

Abstract
Background
Holoprosencephaly (HPE) is the most common structural malformation of the human forebrain. There are several important HPE mutational target genes, including the transcription factor SIX3, which encodes an early regulator of Shh, Wnt, Bmp and Nodal signalling expressed in the developing forebrain and eyes of all vertebrates.
Objective
To characterise genetic and clinical findings in patients with SIX3 mutations.
Methods
Patients with HPE and their family members were tested for mutations in HPE-associated genes and the genetic and clinical findings, including those for additional cases found in the literature, were analysed. The results were correlated with a mutation-specific functional assay in zebrafish.
Results
In a cohort of patients (n = 800) with HPE, SIX3 mutations were found in 4.7% of probands and additional cases were found through testing of relatives. In total, 138 cases of HPE were identified, 59 of whom had not previously been clinically presented. Mutations in SIX3 result in more severe HPE than in other cases of non-chromosomal, non-syndromic HPE. An over-representation of severe HPE was found in patients whose mutations confer greater loss of function, as measured by the functional zebrafish assay. The gender ratio in this combined set of patients was 1.5:1 (F:M) and maternal inheritance was almost twice as common as paternal. About 14% of SIX3 mutations in probands occur de novo. There is a wide intrafamilial clinical range of features and classical penetrance is estimated to be at least 62%.
Conclusions
Our data suggest that SIX3 mutations result in relatively severe HPE and that there is a genotype–phenotype correlation, as shown by functional studies using animal models.
Holoprosencephaly (HPE) is the most common structural malformation of the human forebrain and occurs after failed or abbreviated midline cleavage of the developing brain (cortex and subcortical structures) during the third and fourth weeks of gestation. HPE occurs in up to 1 in 250 gestations, but only 1 in 8000 live births.1,2 Classically, three degrees of severity, defined by the extent of brain malformation, have been described. In the most severe form, alobar HPE, there is a single ventricle and no interhemispheric fissure. The olfactory bulbs and tracts and the corpus callosum are typically absent and dark-grey nuclei are not separated. In semilobar HPE, the most common type of HPE in neonates who survive to clinical examination, findings include partial cortical separation with absent or hypoplastic olfactory structures and corpus callosum. In lobar HPE, findings include separate ventricles but incomplete frontal cortical separation. Recently, an additional milder form, called middle interhemispheric variant (MIHV) has been delineated, in which the posterior frontal and parietal lobes are incompletely separated and the corpus callosum may be hypoplastic.3–6 Of patients with HPE who survived the neonatal period, alobar, semilobar and lobar HPE occurred in 21%, 60% and 19%, respectively.7 A separate study showed that of patients with non-chromosomal, non-syndromic HPE (including both living children and deceased fetuses), alobar, semilobar and lobar HPE occurred in 22%, 45% and 33%, respectively.8
Clinical features may be qualitatively predicted by the specific neuroanatomical abnormalities present. These features can include characteristic craniofacial anomalies, ophthalmological abnormalities such as colobomata or microphthalmia, severe mental retardation or developmental delay, pituitary dysfunction including diabetes insipidus, oromotor dysfunction, dysautonomia and seizures. Severely affected patients do not typically survive beyond early infancy; however, less severely affected patients may have normal life-spans. Though there are exceptions, more severe brain anomalies correlate with more severe clinical sequelae and shorter life-spans.7–10
Craniofacial findings tend to correlate with the type and severity of brain anomalies. In HPE caused by single-gene mutations, facial findings may additionally correlate with the causative gene. Patients with alobar HPE may be found to have cyclopia or synophthalmia (fusion of the optic vesicles and incomplete development of separate eyes), proboscis (a tubular nasal appendage appearing above the fused eyes), severe microcephaly and bilateral cleft lip and palate. Features in less severely affected patients may include microcephaly, hypotelorism, a flat nasal bridge and cleft lip or palate. Findings at the least severe end of the phenotypically recognisable spectrum (sometimes termed a “microform” of HPE, seen in patients without the cardinal central nervous system findings of HPE) may include solitary maxillary central incisor, hypotelorism and microcephaly. However, exceptions are often seen; a patient with severe HPE may have relatively subtle facial dysmorphisms and can have macrocephaly (as opposed to the more common microcephaly) due to hydrocephalus.4,9 Overall, facial anomalies have been reported in approximately 80% of patients with HPE and often lead to the diagnosis.8
HPE is aetiologically associated with teratogens such as maternal diabetes mellitus and alcohol, and has been reported in cases of prenatal exposure to pharmaceutical agents such as retinoic acid and the statin class of drugs, and to infections including cytomegalovirus, toxoplasmosis, and rubella.9,11,12 Up to half of patients with HPE have a numerical or structural chromosomal abnormality, whereas up to a quarter have HPE as part of a recognisable syndrome.13–15 HPE may also be due to single-gene mutations; < 25% of cases of HPE result from single-gene mutations in the currently commercially tested HPE-associated genes SHH, ZIC2, SIX3 and TGIF.8
Sonic Hedgehog (SHH) was the first causative gene identified in human HPE.16 Since then, single mutations in at least 10 other genes have been purported to cause HPE.9,17–24 In families in which these mutations were found, HPE seems to be inherited in an autosomal dominant fashion. Large kindreds segregating HPE-associated mutations show both incomplete penetrance and highly variable expressivity, suggesting additional environmental or genetic influences superimposed on the haploinsufficent state.23 According to this “multiple-hit” model, a mutation in a HPE-associated gene is necessary but not sufficient for HPE, and other genes or environmental factors are required for complete phenotypic feature.25
SIX3 has been reported as the third most common cause of HPE due to single-gene mutations.8 The vertebrate Six genes encode a family of related transcription factors that are orthologues of the sine oculis (“without eyes”) gene in Drosophila, and are expressed in the developing fly’s visual system, suggesting partially conserved phylogenetic roles. Indeed, vertebrate Six3 has been shown to be involved in midline forebrain and eye formation in several organisms including mouse, chick, Xenopus and zebrafish.26–28 Proteins encoded by the Six genes characteristically contain a DNA-binding homeobox domain, and an upstream SIX domain that can recruit additional factors to accomplish transcriptional activation or repression.29,30 Known biological properties of vertebrate Six3 include transcriptional repression of BMP, Wnt and Nodal targets through complex(s) formed with the general co-repressor Groucho.31–33 It also forms a complex with a different factor, geminin, which can influence a cell’s fate towards differentiation rather than proliferation in the early expanding forebrain territory.34 Presumably through a distinct set of co-factors, the Six3 transcription factor can instead activate lens specification genes during eye formation.35 Finally, Six3 acts as a direct regulator of Sonic hedgehog expression in the ventral forebrain.36,37
SIX3 was first identified as a candidate gene because of patients with HPE who had cytogenetic anomalies involving 2p21. SIX3, which has the appropriate spatial and temporal expression pattern to result in HPE, was the most attractive candidate gene in the interval.19,38
We report our findings in 59 new patients with SIX3 mutations and an additional 79 cases collected from the literature, and correlate the clinical phenotypes with SIX3 genotypes and functional studies in the zebrafish.19,38–51 The clinical findings echo the incomplete penetrance and highly variable expressivity seen in HPE due to mutations in other genes. Recent work in animal models specifically supports the multiple-hit model in mammalian SIX3-associated HPE.36 It has been suggested that SIX3 mutations result in a more severe holoprosencephaly phenotype than do mutations in the other HPE-related genes although a mechanism that would explain this is not yet clear.4,9,48
METHODS
The study protocol was approved by the National Human Genome Research Institute, and informed consent was obtained from all participants or their guardians.
