

Abstract
HIV-1 has been the target of intensive research at the molecular and biochemical levels for >25 years. Collectively, this work has led to a detailed understanding of viral replication and the development of 24 approved drugs that have five different targets on various viral proteins and one cellular target (CCR5). Although most drugs target viral enzymatic activities, our detailed knowledge of so much of the viral life cycle is leading us into other types of inhibitors that can block or disrupt protein-protein interactions. Viruses have compact genomes and employ a strategy of using a small number of proteins that can form repeating structures to enclose space (i.e. condensing the viral genome inside of a protein shell), thus minimizing the need for a large protein coding capacity. This creates a relatively small number of critical protein-protein interactions that are essential for viral replication. For HIV-1, the Gag protein has the role of a polyprotein precursor that contains all of the structural proteins of the virion: matrix, capsid, spacer peptide 1, nucleocapsid, spacer peptide 2, and p6 (which contains protein-binding domains that interact with host proteins during budding). Similarly, the Gag-Pro-Pol precursor encodes most of the Gag protein but now includes the viral enzymes: protease, reverse transcriptase (with its associated RNase H activity), and integrase. Gag and Gag-Pro-Pol are the substrates of the viral protease, which is responsible for cleaving these precursors into their mature and fully active forms (see Fig. 1A).
Keywords: Drug Resistance, HIV-1, Protease, Protein Processing, Virus Assembly
Introduction
The Gag and Gag-Pro-Pol precursors assemble at the plasma membrane of the cell, with the membrane ultimately being pinched off from the cell surface to create a membrane-bound virion with a diameter of ∼120 nm, representing a volume of ∼0.9 attoliters (Fig. 1). The host ESCRT (endosomal sorting complex required for transport) pathway that is subverted to drive the membrane fission event needed for virion budding has been reviewed in detail (1–3). The virion assembly process that takes place at the cell membrane results in a finite number of each viral protein within the particle. The budded particle has ∼2400 Gag molecules embedded in the membrane via the N-terminal matrix (MA)3 protein domain, which, in a 120-nm sphere, gives Gag a concentration of ∼4.4 mm, with a crude estimate that the Gag molecules occupy 50–60% of the volume of the sphere (4). There are also ∼120 Gag-Pro-Pol molecules (5). The embedded protease (PR) must dimerize, release itself from the Gag-Pro-Pol precursor, and then cleave the other PR cleavage sites in Gag and Gag-Pro-Pol (6). From these cleaved products, the nucleocapsid (NC) condenses and stabilizes the viral dimeric RNA, and ∼1500 copies of the processed capsid (CA) protein reform to make the mature conical capsid structure around viral RNA to create an infectious particle (7). In this minireview, we examine outstanding issues surrounding the HIV-1 PR, the role of protein processing and rearrangement in the assembly pathway, the impact of PR inhibitor resistance on viral fitness and assembly, and the fact that all of this biochemistry takes place within the confines of a particle that is only 120 nm wide.
FIGURE 1.

HIV-1 assembly pathway. A, schematic diagram representing the Gag and Gag-Pro-Pol polyproteins: MA (blue), CA (dark green), SP1 (light green), NC (brown), SP2 (orange), p6 (salmon), PR (purple), RT (cyan), and IN (navy). B, summary of the HIV-1 budding process at the plasma membrane. C, sequential proteolytic processing of HIV-1 Gag polyprotein. Listed above each cleavage event is the processing site targeted within the intermediate.
A Closed System
The activity of all of the viral enzymes appears to take place within a closed system, with a finite number of protein molecules available for each process. This is true for protein processing during the production of the virus particle and for viral DNA synthesis after the particle infects the next cell. Modest changes in the number of one of the viral enzymes or the number of active molecules can have surprising effects on particle assembly, maturation, and infectivity, and from this, we can infer that certain steps in replication require more than one molecule (or molecular complex) of an enzyme, whereas others require only one. The number of active enzyme molecules in a virus particle can be manipulated by titrating in an inhibitor, titrating in an inactive subunit through phenotypic mixing, or reducing enzymatic activity with mutations that confer a fitness loss. Furthermore, reductions in PR activity can have pleiotropic effects because the PR is responsible for cleaving the Gag-Pro-Pol precursor to generate active reverse transcriptase (RT) and integrase (IN). It is now clear that for replication steps that require multiple copies of an enzyme, the partial loss of enzymatic activity, to the point where this activity is limiting for replication, results in a virus particle that has enhanced sensitivity to further inhibition by an inhibitor.
