

Abstract
Genetic changes in the SMARCB1 tumor suppressor gene have recently been reported in tumors and blood from families with schwannomatosis. Exon scanning of all nine SMARCB1 exons in genomic DNA from our cohort of families meeting the criteria for ‘definite’ or ‘presumptive’ schwannomatosis previously revealed constitutional alterations in 13 of 19 families (68%). Screening of four new familial schwannomatosis probands identified one additional constitutional alteration. We confirmed the presence of mRNA transcripts for two missense alterations, four mutations of conserved splice motifs and two additional mutations, in less conserved sequences, which also affect splicing. Furthermore, we found that transcripts for a rare 3′-untranslated region (c.*82C > T) alteration shared by four unrelated families did not produce splice variants but did show unequal allelic expression, suggesting that the alteration is either causative itself or linked to an unidentified causative mutation. Overexpression studies in cells lacking SMARCB1 suggest that mutant SMARCB1 proteins, like wild-type SMARCB1 protein, retain the ability to suppress cyclin D1 activity. These data, together with the expression of SMARCB1 protein in a proportion of cells from schwannomatosis-related schwannomas, suggest that these tumors develop through a mechanism that is distinct from that of rhabdoid tumors in which SMARCB1 protein is completely absent in tumor cells.
INTRODUCTION
Schwannomatosis (MIM#162091) is a third major form of neurofibromatosis, that is clinically and genetically distinct from NF1 (MIM#162200) and NF2 (MIM#101000) (1). The most common clinical symptom of schwannomatosis is intractable pain, although the mechanism by which this occurs is not well understood. Tumors from familial schwannomatosis patients frequently harbor somatic truncating mutations at the NF2 (MIM#607379) gene on the long arm of chromosome 22 and loss of the wild-type NF2 allele. However, they do not carry germline NF2 mutations (2). Recently, a number of constitutional alterations have been reported in familial and some sporadic schwannomatosis patients in the SMARCB1 (MIM#601607) gene (3–6), which is situated 5.8 Mb proximal to NF2.
Loss of SMARCB1 has been linked previously to development of rhabdoid tumors (RTPS1; MIM#609322) (7). Rhabdoid tumors are highly malignant, appear in the first few years after birth, and are almost always lethal. Several RTPS1 families have been described (8) to include family members who are constitutional carriers of a SMARCB1 mutation, but who never develop schwannomas. Recently, a multigenerational family was described with multiple members affected by either malignant rhabdoid tumors or by schwannomatosis, all of whom share a common germline SMARCB1 exon 6 duplication mutation (9). A second family affected by RTPS1 and schwannoma has also been described with a c.472C > T SMARCB1 mutation (10).
The existence of adult mutation carriers in RTPS1 families has led to the hypothesis that the risk of rhabdoid tumor development from these mutations is time dependent (6,11), in which case the development of schwannomas later in life becomes feasible. However, the majority of RTPS1 and schwannomatosis families remain distinct, making it more likely that the type or location of the mutation determines the resulting disease status.
Almost all of the constitutional SMARCB1 alterations found in familial schwannomatosis patients are predicted to be non-truncating. In contrast, the mutations found in RTPS1 are mainly truncating mutations and large deletions, which lead to a complete loss of SMARCB1 protein. This difference in mutation type may underlie the difference in phenotype presented by these two conditions.
To address this issue, we have carried out analysis of SMARCB1 transcripts from 14 schwannomatosis families, shown to harbor constitutional alterations (6), in order to verify expression of the non-truncated products predicted from each mutation.
RESULTS
Immunohistochemistry of schwannomas reveals mosaic pattern of SMARCB1 expression
Immunostaining for schwannomas from families 1, 5 and 10 which harbor the c.*82C > T mutation and family 11 harboring the c.364G > T mutation have been published previously (12) and show a mosaic pattern of staining for SMARCB1 protein, consistent with loss of expression in a subset of tumor cells. Tumor sections from families 9 and 19 were subsequently analyzed by immunostaining for SMARCB1 and also revealed a mosaic pattern of mixed positive and negative nuclei in all four schwannomas from a member of family 19 (c.795 + 1G > T) and a single schwannoma from family 9 (c.158G > T). Representative staining is shown for a tumor from family 19 in Figure 1. No tumors were available for testing from other families in this study.
Figure 1.