Of the 65 cases for whom inheritance was known, 57.8% had maternal inheritance, 26.6% had paternal inheritance, 13.8% were de novo and the condition in 2 maternal half-siblings in one family seems to be due either an undetected germline mutation or parental mosaicism (the mother had negative mutation testing on peripheral blood analysis). We found no significant association between HPE severity and the parent of origin.
Of the 113 patients for whom gender was known, 40.7% were male and 59.3% were female, giving male:female ratio of 1:1.5. By χ2 analysis, there was a statistically significant difference in the increased prevalence of affected females overall (χ2 = 3.903, p = 0.0482). Of the 61 kindreds in which the gender of the proband was known, 59.0% were female and 41.0% were male, giving a male:female ratio of 1:1.4. There was no significant difference in the increased prevalence of affected female probands (χ2 = 1.984, p = 0.159).
Of those kindreds with molecularly identified specific mutations in the proband, 40.3% had multiple affected family members identified, though familial testing or clinical description was not available in all cases. Of the cases with described phenotypes, the penetrance of recognised phenotypic effect of mutations in SIX3 was estimated to be at least 82% (n = 111). Within kindreds with multiple affected members and where clinical description was available, penetrance was estimated at 73% (n = 61). However, testing was often performed only on individuals with the phenotype and their parents. Many others were ascertained only because of the presence of a relative with severe HPE and only on later examination were considered to have microform HPE (eg, hypotelorism). Considering these patients to be “unaffected” results in a penetrance estimate of 62% (n = 61) (fig 1, table 1).
Figure 1.

Patients with mutations in SIX3, arranged by HPE type (neuroimaging does not correspond with patients). All patients had point mutations in SIX3 with the exception of the patient shown in the lower row, second from left, who had a complex cytogenetic rearrangement including deletion of 2p21. MRI on patients in row 2 courtesy of the Carter Centers.19,48 MIHV, middle interhemispheric variant.
Table 1.
Patient characteristics, mutations and functional data
| Patient* | HPE type | DNA alteration (location) in SIX3 |
Predicted protein alteration in SIX3 |
Functional studies†47 |
Gender | Proband/ relative |
Ref. |
|---|---|---|---|---|---|---|---|
| 1 | Not specified | c.109G→T (NTD) | p.G37C | 0.5–0.9 | Female | Proband | NA |
| 2‡ | Lobar | c.109G→T (NTD) | p.G37C | 0.5–0.9 | Male | Proband | 42 |
| 3a‡ | MIHV | c.109G→T (NTD) | p.G37C | 0.5–0.9 | Female | Proband | NA |
| 3b‡ | None | c.109G→T (NTD) | p.G37C | 0.5–0.9 | Female | Mother | NA |
| 4a‡ | Alobar | c.206G→A (NTD) | c.G69D | >0.9 | Female | Proband | 48 |
| c.406dupGC (SD) | FS | <0.5 | |||||
| 4b‡ | None | c.206G→A (NTD) | c.G69D | >0.9 | Male | Father | 48 |
| c.406dupGC (SD) | FS | <0.5 | 48 | ||||
| 4c | None | c.206G→A (NTD) | c.G69D | >0.9 | Female | Paternal grandmother |
48 |
| c.406dupGC (SD) | FS | <0.5 | Female | 48 | |||
| 5a‡ | Alobar | c. 214G→C (NTD) | p.A72P | <0.5 | Female | Proband | 48 |
| 5b‡ | None | c. 214G→C (NTD) | p.A72P | <0.5 | Female | Mother | 48 |
| 6 | Alobar | c.235A→G (NTD) | p.M79V | Unknown | Not specified | Proband | 47 |
| 7a‡ | Alobar | Not specified (SD) | p.V92G | <0.5 | Male | Proband | 45 |
| 7b‡ | Alobar | Not specified (SD) | p.V92G | <0.5 | Male | Brother | 45 |
| 7c‡ | Alobar | Not specified (SD) | p.V92G | <0.5 | Male | Brother | 45 |
| 7d‡ | Microform | Not specified (SD) | p.V92G | <0.5 | Female | Mother | 45 |
| 8‡ | Semilobar | c.275T→G (SD) | p.V92G | <0.5 | Female | Proband | 43 |
| 9‡ | Semilobar | c.278C→A (SD) (PTCH1: c.1165G→A) |
p.A93D (PTCH1: p.A393T) |
<0.5 | Female | Proband | 22 42 |
| 10a‡ | Alobar | c.311A→G (SD) | p.D104G | 0.5–0.9 | Male | Proband | 48 |
| 10b‡ | None | c.311A→G (SD) | p.D104G | 0.5–0.9 | Male | Father | 48 |
| 11‡ | Not specified | c.313A→G (SD) | p.I105V | >0.9 | Not specified | Proband | 43 |
| 12‡ | Lobar | c.338G→A (SD) | p.W113X | <0.5 | Male | Proband | 42 |
| 13a‡ | Not specified | c.339G→A (SD) | p.W113X | <0.5 | Male | Proband | NA |
| 13b‡ | Not specified | Not tested | Not tested | Not tested | Male | Brother | NA |
| 13c‡ | Microform | c.339G→A (SD) | p.W113X | <0.5 | Female | Mother | NA |
| 14a‡ | Alobar | c.339G→T (SD) | p.W113C | <0.5 | Female | Proband | 42 |
| 14b‡ | Microform | c.339G→T (SD) | p.W113C | <0.5 | Female | Mother | 42 |
| 14c‡ | Microform | c.339G→T (SD) | p.W113C | <0.5 | Female | Maternal aunt | 42 |
| 14d‡ | Microform | c.339G→T (SD) | p.W113C | <0.5 | Male | Maternal uncle | 42 |
| 14e‡ | None | c.339G→T (SD) | p.W113C | <0.5 | Female | Maternal aunt | 42 |
| 14f‡ | Microform | c.339G→T (SD) | p.W113C | <0.5 | Female | Maternal grandmother |
42 |
| 14g‡ | Microform | Linkage only | Linkage only | Linkage only | Male | Maternal great uncle |
42 |
| 14h‡ | None | Linkage only | Linkage only | Linkage only | Female | Maternal first cousin |
42 |
| 14i‡ | Not specified | Linkage only | Linkage only | Linkage only | Female | Maternal half- sister |
42 |