The simplest example is the sensitivity of RT to non-nucleoside RT inhibitors (NNRTIs). When RT activity is partially inhibited by including an intermediate level of an NNRTI (8–10), by creating a mixture of wild-type and mutant RT subunits (8, 11), or by reducing PR activity to decrease the amount of processed RT subunits (12, 13), the remaining viral infectivity is hypersensitive to inhibition by adding additional NNRTI. The interpretation is that viral DNA synthesis requires more than one RT heterodimer to be successful; as the number of RT molecules in the replication complex decreases, the probability of successfully completing DNA synthesis is reduced, i.e. it is easier to get to the threshold of too little RT if some of it is already missing. Thus, multiple RT heterodimers must be associated with the replication complex where viral DNA synthesis occurs in a newly infected cell, and DNA synthesis requires the participation of multiple RT complexes, making the process distributive.
The same phenomenon is seen with PR activity in the formation of an infectious particle. Partial reduction of PR activity through titration with an inhibitor (10), by mixing with an inactive subunit (11, 13), or by incorporating mutations that reduce enzymatic activity/fitness (13) results in enhanced sensitivity to further inhibition with a protease inhibitor (PI). Thus, the maturation process requires multiple PR dimers, and as the total number of active PR dimers in the assembling virion decreases, it becomes easier to titrate the remaining activity to reach a threshold of too little enzymatic activity to make an infectious particle.
The opposite phenomenon is seen with the viral IN tetramer, where reduction in IN activity through partial titration with an inhibitor (10) or including an inactive subunit (11) does not enhance sensitivity to further inhibition, presumably due to the fact that a single IN tetramer binds to the ends of viral DNA and does not exchange with free IN within the replication complex even if the bound form is inactive. Also, chain-terminating nucleoside/nucleotide RT inhibitors (NRTIs) cap the growing DNA chain instead of inhibiting RT itself; thus, viral infectivity does not become increasingly sensitive to NRTIs as the amount of RT activity is decreased because reducing the amount of RT does not change the probability of selecting a normal nucleotide or a chain-terminating nucleotide for incorporation. The exception among NRTIs is azidothymidine (AZT), which, when incorporated, cannot translocate from the nucleotide-binding site on RT to the primer site because of steric hindrance by the large 3′-azido group (14, 15). In this position, the chain-terminating nucleotide can be excised by RT by forming a dinucleotide with ATP (14, 16). The increased sensitivity to AZT in virus with decreased PR activity, first seen with PR fitness mutants (12) and then with phenotypic mixing with an active site mutant (13), shows that the RT heterodimer that incorporates AZT is not necessarily the same one that excises it.
Assembly and Processing
As depicted in Fig. 1B, a small number of Gag molecules traffic dimers of the RNA genome to the plasma membrane (1, 17). Once at the membrane, additional Gag proteins are recruited through Gag-Gag interactions and nonspecific Gag-RNA interactions, utilizing Gag molecules from both the cytosol and those already attached to the membrane (17–19). Gag-Pro-Pol is recruited to sites of assembly simultaneously. Although Gag oligomerization initiates budding, the process is facilitated and completed by the ESCRT family of proteins (1, 4). Each immature virion will contain ∼2400 Gag monomers (4) and ∼120 Gag-Pro-Pol molecules (5). For the emerging virus particle to become infectious, the HIV-1 PR must catalyze a series of cleavage events that trigger structural and morphological changes that result in the condensation of the NC-RNA core, the formation of the CA shell, and the release of viral enzymes from their precursors (Fig. 1C). For a thorough discussion of the architecture of the HIV-1 viral core, we direct you to a number of recent publications (see Ref. 20).
The HIV-1 PR is an aspartic proteinase and functions as a homodimer (Fig. 2A) (21). Each monomer contributes an aspartic acid residue to coordinate a water molecule during the proteolysis reaction. Most aspartic proteinases exist as pseudodimers in eukaryotes, but the retroviral PR originates as a monomer within the Gag-Pro-Pol polyprotein. The need for a dimer to form the active site necessitates the interaction of two Gag-Pro-Pol molecules for proteolysis to begin. Relative to the excised mature PR, dimers formed between monomers still embedded in Gag-Pro-Pol are much less stable (22, 23) and exhibit poorer enzymatic activity (23). The instability results in the embedded dimers sampling a wide variety of conformations (22, 24, 25), potentially adopting the mature dimer interface only 3–5% of the time (25). Because of this low enzymatic activity, the first cleavage events performed by the embedded PR are intramolecular (26), with the primary result being the removal of the transframe region from the N terminus of the PR domain. These processing events occur at three locations: the spacer peptide 1 (SP1)/NC site, an internal site within the p6* domain (the transframe protein in Gag that is expressed only after the frameshift), and finally the transframe/PR interface. Removing the transframe region from the N terminus of the PR greatly improves dimer stability because the first four amino acids of each PR monomer interact with the final four residues to create a four-stranded β-sheet (23). With improved stability, the PR exhibits increased enzymatic activity, which is likely necessary for efficient intermolecular cleavages to occur (26–28). These more functional PR dimers will carry out the bulk of the remaining processing events, including removal of RT from the C-terminal end of the PR and cleavage of the Gag polyprotein.