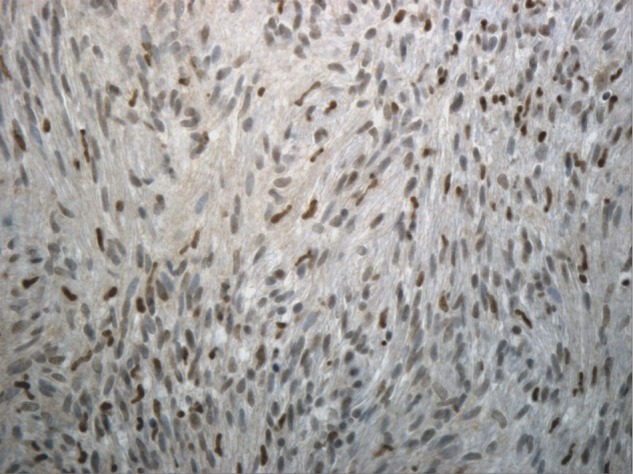
Immunostaining for SMARCB1 shows a mosaic pattern of SMARCB1 expression in a schwannoma from family 19 with a mixture of positive (brown) and negative (blue) nuclei in cells.
cDNA analysis of familial schwannomatosis samples
To confirm the predicted effects of constitutional mutations identified previously (6), we analyzed cDNA derived from mRNA of lymphoblastoid cell lines carrying each of the 10 germline alterations found in 14 families. The results are summarized in Table 1. Representative chromatograms of the sequencing results for each of the seven mutant transcripts predicted to produce a viable protein and the three predicted to undergo nonsense-mediated decay are shown in Figure 2A–J.
Table 1.
Germline mutations found in familial schwannomatosis kindreds and the effects observed in mRNA (family 23 has both schwannomatosis and RTPS1)
| Family ID | Exon | Germline mutation | Mutant amplicons identified |
|---|---|---|---|
| 4 | 1 | c.41C > A | r.41C > A |
| 9 | 2 | c.158G > T | r.158G > U |
| PA-1 | 2 | c.158G > T | r.158G > U |
| E | 3 | c.362 + 1G > A | r.318_362del |
| 11 | 4 | c.364G > T | r.363_500del |
| 23 | 4 | c.472C > T | r.363_500del, r.390_500del, r.796_986del |
| PA-3 | 4 | c.500G > A | r.390_500del |
| V | 4 | c.500 + 5G > T | r.390_500del |
| P/Qu | 6 | c.629-5T > G | r.500ins501-145_501-1 |
| 19 | 6 | c.795 + 1G > T | r.795_796ins795 + 1_795 + 45ins, r.628_795del |
| 1 | 9 | c.*82C > T | r.*82C > U |
| 3 | 9 | c.*82C > T | r.*82C > U |
| 5 | 9 | c.*82C > T | r.*82C > U |
| 10 | 9 | c.*82C > T | r.*82C > U |
Figure 2.

Chromatograms showing SMARCB1 mutant transcripts identified in the study. (A) Exon 1 missense mutation; (B) exon 2 missense mutation found in families 9 and PA1; (C) exon 3 splice mutation that removes the first 45 bp of exon 3; (D) in-frame deletion of the entire exon 4 sequence found in families 11 and 23;(E) exon 4 splice mutation leading to the deletion of the last 111 bp of exon 4 in families PA-3, V and 23; (F) exon 6 splice mutation causing inclusion of the first 45 bp of intron 6; (G) family 19 transcript deleting exon 6, predicted to cause nonsense-mediated decay; (H) family 23 deletion of exon 7, predicted to cause nonsense-mediated decay; (I) family P/Qu mutant transcript including 145 bp of intron 5, predicted to cause nonsense-mediated decay; (J) 3′UTR change found in families 1, 3, 5 and 10.
Missense mutations
Each of the missense alterations, c.41C > A (p.Pro14His) and c.158G > T (p. Arg53Leu), found in family 4, and families 9 and PA-3, respectively, was detected in the cDNA transcript generated from its corresponding lymphoblast cell line (Fig. 2A and B), indicating that the mutant allele for both of these alterations is expressed at the mRNA level.