| 14j | Not specified | Not tested | Not tested | Not tested | Female | Maternal first cousin |
42 |
| 14k‡ | Not specified | Not tested | Not tested | Not tested | Male | Maternal first cousin |
42 |
| 14l‡ | Not specified | Not tested | Not tested | Not tested | Female | Maternal first cousin |
42 |
| 14m | Not specified | Not tested | Not tested | Not tested | Female | Maternal first cousin, once removed |
42 |
| 14n | Not specified | Not tested | Not tested | Not tested | Male | Maternal great uncle |
42 |
| 14o | Not specified | Not tested | Not tested | Not tested | Not specified | Maternal great- grandparent |
42 |
| 15‡ | Semilobar | c.341C→T (SD) | p.S114L | <0.5 | Female | Proband | 42 |
| 16a‡ | Lobar | c.385G→T (SD) | p.E129X | <0.5 | Male | Proband | 42 |
| 16b‡ | Alobar | c.385G→T (SD) | p.E129X | <0.5 | Female | Sister | 42 |
| 16c‡ | Microform | c.385G→T (SD) | p.E129X | <0.5 | Male | Father | 42 |
| 17 | Not specified | c.389C→A (SD) | p.S130X | <0.5 | Male | Proband | 42 |
| 18a‡ | Alobar | c.404G→C (SD) | p.R135P | <0.5 | Male | Proband | 48 |
| 18b‡ | None | c.404G→C (SD) | p.R135P | <0.5 | Female | Mother | 48 |
| 19 | Not specified | c.404_407dupGCGC (SD) | FS | <0.5 | Female | Proband | 42 |
| 20‡ | Semilobar | c.405_409CGCCG (SD) | FS | <0.5 | Male | Proband | 42 |
| 21a | Not specified | c.463_465delCAC (SD) | p.H155del | Unknown | Not specified | Proband | 47 |
| 21b | Not specified | c.463_465delCAC (SD) | p.H155del | Unknown | Female | Mother | 47 |
| 22‡ | Not specified | c.469T→A (SD) | p.F157I | <0.5 | Male | Proband | 42 |
| 23a‡ | Alobar | c.507delG (SD) | FS | <0.5 | Female | Proband | 48 |
| 23b‡ | None | c.507delG (SD) | FS | <0.5 | Male | Father | 48 |
| 24a‡ | Lobar | c.515C→T (SD) | p.A172V | 0.5–0.9 | Male | Proband | 42 |
| 24b‡ | Semilobar | Not tested | Not tested | Not tested | Female | Sister | 42 |
| 24c‡ | Not specified | Not tested | Not tested | Not tested | Female | Mother | 42 |
| 25a | Alobar | c.518A→C (SD) | p.H173P | 0.5–0.9 | Female | Proband | 45 |
| 25b | None | c.518A→C (SD) | p.H173P | 0.5–0.9 | Female | Mother | 45 |
| 26a | Semilobar | c.518A→C (SD) | p.H173P | 0.5–0.9 | Female | Proband | 43 |
| 26b | None | c.518A→C (SD) | p.H173P | 0.5–0.9 | Female | Mother | 43 |
| 26c | None | c.518A→C (SD) | p.H173P | 0.5–0.9 | Female | Maternal aunt | 43 |
| 26d | Not specified | Not tested | Not tested | Not tested | Not specified | Maternal first cousin |
43 |
| 26e | Not specified | Not tested | Not tested | Not tested | Not specified | Maternal first cousin |
43 |
| 26f | Not specified | Not tested | Not tested | Not tested | Not specified | Maternal first cousin |
43 |
| 27‡ | Not specified | c.520T→C (SD) | p.Y174H | 0.5–0.9 | Female | Proband | 42 |
| 28a‡ | Alobar | c.542_576dup (SD) | FS | Unknown | Female | Proband | 43 45 |
| 28b‡ | None | c.542_576dup (SD) | FS | Unknown | Male | Father | 43 45 |
| 29‡ | Semilobar | c.551delC (SD) | FS | <0.5 | Female | Proband | 42 |
| 30a‡ | Alobar | c.556_557dupGG (SD) | FS | <0.5 | Female | Proband | 44 45 |
| 30b‡ | Alobar | c.556_557dupGG (SD) | FS | <0.5 | Male | Brother | 44 45 |
| 30c‡ | Alobar | c.556_557dupGG (SD) | FS | <0.5 | Female | Sister | 44 45 |
| 30d‡ | Not specified | c.556_557dupGG (SD) | FS | <0.5 | Female | Sister | 44 45 |
| 30e‡ | Microform | c.556_557dupGG (SD) | FS | <0.5 | Male | Father | 44 45 |
| 31a‡ | Alobar | Not tested | Not tested | Not tested | Female | Proband | 42 |
| 31b‡ | Semilobar | c.582dup C (SD) | FS | <0.5 | Female | Sister | 42 |
| 31c‡ | Microform | c.582dup C (SD) | FS | <0.5 | Male | Father | 42 |
| 32a‡ | Semilobar | c.605C→T (SD) | p.T202I | <0.5 | Female | Proband | 43 45 |
| 32b‡ | None | c.605C→T (SD) | p.T202I | <0.5 | Female | Mother | 43 45 |
| 33a‡ | Semilobar | c.619G→T (HD) | p.E207X | <0.5 | Male | Proband | 42 |
| 33b‡ | Alobar | c.619G→T (HD) | p.E207X | <0.5 | Female | Sister | 42 |
| 34 | Not specified | c.637T→G (HD) | p.F213V | <0.5 | Male | Proband | 42 |
| 35‡ | Lobar | c.652C→T (HD) | p.R218W | 0.5–0.9 | Female | Proband | 42 |
| 36‡ | Not specified | c. 653G→C (HD) | p.R218P | <0.5 | Female | Proband | 42 |
| 37‡ | Not specified | c.676C→G (HD) | p.L226V | .0.9 | Female | Proband | 19 52 |
| 38 | Not specified | c.680A→C (HD) | p.Q227P | 0.5–0.9 | Not specified | Proband | 42 |
| 39‡ | Microform | c.686C→T (HD) | p.P229L | Unknown | Male | Proband | 46 |
| 40‡ | Alobar | c.692C→G (HD) | p.P231R | <0.5 | Female | Proband | 45 |
| 41‡ | Alobar | c.698_706del (HD) | p.N233_S235del | <0.5 | Female | Proband | 19 39 |
| 42 | Not specified | c.GC718_719AA (HD) | p.A240K | <0.5 | Not specified | Proband | 42 |
| 43a‡ | Not specified | c.721C→T (HD) | p.Q241X | Unknown | Male | Proband | 42 |
| 43b | Not specified | Not tested | Not tested | Unknown | Not specified | Sibling | 42 |
| 43c‡ | None | c.721C→T (HD) | p.Q241X | Unknown | Female | Mother | 42 |
| 44 | Not specified | c.730G→T (HD) | p.G244C | <0.5 | Not specified | Proband | 42 |
| 45 | Not specified | c.736delA (HD) | FS | Unknown | Not specified | Proband | 47 |
| 46 | Not specified | c.743_745delCAG (HD) (TGIF: c.478C→T) |
p.248K delQ (TGIF: p.160S) |
Unknown | Female | Proband | NA |
| 47a‡ | Microform | c.749T→C (HD) | p.V250A | 0.5–0.9 | Male | Proband | 19 |