FIGURE 2.
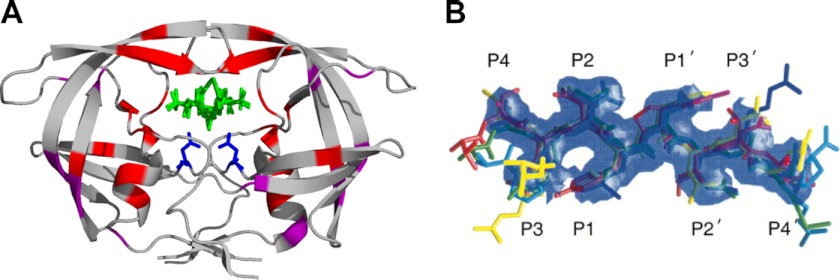
Structure of the HIV-1 PR and its substrates. A, structure of HIV-1 PR with the resistance mutations. The dimeric PR is shown (Protein Data Bank code 3EL1; rendered with PyMOL) bound to an inhibitor (green). The active site aspartic acid (Asp-25) from each subunit is shown in blue. The positions of PI resistance mutations proximal to the active site are shown in red. The positions of compensatory mutations are shown in purple. B, substrate envelope of HIV-1 PR. The substrate envelope was calculated from the overlapping van der Waals volume of four or more substrate peptides. The colors of the substrate peptides are red (MA/CA), green (CA/SP1), blue (SP1/NC), cyan (SP2/p6), magenta (RT/RNase H), and yellow (RNase H/IN). This figure was reprinted from Ref. 101 with permission.
Processing of the Gag polyprotein proceeds in a particular order, with the five cleavage events roughly occurring in three different phases, as measured using an in vitro derived Gag substrate and purified PR (29–31). The initial site targeted is the SP1/NC site. In the second phase of processing, the spacer peptide 2 (SP2)/p6 and MA/CA sites are cleaved. Finally, the spacer peptides SP1 and SP2 are removed from CA and NC, respectively (Fig. 1C) (31). Although processing has proven difficult to observe in the virion, mutant particles defective for cleavage at particular sites generated intermediates consistent with the proposed order of events determined in vitro (32). Furthermore, these mutants or others that alter the processing order severely disrupt infectivity (32–35), indicating that the timing and order of cleavage are important for the assembly of the mature viral core. The mechanisms governing ordered cleavage have been difficult to uncover, largely because no discernible pattern can be found among the cleavage site amino acid sequences (36). Instead of a consensus sequence, the substrate specificity demonstrated by the HIV-1 PR appears to be dependent upon a conserved shape (37). All of the peptide substrates derived from the cleavage sites within the Gag polyprotein were shown to adopt an asymmetric extended β-strand conformation when bound in the active site of the enzyme, creating a consensus volume termed the “substrate envelope” (Fig. 2B). Molecular modeling suggests that the cleavage sites share particular hydrogen bonding patterns between the peptide backbone and the PR, but the hydrogen bonds and hydrophobic interactions between substrate side chains and the PR are not conserved among the different cleavage sites. Thus, the differences in the side chain hydrogen bonding and hydrophobic interactions may contribute to the unique processing rate for each cleavage site (38).
Assembly, budding, and proteolytic processing of Gag and Gag-Pro-Pol are intricately linked events, although the relative timing of each and the importance of that timing are questions that are still being explored. Following initiation of assembly at the membrane, complete virions are observed at the surface of the cell within 5–10 min (18, 39), and virion release occurs 15–20 min later (18). It has been difficult to observe processing events in released virions, and early activation of PR activity by creating a tethered dimer within a single Gag-Pro-Pol molecule or delay of particle formation relative to processing negatively impacts particle formation (40, 41); these observations suggest that activation of the PR is delayed until later in the assembly process but that processing must be completed either during budding or relatively quickly after budding. PR activity is not required for the initiation of assembly (18), but there is some evidence that the presence of a PR with decreased activity can slow the rate of virion release (42). The excess of Gag over Gag-Pro-Pol (20:1) suggests that the vast majority of Gag-Pro-Pol will have primary interactions with Gag molecules. Thus, the infrequent juxtaposition of two Gag-Pro-Pol precursors to create a homodimer, the stochastic spacing of Gag-Pro-Pol precursors in the budding Gag shell spatially limiting the number of Pro-Pol interactions, and the poor stability of the immature PR dimer in the Gag-Pro-Pol homodimer all reduce the likelihood of significant PR activity early in the assembly process. The slow or delayed release of the first PR dimer can then initiate cleavage events in trans that would include the release of PR monomers that could dimerize and have high levels of activity. Still, it must be acknowledged that we do not know when processing is initiated, although most textbook conceptualizations of processing have it occurring after budding. We are likely to know the answer to this question when fluorescent proteins are incorporated into the virion that can also serve as substrates for the PR.