Splicing alterations
Four mutations were detected in conserved splice sites in four kindreds. An intron 3 mutation (c.362 + 1G > A) amplified a faint band containing a splice variant that results in an in-frame deletion of the last 45 bp of exon 3 (Fig. 2C). An exon 4 alteration, c.364G > T, caused skipping of exon 4, without changing the reading frame (Fig. 2D). Another mutation at the ultimate base of exon 4, c.500G > A, produced altered splicing that resulted in the in-frame loss of the last 111 bp of the exon (c.390_500del, Fig. 2E). The intron 6 change, c.795 + 1G > T produced two alternative splice variants. The first involved the in-frame inclusion of the first 45 bp of intron 6 (Fig. 2F) while the second, which was much less abundant, involved skipping of exon 6, with the resulting frame-shift predicted to lead to a premature stop codon and mRNA decay (Fig. 2G).
Sequence analysis of lymphocyte DNA from family 23 [family 07-367 in Eaton et al., (10)] identified the nonsense mutation c.472C > T in exon 4. We isolated three alternately spliced cDNA amplicons derived from the corresponding lymphoblast cell line RNA for this family. First, amplification of exons 3–6 detected two different deletions: complete exon 4 loss (c.363_500del) and partial exon 4 deletion (c.390_500del). Amplicons for exons 5–9 then detected a deletion of the whole of exon 7 (Fig. 1H). The exon 4 changes in the transcript were the same as those found in other families and have the potential to produce non-truncated protein. However, the presence of the exon 7 deletion would lead to a premature stop codon and destabilize these mutant transcripts; therefore, they are predicted to undergo nonsense-mediated decay.
Two additional potential splice site alterations were identified further from the consensus splice sequence the intron 4 change, c.500 + 5G > T, and the intron 5 change, c.629-5T > G. Interestingly, the former leads to the same splicing alteration as the c.500G > A mutation, five bases away (Fig. 2E). The intron 5 alteration, c.629-5T > G, in family P/Qu, seems to disrupt the exon 6 splice acceptor sequence, leading to the insertion of 145 bp of the 3′ end of intron 5 (Fig. 2I), using a cryptic splice acceptor, ag/TTT. This alteration results in a premature stop codon within the intronic sequence and is predicted to lead to mRNA decay.
Variants of uncertain significance
Four unrelated families contained the same alteration, c.*82C > T, within their 3′UTR (Fig. 2J). This alteration was not found in an unaffected panel of 100 alleles and is not recorded in the dbSNP database (6,13). None of the four families produced splice variants in the cDNA analysis screen, but the alteration was observed in the transcript.
Allelic expression analysis of the c.*82C > T alteration
Allelic expression analysis has previously been used to quantify under- or overexpression of mutant NF2 transcripts from NF2 patient-derived lymphoblast cell RNAs (14). We used this technique to measure expression levels of the c.*82C > T alteration.
The full-length SMARCB1 transcripts were amplified, sequenced and the level of expression of the mutant ‘T’ allele in cDNA was calculated, relative to the genomic sequence, using the Mutation Surveyor DNA analysis program (SoftGenetics, State College, PA, USA). The results revealed unequal allelic expression, with a decreased expression of the mutant allele by ∼27% compared with the wild-type. To ensure that differences in expression for the c.*82C > T change were not due to differential expression in lymphoblastoid cells, two known SNPs (rs34399789 and rs2229354), identified on an unaffected allele in two families, were tested for unequal expression in unaffected family members. These polymorphisms both showed equal expression in full-length SMARCB1 cDNA. The results suggest that the c.*82C > T alteration is associated with reduced expression and is either causative or linked to a causative mutation.
The SMARCB1 mutant 3′UTR alters mRNA stability
To substantiate the effect of the mutant SMARCB1 3′UTR on RNA expression levels, we transfected HEK293T cells with a pGL3-promoter luciferase vector containing a wild-type SMARCB1 3′UTR, or a c.*82C > T mutant 3′UTR. Forty-eight hours after transfection, the level of luciferase expression was significantly reduced under the regulation of the c.*82C > T mutant 3′UTR compared with the wild-type 3′UTR (Fig. 3A). To distinguish between the possibilities of the c.*82C > T mutation affecting either translational efficiency or stability of the mRNA, we performed qRT–PCR on RNA extracted from HEK293T cells transfected with luciferase constructs bearing the wild-type or the c.*82C > T 3′UTRs, at 48 and 96 h post-transfection. At 48 and 96 h, the c.*82C > T mutant mRNA levels were reduced by ∼20 and ∼45%, respectively, compared with the wild-type (Fig. 3B). Together, these results suggest that the c.*82C > T mutant 3′UTR leads to a reduced expression of the SMARCB1 transcript, possibly by destabilizing the mRNA.