| 47b‡ | None | c.749T→C (HD) | p.V250A | 0.5–0.9 | Female | Mother | 19 |
| 47c‡ | None | c.749T→C (HD) | p.V250A | 0.5–0.9 | Female | Maternal aunt | 19 |
| 47d‡ | Alobar | c.749T→C (HD) | p.V250A | 0.5–0.9 | Not specified | Sibling | 19 |
| 47e‡ | Alobar | c.749T→C (HD) | p.V250A | 0.5–0.9 | Not specified | Sibling | 19 |
| 47f | Alobar | c.749T→C (HD) | p.V250A | 0.5–0.9 | Not specified | Sibling | 19 |
| 48 | Not specified | c.762T→A (HD) | p.F254L | 0.5–0.9 | Male | Proband | 42 |
| 49‡ | Not specified | c.769C→T (HD) | p.R257W | <0.5 | Female | Proband | 42 |
| 50a | Semilobar | c. 769C→T (HD) | p.R257W | <0.5 | Female | Proband | 43 45 |
| 50b | Alobar | Not tested | Not tested | Not tested | Male | Brother | 43 45 |
| 50c‡ | None | c. 769C→T (HD) | p.R257W | <0.5 | Male | Father | 43 45 |
| 51a | Lobar | c. 769C→G (HD) | p.R257G | Unknown | Not specified | Proband | 47 |
| 51b | Not specified | c. 769C→G (HD) | p.R257G | Unknown | Male | Father | 47 |
| 52‡ | Semilobar | c.770G→C (HD) | p.R257P | >0.9 | Female | Proband | 19 52 |
| 53 | Not specified | c.773G→C (HD) | p.R258L | <0.5 | Not specified | Proband | 42 |
| 54 | Not specified | c.785G→A (HD) | p.R262H | 0.5–0.9 | Not specified | Proband | 42 |
| 55a | Not specified | Not tested | Not tested | Unknown | Not specified | Proband | 47 |
| 55b | None | Not tested | Not tested | Unknown | Not specified | Obligate carrier | 47 |
| 55c | Not specified | c.806G→C (CTD) | p.R269T | Unknown | Male | Grandfather | 47 |
| 56a‡ | Semilobar | c.806G→T (CTD) | p.R269M | Unknown | Female | Proband | 47 51 |
| 56b‡ | Alobar | Not tested | Not tested | Unknown | Male | Sibling | 47 |
| 56c‡ | Lobar | c.806G→T (CTD) | p.R269M | Unknown | Male | Sibling | 47 |
| 56d‡ | Microform | c.806G→T (CTD) | p.R269M | Unknown | Female | Mother | 47 51 |
| 57 | Semilobar | c.806+1G→T (CTD) | Splice | Unknown | Not specified | Proband | 47 |
| 58‡ | Not specified | c.G807C (CTD) | p.R269S | 0.5–0.9 | Male | Proband | 42 |
| 59 | Not specified | c.820_832delinsCTGGACCT (CTD) |
p.A274X | Unknown | Male | Proband | NA |
| 60 | Semilobar | c.850G→C (CTD) (ZIC2: c.910TRC) |
p.A284P (ZIC2: p.W304R) |
Unknown | Female | Proband | NA |
| 61‡ | Semilobar | c.890C→T (CTD) | p.P297L | 0.5–0.9 | Female | Proband | 42 |
| 62‡ | Semilobar | c.944C→T (CTD) | p.T315I | Unknown | Female | Proband | 48 |
| 63 | Not specified | Deletion | NA | Unknown | Not specified | Proband | 47 |
| 64 | Not specified | Microdeletion by qPCR | NA | Unknown | Not specified | Proband | 40 41 |
| 65 | Not specified | Microdeletion by qPCR | NA | Unknown | Not specified | Proband | 40 41 |
| 66 | Not specified | Microdeletion by qPCR | NA | Unknown | Not specified | Proband | 41 |
| 67 | Not specified | Microdeletion by qPCR | NA | Unknown | Female | Proband | 41 |
| 68‡ | Semilobar | del(2)(p2101p2109) | NA | Unknown | Female | Proband | 38 49 |
| 69‡ | None | del(2)(p21) | NA | Unknown | Male | Proband | 38 |
| 70‡ | Semilobar | del(2)(p21p23) | NA | Unknown | Male | Proband | 38 |
| 71‡ | Semilobar | del(2)(p16p22) | NA | Unknown | Female | Proband | 38 |
| 72 | Lobar | del(2)(p21p22.2) | NA | Unknown | Female | Proband | 38 50 |
| 73‡ | Alobar | del(2)(p21p22.1) | NA | Unknown | Male | Proband | 38 |
| 74‡ | Semilobar | inv ins(2)(p21q24q13) | NA | Unknown | Female | Proband | 38 |
| 75‡ | Semilobar | t(1;2)(p21;p21) | NA | Unknown | Male | Proband | 38 |
| 76 | Alobar | t(1;2)(p22.3;p21) | NA | Unknown | Male | Proband | 38 |
| 77‡ | Lobar | del(2)(p16.3p21)§ | NA | Unknown | Male | Proband | NA |
CTD, C-terminal domain; HD, homeodomain; HPE, holoprosencephaly; MIHV, middle interhemispheric variant; NA, not applicable (unpublished when seeming in the Reference column); NTD, N-terminal domain; qPCR, quantitative PCR: SD, SIX domain.
Each kindred is numbered separately; within a kindred, individual members have separate letter identifier.
Protein Activity Index.
Clinical information available.
Complex rearrangement additionally resulting in chromosome 2 inversion, translocation involving chromosomes 7, 13 and 18 and deletions of regions of chromosomes 7 and 18.
Mutations
Of those with molecularly identified mutations, the 62 kindreds encompassed 63 mutations in SIX3 (kindred 4 had two mutations in SIX3), of which 93.7% were unique. Three unrelated kindreds had the same mutation in the N-terminal domain, two had the same mutation in the SIX domain and two had the same mutation in the homeodomain. Three patients had mutations in two HPE genes: one (patient 9) in both SIX3 and PTCH, one (patient 46) in both SIX3 and TGIF and one (patient 60) in both SIX3 and ZIC2 (tables 1, 2; figs 2, 3).
Table 2.
Mutations (n=63)
| n (%) | |
|---|---|
| Location | |
| N-terminal domain | 6 (9.5) |
| SIX domain | 27 (42.9) |
| Homeodomain | 22 (34.9) |
| C-terminal domain | 8 (12.7) |
| Exon 1 | 57 (90.5) |
| Exon 2 | 6 (9.5) |
| Mutation type | |
| Missense | 44 (69.8) |
| Nonsense | 6 (9.5) |
| Duplication→ FS | 6 (9.5) |
| Deletion→FS | 3 (4.8) |
| Splice mutation | 1 (1.6) |
| 9 bp deletion → 3 aa deletion | 1 (1.6) |
| 3 bp deletion → 1 aa deletion | 1 (1.6) |
| Deletion/insertion → nonsense | 1 (1.6) |
aa, Amino acid; FS, frameshift; MIHV, middle interhemispheric variant.