Protease Inhibitors, Resistance, and Viral Fitness
Due to the requirement of the maturation process to produce infectious virions, PR has been a major target for developing antiretroviral inhibitors, with nine PIs approved for clinical use. These PIs are transition state analogs, mostly peptidomimetics, that bind the enzyme with much higher affinity than do the substrates (21). The binding affinity of the PIs for the wild-type enzyme ranges from nm to pm (43) under conditions in which peptide substrates bind with affinities in the high μm range. The effectiveness of the PIs in antiretroviral therapy can be compromised by the emergence of resistance mutations in the PR region. More resistance mutations have been selected by PIs than any other antiretroviral drugs. Although mutations at as many as 46 positions of the 99 residues in PR have been shown to be associated with selection by PIs, only a subset of ∼26 positions have been identified as those most commonly involved in PI resistance (see Refs. 44 and 45 and references therein). High level resistance to a PI typically requires four to six mutations, and PIs are thus considered to have a high genetic barrier (46). The ability to make tight binding transition state analogs that require multiple mutations to confer resistance suggests that it may be possible eventually to treat HIV-1 with a single PI if it were sufficiently potent. Efforts along these lines have been explored with some success (47–49).
There are several mechanistic features of resistance to PI. First, mutations in the active site can change the interaction with the inhibitor either by reducing a contact or creating a steric hindrance (Fig. 2A) (50–55). Such changes are more easily tolerated if a side chain of the drug extends outside of the substrate envelope. However, when such changes also impact interaction with the substrate, this results in a fitness cost in how well the enzyme functions in the context of replication (12, 56–58). Second, the fitness cost associated with these active site mutations can be compensated by mutations that occur outside of the active site but appear to be capable of enhancing PR activity (51, 54, 59–65). Although the active site mutations are absent in the untreated population, the compensatory mutations pre-exist in the population, perhaps compensating for deleterious mutations in PR that can get fixed fortuitously. Third, cleavage sites around SP2 can become limiting for making an infectious virus, and cleavage site sequences can undergo evolution to make them more easily cleaved by the mutant PR (66–70). Fourth, other mutations in Gag have been described that appear to contribute to PI resistance (71) but in unknown ways, suggesting that there are other pathways to at least low level resistance.
Our view is that the concept of fitness is synonymous with the idea of PR acting in a closed system. When PR loses activity on its normal Gag substrate cleavage sites, the probability of completing assembly and processing to yield an infectious particle is reduced, i.e. the virus is less fit to produce the full complement of infectious virus. Thus, in some assays, a single resistance-associated mutation will actually sensitize the virus to an inhibitor when the fitness loss in substrate recognition is greater than the fitness gain in resistance (13). This balance shifts as multiple resistance mutations and compensatory mutations are added. Such fitness cost and pleiotropic effects of a virus with reduced PR activity may be the reason that patients with virus carrying PI resistance mutations (that lower fitness overall) can have slowed disease progression in the setting of drug failure (72).
Assembly Inhibitors
HIV-1 particle assembly is a highly ordered process and involves the association and rearrangement of several thousand viral structural proteins. One key step involves cleavage at the N terminus of CA by the viral PR, followed by the formation of a new β-hairpin structure anchored by a salt bridge between the released N-terminal Pro-1 of CA and an internal aspartic acid side chain in CA (Asp-51 in HIV-1), an essential step in the proper assembly of the capsid cone (73, 74). Disrupting this salt bridge is an attractive drug target, although, as yet, an unrealized target. The fully processed CA makes key intermolecular CA-CA interactions that result in hexameric (and some pentameric) rings that are the basic structural unit of the conical capsid (75). Due to the indispensable nature of these interactions in generating infectious virus particles, there is an ongoing search for molecules that bind CA and inhibit these interactions. A 12-mer peptide (capsid assembly inhibitor (CAI)) and a small molecule (CAP-1) are able to disrupt HIV-1 CA assembly. CAI, a helical peptide selected in a phage display, binds to a hydrophobic cleft within the C-terminal domain (CTD) of CA (76, 77), and CAP-1 bind to the N-terminal domain (NTD) of CA, forming a hydrophobic pocket (78, 79). Recently, new CA inhibitors have been identified in high throughput screening assays. PF74, a small molecule, binds to the NTD of HIV-1 CA, near the CAP-1-binding site, and inhibits both early and late events of viral replication (80, 81). Two more series of inhibitors have been identified based on benzodiazepines (BDs) and benzimidazoles (BMs), which also bind to the same NTD of CA as CAP-1 (82). It has been proposed that all of these inhibitors are interfering with a critical NTD-CTD intermolecular interaction of CA-CA that stabilizes the hexameric and pentameric rings (82).