Figure 3.

The SMARCB1 mutant 3′UTR alters mRNA stability. Relative mRNA expression levels of wild-type and mutant SMARCB1 3′untranslated regions in HEK293T cells. (A) Luciferase levels indicate the relative expression under the control of wild-type and c.*82C > T mutated SMARCB1 3′UTRs, normalized to transfection efficiency by GFP fluorescence *P < 0.01. (B) qRT–PCR of wild-type and mutant SMARCB1 3′UTR expression levels at 48 and 96 h post-transfection normalized to both GFP and GAPDH *P < 0.015; **P < 0.001.
Cyclin D1 reporter activity is appropriately suppressed by mutant SMARCB1
To begin to elucidate the mechanism by which mutations in SMARCB1 affect its normal function in cells, we created expression constructs for three of the mutant SMARCB1 transcripts found in our cohort, predicted to result in altered protein sequence. The exon 1 missense, c.41C > A, and exon 2 missense, c.158G > T, mutations were created by site-directed mutagenesis. The splice mutant lacking exon 4 was isolated from full-length cDNA derived from mRNA from a lymphoblastoid cell line with the c.364G > T mutation. These expression constructs were used to determine whether mutant SMARCB1 proteins are able to suppress elevated cyclin D1 expression, as has been shown previously for wild-type SMARCB1 (15). A luciferase reporter construct containing the cyclin D1 promoter region −1745 to +35 was used to transfect MON cells, which lack endogenous SMARCB1 and have elevated levels of cyclin D1 (15). This construct was co-transfected with either a wild-type SMARCB1 construct or a mutant transcript construct. When wild-type SMARCB1 was reintroduced into MON cells, luciferase expression was suppressed by ∼60% (Fig. 4) similar to previous reports (15). Similarly, when MON cells were transfected with mutant SMARCB1 constructs, luciferase expression was suppressed by 53% (exon 1 missense), 74% (exon 2 missense) or 65% (exon 4 deletion). The results show that these schwannomatosis-related mutant SMARCB1 proteins retain the ability to repress cyclin D1 transcription.
Figure 4.

Cyclin D1 activity is appropriately suppressed by both wild-type and mutant SMARCB1 proteins. Introduction of both wild-type and mutant SMARCB1 transcripts (c.41C > A missense, c.158G > T missense or c.364G > T splice mutant) into MON cells which lack endogenous SMARCB1 leads to suppression of luciferase reporter activity under the control of the cyclin D1 promoter. Normalized to transfection efficiency by GFP fluorescence *P < 0.05.
DISCUSSION
Exon scanning analysis of SMARCB1 in our cohort of familial schwannomatosis patients has now identified a total of 14/23 (61%) probands with a germline point mutation. The majority of these mutations are predicted to be non-truncating. This contrasts with the mutational spectrum of RTPS1 in which mutations are predicted to be truncating (7,16). We analyzed cDNA from this select group of familial schwannomatosis patients to confirm the predicted effects of constitutional mutations identified during initial SMARCB1 exon scanning and found that almost all of the constitutional alterations in the SMARCB1 gene produced a mutant transcript that is likely to produce a non-truncated protein.
Families P/Qu and 23 both produced mutant transcripts that are likely to be degraded by nonsense-mediated decay. Family 23 has a family history of both schwannomatosis and RTPS1 and, in addition to the truncating mutation, also has a somatic deletion of the wild-type SMARCB1 allele in a tumor (10). The c.472C > T nonsense mutation is known to predispose to RTPS1, but it is unclear why some members of the same family developed schwannomas rather than rhabdoid tumors. It is possible that a modifier gene is involved. It is also unclear how the mutation, which occurs in exon 4, causes a deletion of exon 7. It is possible that a second, unidentified, mutation exists within intron 6, leading to skipping of exon 7.