Figure 2.

Results of mutation studies.
Figure 3.

Known mutations in SIX3, showing holoprosencephaly and mutation type. MIHV, middle interhemispheric variant. Numbers refer to kindreds.
Clinical features
Patients with mutations in SIX3 (not including cytogenetic cases) had a significantly different distribution of HPE types than previously published cases of non-chromosomal, nonsyndromic HPE (χ2 = 24.179, p<1·10−4).8 Overall, patients with SIX3 mutations had a higher proportion of severe HPE (table 3).
Table 3.
Distribution of HPE types (n = 138)
| HPE type | n (% of total) (% of cases with known HPE type) |
|---|---|
| Alobar | 27 (19.6, 29.3) |
| Semilobar | 22 (15.9, 23.9) |
| Lobar | 9 (6.5, 9.8) |
| MIHV | 1 (0.7, 1.1) |
| Microform | 13 (9.4, 14.1) |
| None* | 20 (14.5, 21.7) |
| Not specified | 46 (33.3) |
MIHV, middle interhemispheric variant.
No evidence for penetrance.
The quality of patient data was highly variable, so it is difficult to accurately calculate the prevalence of specific phenotypic features. However, we present the most commonly reported craniofacial anomalies (table 4). Severe craniofacial findings such as cyclopia and proboscis were only reported with alobar HPE. Overall, the severity of facial dysmorphisms seemed to correlate with the degree of brain anomalies—for example, the degree of hypotelorism was more pronounced in patients with alobar HPE.
Table 4.
Reported* phenotypic findings (n = 91)
| Finding | Prevalence (%) |
|---|---|
| Craniofacial anomalies | |
| Hypotelorism | 44.0 |
| Microcephaly | 36.3 |
| Cleft lip and/or palate | 35.2 |
| Flat nasal bridge/absent nasal septum | 17.6 |
| Philtral agenesis/hypoplasia | 13.2 |
| Coloboma | 9.9 |
| Solitary maxillary central incisor | 8.8 |
| Cyclopia | 6.6 |
It is likely that many of these findings, such as hypotelorism, occur more often than was reported.
In terms of clinical features other than craniofacial anomalies, the most commonly reported findings, in decreasing order of prevalence are: mental retardation or developmental delay, seizure disorder and diabetes insipidus (supplemental tables 1a,b online).
Functional studies
Using the functional analysis developed by Domené et al,42 46 mutations (representing 99 patients with HPE who had neuroimaging performed or who were neurologically normal with SIX3 mutations) had functional studies performed using a zebrafish assay.42 Functional study results were divided into three categories, with protein activity described relative to a normal control (value of 1.0): (1) protein activity <0.5 (alleles with the least functional activity), (2) protein activity 0.5–0.9 (alleles with moderate functional activity) and (3) protein activity >0.9 (alleles with near-normal functional activity). One kindred (kindred 4) had two separate mutations in SIX3 and was categorised as belonging to the functional group with the more severe impairment of the two.
Including those patients who had alobar, semilobar and lobar HPE types with a functional protein index <0.5, there was an unequal distribution of HPE types (χ2 = 12.071, p = 0.0039), with more severe HPE over-represented. Of those with functional protein index <0.5 and HPE on neuroimaging, 60.7% had alobar, 32.1% had semilobar and 7.1% had lobar HPE. Within the group of patients whose mutations had a functional protein index of 0.5–0.9, a significantly unequal distribution of HPE types was not found (χ2 = 0.727, p = 0.8054). (fig 4, table 5).
Figure 4.
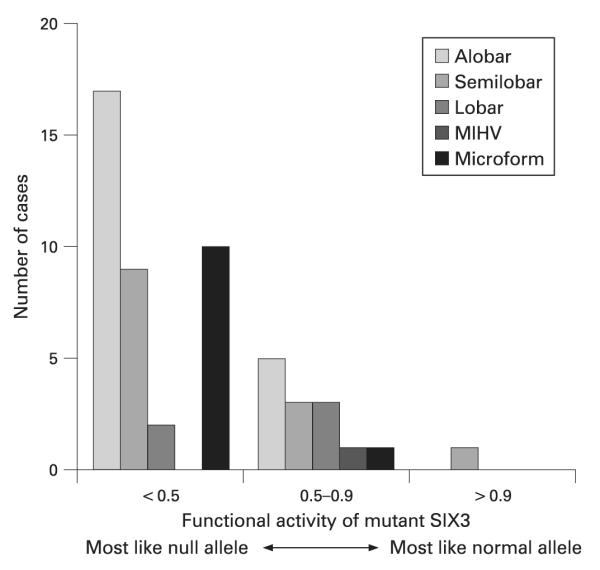
Functional results and HPE types, showing overrepresentation of severe HPE of whose mutation resulted in the greatest functional impairment by zebrafish assay.42 MIHV, middle interhemispheric variant.
Table 5.
Functional result vs. HPE type (n = 99)
| <0.5 | 0.5–0.9 | >0.9 | Total | |
|---|---|---|---|---|
| Alobar | 17 | 5 | 0 | 22 |
| Semilobar | 9 | 3 | 1 | 13 |
| Lobar | 2 | 3 | 0 | 5 |
| MIHV | 0 | 1 | 0 | 1 |
| Microform | 10 | 1 | 0 | 11 |
| None | 9 | 7 | 0 | 16 |
| Not specified | 19 | 10 | 2 | 31 |
| Total | 66 | 30 | 3 | 99 |
MIHV, middle interhemispheric variant.
Including those with the most severe types of HPE (alobar, semilobar and lobar) and functional protein index either <0.5 and 0.5–0.9, we performed the linear trend alternative to independence test,52 using SAS V9.1 software. As there was only one person with functional protein index >0.9 whose HPE type was known, this category was not included in the statistical analysis. Our results showed that, within the group of patients whose mutations had a functional protein index <0.9 there was a significant correlation between HPE types and the functional protein index (χ2 = 12.99, p = 0.0003). That is, the functional protein index is an explanatory variable with ordered categories for traditional HPE types.
DISCUSSION
We present 138 cases of HPE with mutations in SIX3, many of whom have not been previously clinically described; this is the largest comprehensive evaluation of a cohort of patients with mutations in a gene involved in HPE. Analysis of this group allows several conclusions to be made.
First, as has been previously posited, our study shows that mutations in SIX3 correlate with more severe HPE than with non-chromosomal, non-syndromic HPE overall.4,9,48 Despite the overall high severity, the clinical features are highly variable. Within large kindreds, people with SIX3 mutations include both phenotypically normal people and people with severe holoprosencephaly incompatible with life. The basis of this variable expressivity is largely unknown. One potential explanation is a multi-hit mechanism, examples of which are the three cases where mutations were found in SIX3 and in one other HPE-associated gene. In these patients, mutations in SIX3 may be necessary but not sufficient for HPE. Another insult, either genetic (eg, changes in at least one other HPE-associated gene) or environmental (eg, gestational diabetes mellitus) must also occur.