The structural changes that must occur during virion maturation represent one type of target in inhibiting assembly. Another target is the processing sites themselves. Blocking cleavage at a specific processing site is analogous to blocking viral DNA synthesis with a chain-terminating analog; in each case, the enzyme (PR or RT) is not inhibited, but rather its substrate (a cleavage site or the growing DNA chain) is blocked. Processing at each site in Gag is essential for making an infectious particle (32, 33, 83), although mutations blocking processing at the NC/SP2 site do not completely ablate infectivity (33, 84). Bevirimat, the prototype HIV-1 maturation inhibitor identified in a screen for inhibition of viral replication, specifically inhibits the cleavage event between CA and SP1 within the Gag polyprotein (85, 86). The drug is incorporated into immature particles near the CA/SP1 cleavage site and stabilizes an immature form of the CA lattice, and this interaction with Gag alters its ability to serve as a PR substrate at the CA/SP1 site (87, 88). Recently, direct binding of bevirimat to the CA/SP1 cleavage site in immature Gag particles has been reported (89). The dramatic effect on infectivity of bevirimat binding to the CA/SP1 site is similar to the effect of a processing mutant at this site, especially if a fortuitous cleavage site within SP1 is absent (90). Thus, bevirimat provides a proof of concept for an inhibitor of a specific processing site. Along these lines, cleavage at the MA/CA site must go to near-completion to make an infectious particle, and this has been seen for both MuLV and HIV-1 (33, 34, 91). Inhibiting cleavage at this site by as little as 10% is sufficient to ablate virion infectivity (33). The MA/CA cleavage site is the most sensitive site among all of the cleavage sites in Gag in terms of requiring nearly complete cleavage to allow infectivity (33), suggesting that the MA/CA cleavage site could be an important target for the development of a new class of maturation inhibitors. The extreme sensitivity of this site to underprocessing in the context of forming an infectious particle reinforces the idea that strong trans-dominant effects can be realized in inhibiting the assembly/processing pathway. The EM morphology of the virions showing defects in the CA assembly process is shown in Fig. 3. Note that the inside of the Y132I virion (where cleavage between MA and CA has been blocked) is less electron-dense due to the lack of free CA protein. Virus particles with other CA assembly defects often display similar ring-shaped capsid-like structures, presumably aberrant CA assemblies, regardless of the type of maturation inhibitors used.
FIGURE 3.
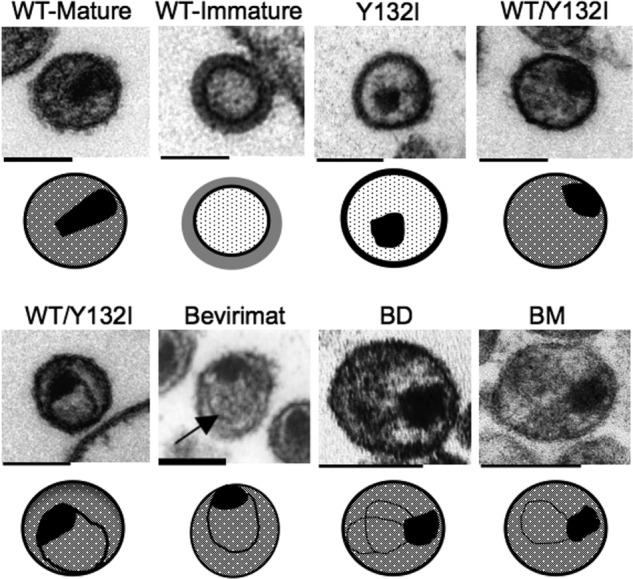
Thin-section EM morphology of virions displaying CA assembly defects caused by a trans-dominant mutation, or treatment with maturation or CA assembly inhibitors. The Y132I mutant contains an Ile mutation at the P1 position blocking PR cleavage at the MA/CA cleavage site. WT/Y132I virus particles were generated by cotransfection of infectious viral DNA with a ratio of 80% WT and 20% Y132I (33). Bevirimat is a maturation inhibitor that blocks PR cleavage at the CA/SP1 cleavage site (85, 88). BD and BM are examples of two classes of CAIs (82). Due to the lack of free CA proteins in the virion, the space between the viral membrane and the core structure in the Y132I virions shows less electron-dense material than that in the wild-type mature virion, where CA is released from Gag, but only a fraction is used in assembling the capsid. Schematic diagrams of each virion are shown below the EM image to compare aberrant CA core structures that resulted from either cleavage site mutation or treatment with the different maturation/CA assembly inhibitors. The black irregular mass is assumed to be NC bound to viral RNA. Scale bars = ∼100 nm. The EM morphologies of WT mature, WT immature, Y132I, and WT/Y132I virions are reprinted from Ref. 33. The EM morphology of the bevirimat-treated virion is reprinted from Ref. 85. The EM morphologies of BD- and BM-treated virions are reprinted from Ref. 82.