The only mutant transcript detected in family P/Qu contained an insertion of the last 145 bp of intron 5, which leads to a stop codon 26 codons into the insertion. This is predicted to lead to mRNA decay and no expression of the mutant copy of SMARCB1. This family did not show loss of heterozygosity (LOH) by microsatellite marker analysis at the SMARCB1 locus, suggesting that the wild-type copy of SMARCB1 is still present in tumors, although it remains possible that other mechanisms may affect this remaining allele. LOH was, however, seen at the NF2 locus—downstream of SMARCB1—in a tumor from family P/Qu, supporting the theory that co-mutation of these two genes is involved in schwannoma formation (4–6). Unfortunately, no tumor material was available for immunohistochemical analysis.
Tumors from schwannomatosis kindreds frequently carry SMARCB1 alterations in conjunction with loss of the wild-type allele. The combination of a constitutional SMARCB1 mutation with a somatically acquired truncating mutation in the NF2 gene and loss of the wild-type copies of both genes indicates cross-talk between tumorigenic pathways and a complex mechanism of schwannoma formation in this disorder. Indeed, a multi-hit hypothesis has been suggested (3–6).
The alteration, c.*82C > T, found in the 3′ UTR of four unrelated kindreds (13) in our cohort, showed no splice variants, but was detected by sequencing of the full-length transcript from lymphoblast cell lines, suggesting that the mutant mRNA is expressed. Allelic expression analysis, and qRT–PCR on cells transfected with wild-type and mutant 3′UTRs, showed unequal expression with decreased levels of the mutant allele in comparison with the wild-type. Families 3 and 5, for which tumor DNA was available, showed LOH for both the SMARCB1 and NF2 loci and showed somatic mutation of the remaining NF2 allele (6). These results, together with the frequency of this alteration in unrelated families, support a pathogenic status for the c.*82C > T mutation and implicate it in the occurrence of schwannomatosis disease.
SMARCB1 regulation of cyclin D1
Loss of SMARCB1 in RTPS1 leads to upregulation of cyclin D1 and progression into the cell cycle. A cyclin D1 repression assay showed that schwannomatosis-related missense and splice mutants are capable of suppressing cyclin D1 activity in a similar way to that shown for the wild-type SMARCB1 protein. This finding suggests that the downstream effects of SMARCB1 alterations are different in schwannomatosis compared with RTPS1, with cyclin D1 and related cell cycle processes being affected primarily in the RTPS1 tumors.
Immunohistochemistry for SMARCB1 revealed a mosaic pattern of mixed positive and negative nuclei in all tumor specimens. In a previous report, this finding was interpreted as loss of protein expression in a subset of tumor cells resulting in a mosaic of null and haploinsufficient cells (12). This could be as a result of transient or unstable expression. However, further work would be required to determine this. It is also unclear why the exon 2 mutation, which appears to produce a more highly expressed transcript, also leads to a mosaic pattern of protein production. It could be that the mutant DNA is transcribed normally, while the mutant mRNA is mis-folded, causing a reduction in translation efficiency.
This data, in conjunction with the different spectrum of mutations in comparison with that seen in RTPS1, suggests that in familial schwannomatosis kindreds, almost all constitutional alterations in the SMARCB1 gene are capable of producing mutant, but viable transcripts, that yield SMARCB1 protein with altered levels of functionality in schwannoma tumor cells. Further work is required to determine the precise mechanism by which mutant SMARCB1 protein can promote the pathogenesis of schwannomas.
MATERIALS AND METHODS
Patient material
We studied 13 of 19 carefully characterized schwannomatosis kindreds described previously (6) and four additional familial probands. Lymphoblastoid cell lines were established from peripheral blood samples as described previously (17). High-molecular-weight DNA was extracted from peripheral blood leukocytes, cultured lymphoblasts, frozen pulverized tumor, cultured tumor and normal tissues obtained at autopsy using a PureGene DNA isolation kit (Gentra Systems, Minneapolis, MN, USA). This study was approved by the Institutional Review Board of Partners HealthCare and informed consent was obtained from the patients participating in the study. For patients who had died, an autopsy permit was used as consent.