Second, mutations in SIX3 result in holoprosencephaly through a loss-of-function mechanism. Of patients whose mutations result in the greatest functional impairment in the zebrafish assay, protein activity correlates with human HPE severity.42 This analysis allows for a more refined genotype–phenotype discussion. Instead of correlating the gene involved or the location or type of the mutation with severity of phenotype, we can begin to predict features based on functional analyses.
Third, these patients do confirm the idea that in HPE, “the face predicts the brain”. That is, more severe facial dysmorphisms (such as cyclopia) tend to correlate with more severe HPE. Although this adage does not apply to all cases of HPE, it does seem to hold true in HPE due to SIX3 mutations.
Fourth, we estimated penetrance at 82% of all cases, 73% of cases from well-described families with multiple mutation carriers and 62% of those diagnosed on clinical grounds alone. In the practice of clinical genetics, this lowest estimate could be considered the most accurate. However, this latter penetrance estimate may be inaccurate for at least two reasons. First, the quality of clinical data was variable and subtle signs of midline defects may have been missed, resulting in an under-estimate of penetrance. Second, as full testing was not available on family members in many cases, there may have been many cases with SIX3 mutations who were not ascertained. This would result in an overestimate of penetrance. If more members of families could be tested, it would be possible to better calculate the penetrance, quantify expressivity and calculate the rate of sporadic mutations.
Fifth, it is interesting that more female patients have been reported with SIX3 mutations. The female predominance has been reported in some, but not all studies of HPE.4,8 The fact that the difference does reach significance when all mutation carriers are considered lends credence to the idea that being female is somehow protective. One explanation is that the SIX3 mutations in males may be more likely to be embryonic-lethal. However, in patients who survived long enough to have the type of HPE identified, the correlation between gender and severity of HPE was not significant.
One shortcoming of this report is that most of the patients discussed here were not seen in person, although we did perform the laboratory analysis to identify the SIX3 mutation in approximately half the cases. Details of the data available from referring clinicians varied greatly; in some cases, extensive medical records were sent, whereas in other cases, relatively little was available. For this reason, it is difficult to make certain genotype–phenotype correlations. However, it can be noted that in cases for which more clinical information was available, the severity of features often seemed more impressive. Thus, these data may under-represent the severity of the clinical features of SIX3 mutations. On the other hand, many people who had either no or very subtle features were ascertained only because they were related to a person with the same mutation but much more severe presenting signs. Following this logic, these data may over-represent severity.
Despite the challenges interpreting the large and varied data, the number of patients and families described here greatly enriches our understanding of the spectrum of features in patients with mutations in SIX3. These considerations argue for the importance of a combined and comprehensive approach to clinical and genetic studies of complex genetic disorders such as HPE.
Acknowledgements
We would like to express our deep gratitude to the patients and families who participated in these studies. We also thank all of the members of the Carter Centers for Brain Research in Holoprosencephaly and Related Malformations. We appreciate the contribution of Dr D A T Cummings for his assistance with statistical analysis and J Fekecs for her assistance with illustrations.
Funding: This research was supported by the Division of Intramural Research, National Human Genome Research Institute, National Institutes of Health, Department of Health and Human Services, USA
Footnotes
Competing interests: There are no competing interests.
Patient consent: Obtained.
Additional data are published online only at http://jmg.bmj.com/content/vol46/issue6
REFERENCES
- 1.Matsunaga E, Shiota K. Holoprosencephaly in human embryos: epidemiologic studies of 150 cases. Teratology. 1977;16:261–72. doi: 10.1002/tera.1420160304. [DOI] [PubMed] [Google Scholar]
- 2.Leoncini E, Baranello G, Orioli IM, Annerén G, Bakker M, Bianchi F, Bower C, Canfield MA, Castilla EE, Cocchi G, Correa A, De Vigan C, Doray B, Feldkamp ML, Gatt M, Irgens LM, Lowry RB, Maraschini A, Mc Donnell R, Morgan M, Mutchinick O, Poetzsch S, Riley M, Ritvanen A, Gnansia ER, Scarano G, Sipek A, Tenconi R, Mastroiacovo P. Frequency of holoprosencephaly in the International Clearinghouse Birth Defects Surveillance Systems: Searching for population variations. Birth Defects Res A Clin Mol Teratol. 2008;82:585–91. doi: 10.1002/bdra.20479. [DOI] [PubMed] [Google Scholar]
- 3.Muenke M, Beachy PA. Genetics of ventral forebrain development and holoprosencephaly. Curr Opin Genet Dev. 2000;10:262–9. doi: 10.1016/s0959-437x(00)00084-8. [DOI] [PubMed] [Google Scholar]
- 4.Cohen MM., Jr Holoprosencephaly: clinical, anatomic, and molecular dimensions. Birth Defects Res A Clin Mol Teratol. 2006;76:658–73. doi: 10.1002/bdra.20295. [DOI] [PubMed] [Google Scholar]
- 5.Barkovich AJ, Simon EM, Clegg NJ, Kinsman SL, Hahn JS. Analysis of the cerebral cortex in holoprosencephaly with attention to the sylvian fissures. AJNR Am J Neuroradiol. 2002;23:143–50. [PMC free article] [PubMed] [Google Scholar]
- 6.Barkovich AJ, Quint DJ. Middle interhemispheric fusion: an unusual variant of holoprosencephaly. AJNR Am J Neuroradiol. 1993;14:431–40. [PMC free article] [PubMed] [Google Scholar]
- 7.Hahn JS, Barkovich AJ, Stashinko EE, Kinsman SL, Delgado MR, Clegg NJ. Factor analysis of neuroanatomical and clinical characteristics of holoprosencephaly. Brain Dev. 2006;28:413–9. doi: 10.1016/j.braindev.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 8.Lazaro L, Dubourg C, Pasquier L, Le Duff F, Blayau M, Durou MR, de la Pintière AT, Aguilella C, David V, Odent S. Phenotypic and molecular variability of the holoprosencephalic spectrum. Am J Med Genet. 2004;129A:21–4. doi: 10.1002/ajmg.a.30110. [DOI] [PubMed] [Google Scholar]