Another potential step in the life cycle that is impacted by PR processing is the condensation of the viral dimeric RNA. In the absence of processing, viral RNA is in a low stability dimeric form in the virion (92). With processing, the RNA is in a much more stable dimeric form. Condensation of the RNA is mediated by the NC region after it is released from Gag (93). During the maturation process, NC is found within four different proteins: full-length Gag, NC/SP2/p6 (p15), NC/SP2, and fully released NC. These different versions of the NC protein may have distinct functions at different steps in the life cycle (94), providing a role for processing in the regulation of RNA binding by the NC domain. Furthermore, there is evidence that nucleic acid binding can regulate the efficiency of cleavage at the SP2/p6 site using a p15 substrate (95, 96). Thus, on several levels, processing around the NC domain of Gag is involved in regulation of the protein activity.
Looking Ahead
Answering the question of when processing occurs in the budding pathway is central to our understanding of virion morphogenesis, and we are likely to know the answer to this question with the application of new technologies. Our detailed understanding of the role of protein processing in the regulation of protein function for the proteins present in Gag is creating opportunities to design assays amenable for use in high throughput screens to search for lead compounds that can inhibit the assembly of an infectious particle (97–99). The 25 years of studying the biochemistry of the HIV-1 virion was built on an earlier 15 years of studying other retroviruses, starting with the identification of a Gag precursor in avian myeloblastosis virus (100). We are now at a point where we understand critical steps in virion assembly at the molecular level and can conceptualize new ways of disrupting these essential processes.
Acknowledgments
We thank Dr. Celia Schiffer and her colleagues for many helpful discussions.
This work was supported, in whole or in part, by National Institutes of Health Grants P01 GM066524, R37 AI44667, and R21 NS073052. This is the fourth article in the Thematic Minireview Series on Understanding Human Immunodeficiency Virus-Host Interactions at the Biochemical Level.
- MA
- matrix
- PR
- protease
- NC
- nucleocapsid
- RT
- reverse transcriptase
- IN
- integrase
- NNRTI
- non-nucleoside RT inhibitor
- PI
- protease inhibitor
- NRTI
- nucleoside/nucleotide RT inhibitor
- AZT
- azidothymidine
- SP1
- spacer peptide 1
- SP2
- spacer peptide 2
- CA
- capsid
- CAI
- capsid assembly inhibitor
- CTD
- C-terminal domain
- NTD
- N-terminal domain
- BD
- benzodiazepine
- BM
- benzimidazole.
REFERENCES
- 1. Bieniasz P. D. (2009) The cell biology of HIV-1 virion genesis. Cell Host Microbe 5, 550–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Weiss E. R., Göttlinger H. (2011) The role of cellular factors in promoting HIV budding. J. Mol. Biol. 410, 525–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sundquist W. I., Krausslich H. G. (2012) HIV-1 Assembly, Budding, and Maturation. Cold Spring Harb. Perspect. Med. 2, a006924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Carlson L. A., Briggs J. A., Glass B., Riches J. D., Simon M. N., Johnson M. C., Müller B., Grünewald K., Kräusslich H. G. (2008) Three-dimensional analysis of budding sites and released virus suggests a revised model for HIV-1 morphogenesis. Cell Host Microbe 4, 592–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jacks T. (1990) Translational suppression in gene expression in retroviruses and retrotransposons. Curr. Top. Microbiol. Immunol. 157, 93–124 [DOI] [PubMed] [Google Scholar]
- 6. Swanstrom R., Wills J. W. (1997) in Retroviruses (Coffin J. M., Hughes S. H., Varmus H. E., ed) pp. 263–334, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 7. Briggs J. A., Simon M. N., Gross I., Kräusslich H. G., Fuller S. D., Vogt V. M., Johnson M. C. (2004) The stoichiometry of Gag protein in HIV-1. Nat. Struct. Mol. Biol. 11, 672–675 [DOI] [PubMed] [Google Scholar]