Multiplex ligation-dependent probe amplification
Multiplex ligation-dependent probe amplification was carried out as described by Boyd et al. (6), using a SALSA multiplex ligation-dependent probe amplification P258 SMARCB1 kit (MRC-Holland, Amsterdam, the Netherlands). Briefly, 20–500 ng DNA were used for hybridization, ligation and amplification of the SMARCB1 exon probes according to the manufacturer's instructions. The amplification products were analyzed using an ABI 3730 DNA Analyzer, with Biomek FX robotics and with GeneScan 500 LIZ (Applied Biosystems, Foster City, CA, USA) as the internal size standard. Relative probe signals were calculated by dividing each measured peak by the sum of all peak areas for that sample. DNA from four unaffected individuals was used for control samples.
cDNA analysis
mRNA was extracted from frozen cell pellets of established lymphoblastoid cell lines for each kindred, using the PolyATract mRNA extraction kit (Promega, Madison, WI, USA). Poly(A)+ mRNA was reverse transcribed to cDNA with oligo(d)T primers, using the Superscript III cDNA first strand synthesis kit (Invitrogen, Carlesbad, CA, USA). Full-length SMARCB1 and three overlapping fragments of the SMARCB1 transcript were PCR amplified from the cDNAs using nested primers. For the full-length transcript, the primers 5′-CAGCCCTCCTGATCCCT-3′ and 5′-CCCAATCTTCTGAGATGCTC-3′ were used. The reverse primer 5′-ACAAATGGAATGTGTGCCGG-3′ was used when the 3′UTR SNPs were amplified. Exons 1 through 4 were amplified with primers 5′-CAGCCCTCCTGATCCCT-3′ and 5′-TCACAGCTGGGTCATGGTC-3′, exons 3–6 were amplified by 5′-CACGGATACACGACTCTAGC-3′ and 5′-CACTCAAACTGGTCCACC-3′ and for exons 5–9, 5′-CCATGCTCCACAACCATC-3′ and 5′-CCCAATCTTCTGAGATGCTC-3′were used. PCR products were electrophoresed on 2% agarose gels, with a normal control. Aberrantly sized fragments were excised and analyzed by direct sequencing on an ABI Prism 3730 DNA analyzer.
Quantitative analysis of the c.*82C > T mutation (previously denoted c.1240C > T) was carried out using the Mutation Surveyor program v3.20 (Softgenetics LLC, State College, PA, USA) by comparison of relative levels of C and T alleles in cDNA with reference to levels in genomic DNA.
Construction of wild-type and mutant SMARCB1 expression vectors
Mutagenic primers were designed to introduce the c.41C > A and c.158G > T point mutations identified in our cohort into the wild-type human SMARCB1 sequence obtained from Origene (Rockville, MD, USA). Site-directed mutagenesis was carried out using the Quick-change site-directed mutagenesis kits (Stratagene, Carlesbad, CA, USA). Mutagenic primers for c.41C > A were GACCTTCGGGCAGAAGCACGTGAAGTTCCAGCTGG and CCAGCTGGAACTTCACGTGCTTCTGCCCGAAGGTC. Mutagenic primers used for c.158G > T were CCCTCACTCTGGAGGCTACTAGCCACTGTGGAAG and CTTCCACAGTGGCTAGTAGCCTCCAGAGTGAGGG.
The splice mutant lacking exon 4 was obtained by PCR amplification of full-length cDNA generated from mRNA of a lymphoblastoid cell line from family 11, using a forward primer containing an NheI restriction site, GCATGCTAGCATGATGATGATGGCGCTGAGC, and a reverse primer containing a HindIII restriction site, TTTAAGCTTCCAGGCCGGGGCCGTGTT. Each mutant transcript was sub-cloned into a pcDNA plasmid, containing a C-terminal GFP tag.
For the cyclin D1 repression experiment, the cyclin D1 promoter region −1745 to +35 was sub-cloned into the pGL3-basic plasmid, which contains a luciferase reporter gene (Promega, Madison, WI, USA).
For the 3′UTR experiment, site-directed mutagenesis was used to generate the c.*82C > T mutant, with primers TGGCAAGGACAGAGGTGAGGGGACAGCCCA and TGGGCTGTCCCCTCACCTCTGTCCTTGCCA. cDNAs representing the wild-type and c.*82C > T mutant 3′UTRs were each sub-cloned into the pGL3-promoter vector, downstream of the luciferase coding region.
Cell lines
HEK293T cells were purchased from ATCC (Manassas, VA , USA). MON tumor cells were obtained from the laboratory of Dr Olivier Delattre.