- 9.Dubourg C, Bendavid C, Pasquier L, Henry C, Odent S, David V. Holoprosencephaly. Orphanet J Rare Dis. 2007;2:8. doi: 10.1186/1750-1172-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Plawner LL, Delgado MR, Miller VS, Levey EB, Kinsman SL, Barkovich AJ, Simon EM, Clegg NJ, Sweet VT, Stashinko EE, Hahn JS. Neuroanatomy of holoprosencephaly as predictor of function: beyond the face predicting the brain. Neurology. 2002;59:1058–66. doi: 10.1212/wnl.59.7.1058. [DOI] [PubMed] [Google Scholar]
- 11.Edison RJ, Muenke M. Mechanistic and epidemiologic considerations in the evaluation of adverse birth outcomes following gestational exposure to statins. Am J Med Genet. 2004;131:287–98. doi: 10.1002/ajmg.a.30386. [DOI] [PubMed] [Google Scholar]
- 12.Edison RJ, Muenke M. Central nervous system and limb anomalies in case reports of first-trimester statin exposure. N Engl J Med. 2004;350:1579–82. doi: 10.1056/NEJM200404083501524. [DOI] [PubMed] [Google Scholar]; N Engl J Med. 2005;352:2759. Erratum in: [Google Scholar]
- 13.Cohen MM., Jr Perspectives on holoprosencephaly: Part I. Epidemiology, genetics, and syndromology. Teratology. 1989;40:211–35. doi: 10.1002/tera.1420400304. [DOI] [PubMed] [Google Scholar]
- 14.Cohen MM, Jr, Sulik KK. Perspectives on holoprosencephaly: Part II. Central nervous system, craniofacial anatomy, syndrome commentary, diagnostic approach, and experimental studies. J Craniofac Genet Dev Biol. 1992;12:196–244. [PubMed] [Google Scholar]
- 15.Croen LA, Shaw GM, Lammer EJ. Holoprosencephaly: epidemiologic and clinical characteristics of a California population. Am J Med Genet. 1996;64:465–72. doi: 10.1002/(SICI)1096-8628(19960823)64:3<465::AID-AJMG4>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 16.Roessler E, Belloni E, Gaudenz K, Jay P, Berta P, Scherer SW, Tsui LC, Muenke M. Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat Genet. 1996;14:357–60. doi: 10.1038/ng1196-357. [DOI] [PubMed] [Google Scholar]
- 17.Brown SA, Warburton D, Brown LY, Yu CY, Roeder ER, Stengel-Rutkowski S, Hennekam RC, Muenke M. Holoprosencephaly due to mutations in ZIC2, a homologue of Drosophila odd-paired. Nat Genet. 1998;20:180–3. doi: 10.1038/2484. [DOI] [PubMed] [Google Scholar]
- 18.Wallis D, Muenke M. Mutations in holoprosencephaly. Hum Mutat. 2000;16:99–108. doi: 10.1002/1098-1004(200008)16:2<99::AID-HUMU2>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 19.Wallis DE, Roessler E, Hehr U, Nanni L, Wiltshire T, Richieri-Costa A, Gillessen-Kaesbach G, Zackai EH, Rommens J, Muenke M. Mutations in the homeodomain of the human SIX3 gene cause holoprosencephaly. Nat Genet. 1999;22:196–8. doi: 10.1038/9718. [DOI] [PubMed] [Google Scholar]
- 20.Gripp KW, Wotton D, Edwards MC, Roessler E, Ades L, Meinecke P, Richieri-Costa A, Zackai EH, Massagué J, Muenke M, Elledge SJ. Mutations in TGIF cause holoprosencephaly and link NODAL signalling to human neural axis determination. Nat Genet. 2000;25:205–8. doi: 10.1038/76074. [DOI] [PubMed] [Google Scholar]
- 21.de la Cruz JM, Bamford RN, Burdine RD, Roessler E, Barkovich AJ, Donnai D, Schier AF, Muenke M. A loss-of-function mutation in the CFC domain of TDGF1 is associated with human forebrain defects. Hum Genet. 2002;110:422–8. doi: 10.1007/s00439-002-0709-3. [DOI] [PubMed] [Google Scholar]
- 22.Ming JE, Kaupas ME, Roessler E, Brunner HG, Golabi M, Tekin M, Stratton RF, Sujansky E, Bale SJ, Muenke M. Mutations in PATCHED-1, the receptor for SONIC HEDGEHOG, are associated with holoprosencephaly. Hum Genet. 2002;110:297–301. doi: 10.1007/s00439-002-0695-5. [DOI] [PubMed] [Google Scholar]
- 23.Roessler E, Du YZ, Mullor JL, Casas E, Allen WP, Gillessen-Kaesbach G, Roeder ER, Ming JE, Ruiz i Altaba A, Muenke M. Loss-of-function mutations in the human GLI2 gene are associated with pituitary anomalies and holoprosencephaly-like features. Proc Natl Acad Sci USA. 2003;100:13424–9. doi: 10.1073/pnas.2235734100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roessler E, Ouspenskaia MV, Karkera JD, Vélez JI, Kantipong A, Lacbawan F, Bowers P, Belmont JW, Towbin JA, Goldmuntz E, Feldman B, Muenke M. Reduced NODAL signaling strength via mutation of several pathway members including FOXH1 is linked to human heart defects and holoprosencephaly. Am J Hum Genet. 2008;83:18–29. doi: 10.1016/j.ajhg.2008.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ming JE, Muenke M. Multiple hits during early embryonic development: digenic diseases and holoprosencephaly. Am J Hum Genet. 2002;71:1017–32. doi: 10.1086/344412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gestri G, Carl M, Appolloni I, Wilson SW, Barsacchi G, Andreazzoli M. Six3 functions in anterior neural plate specification by promoting cell proliferation and inhibiting Bmp4 expression. Development. 2005;132:2401–13. doi: 10.1242/dev.01814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kobayashi M, Toyama R, Takeda H, Dawid IB, Kawakami K. Overexpression of the forebrain-specific homeobox gene six3 induces rostral forebrain enlargement in zebrafish. Development. 1998;125:2973–82. doi: 10.1242/dev.125.15.2973. [DOI] [PubMed] [Google Scholar]
- 28.Oliver G, Mailhos A, Wehr R, Copeland NG, Jenkins NA, Gruss P. Six3, a murine homologue of the sine oculis gene, demarcates the most anterior border of the developing neural plate and is expressed during eye development. Development. 1995;121:4045–55. doi: 10.1242/dev.121.12.4045. [DOI] [PubMed] [Google Scholar]
- 29.Kawakami K, Ohto H, Takizawa T, Saito T. Identification and expression of six family genes in mouse retina. FEBS Lett. 1996;393:259–63. doi: 10.1016/0014-5793(96)00899-x. [DOI] [PubMed] [Google Scholar]
- 30.Granadino B, Gallardo ME, Lopez-Rios J, Sanz R, Ramos C, Ayuso C, Bovolenta P, Rodriguez de Cordoba S. Genomic cloning, structure, expression pattern, and chromosomal location of the human SIX3 gene. Genomics. 1999;55:100–5. doi: 10.1006/geno.1998.5611. [DOI] [PubMed] [Google Scholar]