- 8. Ambrose Z., Julias J. G., Boyer P. L., Kewalramani V. N., Hughes S. H. (2006) The level of reverse transcriptase (RT) in human immunodeficiency virus type 1 particles affects susceptibility to nonnucleoside RT inhibitors but not to lamivudine. J. Virol. 80, 2578–2581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shen L., Peterson S., Sedaghat A. R., McMahon M. A., Callender M., Zhang H., Zhou Y., Pitt E., Anderson K. S., Acosta E. P., Siliciano R. F. (2008) Dose-response curve slope sets class-specific limits on inhibitory potential of anti-HIV drugs. Nat. Med. 14, 762–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sampah M. E., Shen L., Jilek B. L., Siliciano R. F. (2011) Dose-response curve slope is a missing dimension in the analysis of HIV-1 drug resistance. Proc. Natl. Acad. Sci. U.S.A. 108, 7613–7618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shen L., Rabi S. A., Sedaghat A. R., Shan L., Lai J., Xing S., Siliciano R. F. (2011) A critical subset model provides a conceptual basis for the high antiviral activity of major HIV drugs. Sci. Transl. Med. 3, 91ra63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. de la Carrière L. C., Paulous S., Clavel F., Mammano F. (1999) Effects of human immunodeficiency virus type 1 resistance to protease inhibitors on reverse transcriptase processing, activity, and drug sensitivity. J. Virol. 73, 3455–3459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Henderson G. J., Lee S. K., Irlbeck D. M., Harris J., Kline M., Pollom E., Parkin N., Swanstrom R. (2012) Interplay between single resistance-associated mutations in the HIV-1 protease and viral infectivity, protease activity, and inhibitor sensitivity. Antimicrob. Agents Chemother. 56, 623–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Boyer P. L., Sarafianos S. G., Arnold E., Hughes S. H. (2001) Selective excision of AZTMP by drug-resistant human immunodeficiency virus reverse transcriptase. J. Virol. 75, 4832–4842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tong W., Lu C. D., Sharma S. K., Matsuura S., So A. G., Scott W. A. (1997) Nucleotide-induced stable complex formation by HIV-1 reverse transcriptase. Biochemistry 36, 5749–5757 [DOI] [PubMed] [Google Scholar]
- 16. Meyer P. R., Matsuura S. E., Mian A. M., So A. G., Scott W. A. (1999) A mechanism of AZT resistance: an increase in nucleotide-dependent primer unblocking by mutant HIV-1 reverse transcriptase. Mol. Cell 4, 35–43 [DOI] [PubMed] [Google Scholar]
- 17. Jouvenet N., Simon S. M., Bieniasz P. D. (2009) Imaging the interaction of HIV-1 genomes and Gag during assembly of individual viral particles. Proc. Natl. Acad. Sci. U.S.A. 106, 19114–19119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ivanchenko S., Godinez W. J., Lampe M., Kräusslich H. G., Eils R., Rohr K., Bräuchle C., Müller B., Lamb D. C. (2009) Dynamics of HIV-1 assembly and release. PLoS Pathog. 5, e1000652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kutluay S. B., Bieniasz P. D. (2010) Analysis of the initiating events in HIV-1 particle assembly and genome packaging. PLoS Pathog. 6, e1001200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ganser-Pornillos B. K., Yeager M., Pornillos O. (2012) Assembly and architecture of HIV. Adv. Exp. Med. Biol. 726, 441–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lefebvre E., Schiffer C. A. (2008) Resilience to resistance of HIV-1 protease inhibitors: profile of darunavir. AIDS Rev. 10, 131–142 [PMC free article] [PubMed] [Google Scholar]
- 22. Agniswamy J., Sayer J. M., Weber I. T., Louis J. M. (2012) Terminal interface conformations modulate dimer stability prior to amino-terminal autoprocessing of HIV-1 protease. Biochemistry 51, 1041–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Louis J. M., Clore G. M., Gronenborn A. M. (1999) Autoprocessing of HIV-1 protease is tightly coupled to protein folding. Nat. Struct. Biol. 6, 868–875 [DOI] [PubMed] [Google Scholar]
- 24. Huang L., Li Y., Chen C. (2011) Flexible catalytic site conformations implicated in modulation of HIV-1 protease autoprocessing reactions. Retrovirology 8, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tang C., Louis J. M., Aniana A., Suh J. Y., Clore G. M. (2008) Visualizing transient events in amino-terminal autoprocessing of HIV-1 protease. Nature 455, 693–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pettit S. C., Gulnik S., Everitt L., Kaplan A. H. (2003) The dimer interfaces of protease and extra-protease domains influence the activation of protease and the specificity of Gag-Pol cleavage. J. Virol. 77, 366–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ludwig C., Leiherer A., Wagner R. (2008) Importance of protease cleavage sites within and flanking human immunodeficiency virus type 1 transframe protein p6* for spatiotemporal regulation of protease activation. J. Virol. 82, 4573–4584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pettit S. C., Everitt L. E., Choudhury S., Dunn B. M., Kaplan A. H. (2004) Initial cleavage of the human immunodeficiency virus type 1 Gag-Pol precursor by its activated protease occurs by an intramolecular mechanism. J. Virol. 78, 8477–8485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Erickson-Viitanen S., Manfredi J., Viitanen P., Tribe D. E., Tritch R., Hutchison C. A., 3rd, Loeb D. D., Swanstrom R. (1989) Cleavage of HIV-1 Gag polyprotein synthesized in vitro: sequential cleavage by the viral protease. AIDS Res. Hum. Retroviruses 5, 577–591 [DOI] [PubMed] [Google Scholar]
