) by Rok and YkrK, a novel type of regulator in Bacillus subtilis. J. Bacteriol. 194:2837–2845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Maul B, Volker U, Riethdorf S, Engelmann S, Hecker M. 1995. sigma B-dependent regulation of gsiB in response to multiple stimuli in Bacillus subtilis. Mol. Gen. Genet. 248:114–120 [DOI] [PubMed] [Google Scholar]
- 19. Miller JH. 1972. Experiments in molecular genetics, p 352–355 Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 20. O'Kane C, Stephens MA, McConnell D. 1986. Integrable alpha-amylase plasmid for generating random transcriptional fusions in Bacillus subtilis. J. Bacteriol. 168:973–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pospiech A, Neumann B. 1995. A versatile quick-prep of genomic DNA from gram-positive bacteria. Trends Genet. 11:217–218 [DOI] [PubMed] [Google Scholar]
- 22. Sakoh M, Ito K, Akiyama Y. 2005. Proteolytic activity of HtpX, a membrane-bound and stress-controlled protease from Escherichia coli. J. Biol. Chem. 280:33305–33310 [DOI] [PubMed] [Google Scholar]
- 23. Sambrook J, Russell D. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 24. Schrogel O, Allmansberger R. 1997. Optimisation of the BgaB reporter system: determination of transcriptional regulation of stress responsive genes in Bacillus subtilis. FEMS Microbiol. Lett. 153:237–243 [DOI] [PubMed] [Google Scholar]
- 25. Schumann W. 2003. The Bacillus subtilis heat shock stimulon. Cell Stress Chaperones 8:207–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shimohata N, Chiba S, Saikawa N, Ito K, Akiyama Y. 2002. The Cpx stress response system of Escherichia coli senses plasma membrane proteins and controls HtpX, a membrane protease with a cytosolic active site. Genes Cells 7:653–662 [DOI] [PubMed] [Google Scholar]
- 27. Siddiqui AA, Jalah R, Sharma YD. 2007. Expression and purification of HtpX-like small heat shock integral membrane protease of an unknown organism related to Methylobacillus flagellatus. J. Biochem. Biophys. Methods 70:539–546 [DOI] [PubMed] [Google Scholar]
- 28. Tseng CL, Chen JT, Lin JH, Huang WZ, Shaw GC. 2011. Genetic evidence for involvement of the alternative sigma factor SigI in controlling expression of the cell wall hydrolase gene lytE and contribution of LytE to heat survival of Bacillus subtilis. Arch. Microbiol. 193:677–685 [DOI] [PubMed] [Google Scholar]
- 29. Tseng CL, Shaw GC. 2008. Genetic evidence for the actin homolog gene mreBH and the bacitracin resistance gene bcrC as targets of the alternative sigma factor SigI of Bacillus subtilis. J. Bacteriol. 190:1561–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yuan G, Wong SL. 1995. Regulation of groE expression in Bacillus subtilis: the involvement of the sigma A-like promoter and the roles of the inverted repeat sequence (CIRCE). J. Bacteriol. 177:5427–5433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang C, Liu MS, Xing XH. 2009. Temperature influence on fluorescence intensity and enzyme activity of the fusion protein of GFP and hyperthermophilic xylanase. Appl. Microbiol. Biotechnol. 84:511–517 [DOI] [PubMed] [Google Scholar]
- 32. Zuber P, Losick R. 1983. Use of a lacZ fusion to study the role of the spoO genes of Bacillus subtilis in developmental regulation. Cell 35:275–283 [DOI] [PubMed] [Google Scholar]