Luciferase reporter assays
For experiments investigating SMARCB1 3′UTR regulation, using a luciferase reporter, HEK293T cells were co-transfected with the pGL3-promoter vector construct with either a wild-type or mutant SMARCB1 3′UTR and an eGFP vector, using Lipofectamine transfection reagent (Invitrogen, Carlesbad, CA, USA). Cells were harvested after 48 h and luciferase activity was measured using One-glow luciferase reagent (Promega, Madison, WI, USA).
For experiments investigating the regulation of cyclin D1, the wild-type SMARCB1 transcript and three mutant transcripts [containing the exon 1 missense mutation (c.41C > A), the exon 2 missense mutation (c.158G > T) or the deletion of exon 4], were subcloned into the pcDNA vector with a C-terminal GFP tag. The cyclin D1 luciferase vector was transfected into MON cells which lack endogenous SMARCB1 along with each of the SMARCB1 constructs, or an empty GFP control vector. Cells were harvested after 48 h and luciferase activity was measured using One-glow luciferase reagent (Promega, Madison, WI, USA). Luciferase levels were normalized to transfection efficiency using GFP fluorescence.
qRT–PCR analysis of luciferase-3′UTR reporters
Total RNA was extracted from transfected cells using the RNeasy kit (Qiagen, Valencia, CA, USA) and treated with DNase (Ambion, Foster City, CA, USA). First-strand cDNA synthesis was performed using random hexamers (GE Healthcare, Piscataway, NJ, USA). Subsequently, mRNA expression levels of luciferase-3′ UTR reporters were assessed using 1 μl of the appropriate cDNA for real-time qRT–PCR using FastStart Universal SyberGreen and a LightCycler 480 machine (Roche, Indianapolis, IN, USA). The forward oligonucleotide primer (5′-GGTCTTACCGGAAAACTCGAC-3′) corresponds to a sequence at the 3′ end of the luciferase cDNA; the reverse primer (5′-CTCTGTCCTTGCCAGAAGATG-3′) is within the 3′UTR of SMARCB1. The results were analyzed using the comparative Ct method (ΔΔCt) and normalized to GAPDH and eGFP expression levels to ensure equal loading and transfection efficiencies.
Immunohistochemistry
Formalin-fixed, paraffin-embedded tissue sections from four schwannomas resected from member of family 19 (c.795 + 1G > T) and a single schwannoma from family 9 were immunostained using a commercial SMARCB1 antibody (BD Transduction Laboratories, Franklin Lakes, NJ, USA) along with appropriate positive (normal cortex) and negative controls (RTPS1). Antigen retrieval was achieved by microwaving in a Borg Decloaker RTU for 45 min (primary antibody concentration 1:50).
FUNDING
This work was supported in part by NINDS grant NS24279 and by the Harvard Medical School Center for Neurofibromatosis and Allied Disorders. J.A.W. is supported by the DOD (W81XWH-09-1-0487). M.J.S. is currently supported by the Children's Tumor Foundation and the Association for International Cancer Research.
ACKNOWLEDGEMENTS
We would like to thank Dr Olivier Delattre for the generous gift of MON cells and Dr Ganjam Kalpana for reagents and technical assistance with the luciferase assays. We would also like to thank Robert Maher for help with formatting of the figures.
Conflict of Interest statement. None declared.