- 31.Inbal A, Kim SH, Shin J, Solnica-Krezel L. Six3 represses nodal activity to establish early brain asymmetry in zebrafish. Neuron. 2007;55:407–15. doi: 10.1016/j.neuron.2007.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kobayashi M, Nishikawa K, Suzuki T, Yamamoto M. The homeobox protein Six3 interacts with the Groucho corepressor and acts as a transcriptional repressor in eye and forebrain formation. Dev Biol. 2001;232:315–26. doi: 10.1006/dbio.2001.0185. [DOI] [PubMed] [Google Scholar]
- 33.Lagutin OV, Zhu CC, Kobayashi D, Topczewski J, Shimamura K, Puelles L, Russell HR, McKinnon PJ, Solnica-Krezel L, Oliver G. Six3 repression of Wnt signaling in the anterior neuroectoderm is essential for vertebrate forebrain development. Genes Dev. 2003;17:368–79. doi: 10.1101/gad.1059403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Del Bene F, Tessmar-Raible K, Wittbrodt J. Direct interaction of geminin and Six3 in eye development. Nature. 2004;427:745–9. doi: 10.1038/nature02292. [DOI] [PubMed] [Google Scholar]
- 35.Liu W, Lagutin OV, Mende M, Streit A, Oliver G. Six3 activation of Pax6 expression is essential for mammalian lens induction and specification. EMBO J. 2006;25:5383–95. doi: 10.1038/sj.emboj.7601398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Geng X, Speirs C, Lagutin O, Inbal A, Liu W, Solnica-Krezel L, Jeong Y, Epstein DJ, Oliver G. Haploinsufficiency of Six3 fails to activate Sonic hedgehog expression in the ventral forebrain and causes holoprosencephaly. Dev Cell. 2008;15:236–47. doi: 10.1016/j.devcel.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jeong Y, Leskow FC, El-Jaick K, Roessler E, Muenke M, Yocum A, Dubourg C, Li X, Geng X, Oliver G, Epstein DJ. Regulation of a remote Shh forebrain enhancer by the Six3 homeoprotein. Nat Genet. 2008;40:1348–53. doi: 10.1038/ng.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schell U, Wienberg J, Köhler A, Bray-Ward P, Ward DE, Wilson WG, Allen WP, Lebel RR, Sawyer JR, Campbell PL, Aughton DJ, Punnett HH, Lammer EJ, Kao FT, Ward DC, Muenke M. Molecular characterization of breakpoints in patients with holoprosencephaly and definition of the HPE2 critical region 2p21. Hum Mol Genet. 1996;5:223–9. doi: 10.1093/hmg/5.2.223. [DOI] [PubMed] [Google Scholar]
- 39.Nanni L, Croen LA, Lammer EJ, Muenke M. Holoprosencephaly: molecular study of a California population. Am J Med Genet. 2000;90:315–19. doi: 10.1002/(sici)1096-8628(20000214)90:4<315::aid-ajmg10>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 40.Bendavid C, Dubourg C, Gicquel I, Pasquier L, Saugier-Veber P, Durou MR, Jaillard S, Frebourg T, Haddad BR, Henry C, Odent S, David V. Molecular evaluation of foetuses with holoprosencephaly shows high incidence of microdeletions in the HPE genes. Hum Genet. 2006;119:1–8. doi: 10.1007/s00439-005-0097-6. [DOI] [PubMed] [Google Scholar]
- 41.Bendavid C, Haddad BR, Griffin A, Huizing M, Dubourg C, Gicquel I, Cavalli LR, Pasquier L, Shanske AL, Long R, Ouspenskaia M, Odent S, Lacbawan F, David V, Muenke M. Multicolour FISH and quantitative PCR can detect submicroscopic deletions in holoprosencephaly patients with a normal karyotype. J Med Genet. 2006;43:496–500. doi: 10.1136/jmg.2005.037176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Domené S, Roessler E, El-Jaick K, Boorech J, Vélez JI, Bale S, Lacbawan F, Muenke M, Feldman B. Mutations in the human SIX3 gene in holoprosencephaly are loss-of-function. Hum Mol Genet. 2008;17:3919–28. doi: 10.1093/hmg/ddn294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dubourg C, Lazaro L, Pasquier L, Bendavid C, Blayau M, Le Duff F, Durou MR, Odent S, David V. Molecular screening of SHH, ZIC2, SIX3, and TGIF genes in patients with features of holoprosencephaly spectrum: Mutation review and genotype-phenotype correlations. Hum Mutat. 2004;24:43–51. doi: 10.1002/humu.20056. [DOI] [PubMed] [Google Scholar]
- 44.Pasquier L, Dubourg C, Blayau M, Lazaro L, Le Marec B, David V, Odent S. A new mutation in the six-domain of SIX3 gene causes holoprosencephaly. Eur J Hum Genet. 2000;8:797–800. doi: 10.1038/sj.ejhg.5200540. [DOI] [PubMed] [Google Scholar]
- 45.Pasquier L, Dubourg C, Gonzales M, Lazaro L, David V, Odent S, Encha-Razavi F. First occurrence of aprosencephaly/atelencephaly and holoprosencephaly in a family with a SIX3 gene mutation and phenotype/genotype correlation in our series of SIX3 mutations. J Med Genet. 2005;42:e4. doi: 10.1136/jmg.2004.023416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.El-Jaick KB, Fonseca RF, Moreira MA, Ribeiro MG, Bolognese AM, Dias SO, Pereira ET, Castilla EE, Orioli IM. Single median maxillary central incisor: new data and mutation review. Birth Defects Res A Clin Mol Teratol. 2007;79:573–80. doi: 10.1002/bdra.20380. [DOI] [PubMed] [Google Scholar]
- 47.Paulussen A, Tserpelis D, Spierts S, Smeets H, Herbergs J. Holoprosencephaly mutations in the Dutch population. Poster presented at the European Society of Human Genetics; Barcelona, Spain. 31 May to 3 June, 2008. [Google Scholar]
- 48.Ribeiro LA, El-Jaick KB, Muenke M, Richieri-Costa A. SIX3 mutations with holoprosencephaly. Am J Med Genet. 2006;140:2577–83. doi: 10.1002/ajmg.a.31377. [DOI] [PubMed] [Google Scholar]
- 49.Wilson WG, Shanks DE, Sudduth KW, Couper KA, McIlhenny J. Holoprosencephaly and interstitial deletion of 2(p2101p2109) Am J Med Genet. 1989;34:252–4. doi: 10.1002/ajmg.1320340226. [DOI] [PubMed] [Google Scholar]
- 50.Sawyer JR, Jones E, Hawks FF, Quirk JG, Jr, Cunniff C. Duplication and deletion of chromosome band 2(p21p22) resulting from a familial interstitial insertion (2;11)(p21;p15) Am J Med Genet. 1994;49:422–7. doi: 10.1002/ajmg.1320490414. [DOI] [PubMed] [Google Scholar]
- 51.Zwetsloot CP, Brouwer OF, Maaswinkel-Mooy PD. Holoprosencephaly: variation of expression in face and brain in three sibs. J Med Genet. 1989;26:274–6. doi: 10.1136/jmg.26.4.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Laflamme C, Filion C, Labelle Y. Functional characterization of SIX3 homeodomain mutations in holoprosencephaly: interaction with the nuclear receptor NR4A3/NOR1. Hum Mutat. 2004;24:502–8. doi: 10.1002/humu.20102. [DOI] [PubMed] [Google Scholar]
- 53.Agresti A. An introduction to categorical data analysis. John Wiley and Sons; New York: 1996. [Google Scholar]