REFERENCES
- 1.MacCollin M., Chiocca E.A., Evans D.G., Friedman J.M., Horvitz R., Jaramillo D., Lev M., Mautner V.F., Niimura M., Plotkin S.R., et al. Diagnostic criteria for schwannomatosis. Neurology. 2005;64:1838–1845. doi: 10.1212/01.WNL.0000163982.78900.AD. [DOI] [PubMed] [Google Scholar]
- 2.MacCollin M., Willett C., Heinrich B., Jacoby L.B., Acierno J.S., Jr, Perry A., Louis D.N. Familial schwannomatosis: exclusion of the NF2 locus as the germline event. Neurology. 2003;60:1968–1974. doi: 10.1212/01.wnl.0000070184.08740.e0. [DOI] [PubMed] [Google Scholar]
- 3.Hulsebos T.J., Plomp A.S., Wolterman R.A., Robanus-Maandag E.C., Baas F., Wesseling P. Germline mutation of INI1/SMARCB1 in familial schwannomatosis. Am. J. Hum. Genet. 2007;80:805–810. doi: 10.1086/513207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sestini R., Bacci C., Provenzano A., Genuardi M., Papi L. Evidence of a four-hit mechanism involving SMARCB1 and NF2 in schwannomatosis-associated schwannomas. Hum. Mutat. 2008;29:227–231. doi: 10.1002/humu.20679. [DOI] [PubMed] [Google Scholar]
- 5.Hadfield K.D., Newman W.G., Bowers N.L., Wallace A., Bolger C., Colley A., McCann E., Trump D., Prescott T., Evans D.G. Molecular characterisation of SMARCB1 and NF2 in familial and sporadic schwannomatosis. J. Med. Genet. 2008;45:332–339. doi: 10.1136/jmg.2007.056499. [DOI] [PubMed] [Google Scholar]
- 6.Boyd C., Smith M.J., Kluwe L., Balogh A., Maccollin M., Plotkin S.R. Alterations in the SMARCB1 (INI1) tumor suppressor gene in familial schwannomatosis. Clin. Genet. 2008;74:358–366. doi: 10.1111/j.1399-0004.2008.01060.x. [DOI] [PubMed] [Google Scholar]
- 7.Versteege I., Sevenet N., Lange J., Rousseau-Merck M.F., Ambros P., Handgretinger R., Aurias A., Delattre O. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. 1998;394:203–206. doi: 10.1038/28212. [DOI] [PubMed] [Google Scholar]
- 8.Sevenet N., Sheridan E., Amram D., Schneider P., Handgretinger R., Delattre O. Constitutional mutations of the hSNF5/INI1 gene predispose to a variety of cancers. Am. J. Hum. Genet. 1999;65:1342–1348. doi: 10.1086/302639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Swensen J.J., Keyser J., Coffin C.M., Biegel J.A., Viskochil D.H., Williams M.S. Familial occurrence of schwannomas and malignant rhabdoid tumour associated with a duplication in SMARCB1. J. Med. Genet. 2009;46:68–72. doi: 10.1136/jmg.2008.060152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eaton K.W., Tooke L.S., Wainwright L.M., Judkins A.R., Biegel J.A. Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr. Blood Cancer. 2011;56:7–15. doi: 10.1002/pbc.22831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Janson K., Nedzi L.A., David O., Schorin M., Walsh J.W., Bhattacharjee M., Pridjian G., Tan L., Judkins A.R., Biegel J.A. Predisposition to atypical teratoid/rhabdoid tumor due to an inherited INI1 mutation. Pediatr. Blood Cancer. 2006;47:279–284. doi: 10.1002/pbc.20622. [DOI] [PubMed] [Google Scholar]
- 12.Patil S., Perry A., Maccollin M., Dong S., Betensky R.A., Yeh T.H., Gutmann D.H., Stemmer-Rachamimov A.O. Immunohistochemical analysis supports a role for INI1/SMARCB1 in hereditary forms of schwannomas, but not in solitary, sporadic schwannomas. Brain Pathol. (Zurich, Switzerland) 2008;18:517–519. doi: 10.1111/j.1750-3639.2008.00155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith M.J., Boyd C.D., MacCollin M.M., Plotkin S.R. Identity analysis of schwannomatosis kindreds with recurrent constitutional SMARCB1 (INI1) alterations. Clin. Genet. 2009;75:501–502. doi: 10.1111/j.1399-0004.2009.01156.x. [DOI] [PubMed] [Google Scholar]
- 14.Jacoby L.B., MacCollin M., Parry D.M., Kluwe L., Lynch J., Jones D., Gusella J.F. Allelic expression of the NF2 gene in neurofibromatosis 2 and schwannomatosis. Neurogenetics. 1999;2:101–108. doi: 10.1007/s100480050060. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Z.K., Davies K.P., Allen J., Zhu L., Pestell R.G., Zagzag D., Kalpana G.V. Cell cycle arrest and repression of cyclin D1 transcription by INI1/hSNF5. Mol. Cell. Biol. 2002;22:5975–5988. doi: 10.1128/MCB.22.16.5975-5988.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Biegel J.A., Zhou J.Y., Rorke L.B., Stenstrom C., Wainwright L.M., Fogelgren B. Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res. 1999;59:74–79. [PubMed] [Google Scholar]
- 17.Anderson M.A., Gusella J.F. Use of cyclosporin A in establishing Epstein-Barr virus-transformed human lymphoblastoid cell lines. In Vitro. 1984;20:856–858. doi: 10.1007/BF02619631. [DOI] [PubMed] [Google Scholar]