- 30. Pettit S. C., Lindquist J. N., Kaplan A. H., Swanstrom R. (2005) Processing sites in the human immunodeficiency virus type 1 (HIV-1) Gag-Pro-Pol precursor are cleaved by the viral protease at different rates. Retrovirology 2, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pettit S. C., Moody M. D., Wehbie R. S., Kaplan A. H., Nantermet P. V., Klein C. A., Swanstrom R. (1994) The p2 domain of human immunodeficiency virus type 1 Gag regulates sequential proteolytic processing and is required to produce fully infectious virions. J. Virol. 68, 8017–8027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wiegers K., Rutter G., Kottler H., Tessmer U., Hohenberg H., Kräusslich H. G. (1998) Sequential steps in human immunodeficiency virus particle maturation revealed by alterations of individual Gag polyprotein cleavage sites. J. Virol. 72, 2846–2854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lee S. K., Harris J., Swanstrom R. (2009) A strongly transdominant mutation in the human immunodeficiency virus type 1 gag gene defines an Achilles heel in the virus life cycle. J. Virol. 83, 8536–8543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Müller B., Anders M., Akiyama H., Welsch S., Glass B., Nikovics K., Clavel F., Tervo H. M., Keppler O. T., Kräusslich H. G. (2009) HIV-1 Gag processing intermediates trans-dominantly interfere with HIV-1 infectivity. J. Biol. Chem. 284, 29692–29703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tritch R. J., Cheng Y. E., Yin F. H., Erickson-Viitanen S. (1991) Mutagenesis of protease cleavage sites in the human immunodeficiency virus type 1 Gag polyprotein. J. Virol. 65, 922–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pettit S. C., Simsic J., Loeb D. D., Everitt L., Hutchison C. A., 3rd, Swanstrom R. (1991) Analysis of retroviral protease cleavage sites reveals two types of cleavage sites and the structural requirements of the P1 amino acid. J. Biol. Chem. 266, 14539–14547 [PubMed] [Google Scholar]
- 37. Prabu-Jeyabalan M., Nalivaika E., Schiffer C. A. (2002) Substrate shape determines specificity of recognition for HIV-1 protease: analysis of crystal structures of six substrate complexes. Structure 10, 369–381 [DOI] [PubMed] [Google Scholar]
- 38. Ozen A., Haliloğlu T., Schiffer C. A. (2011) Dynamics of preferential substrate recognition in HIV-1 protease: redefining the substrate envelope. J. Mol. Biol. 410, 726–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jouvenet N., Bieniasz P. D., Simon S. M. (2008) Imaging the biogenesis of individual HIV-1 virions in live cells. Nature 454, 236–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kräusslich H. G. (1991) Human immunodeficiency virus proteinase dimer as component of the viral polyprotein prevents particle assembly and viral infectivity. Proc. Natl. Acad. Sci. U.S.A. 88, 3213–3217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ott D. E., Coren L. V., Shatzer T. (2009) The nucleocapsid region of human immunodeficiency virus type 1 Gag assists in the coordination of assembly and Gag processing: role for RNA-Gag binding in the early stages of assembly. J. Virol. 83, 7718–7727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kaplan A. H., Manchester M., Swanstrom R. (1994) The activity of the protease of human immunodeficiency virus type 1 is initiated at the membrane of infected cells before the release of viral proteins and is required for release to occur with maximum efficiency. J. Virol. 68, 6782–6786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Anderson J., Schiffer C., Lee S. K., Swanstrom R. (2009) Viral protease inhibitors. Handb. Exp. Pharmacol. 189, 85–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hoffman N. G., Schiffer C. A., Swanstrom R. (2003) Covariation of amino acid positions in HIV-1 protease. Virology 314, 536–548 [DOI] [PubMed] [Google Scholar]
- 45. Rhee S. Y., Taylor J., Fessel W. J., Kaufman D., Towner W., Troia P., Ruane P., Hellinger J., Shirvani V., Zolopa A., Shafer R. W. (2010) HIV-1 protease mutations and protease inhibitor cross-resistance. Antimicrob. Agents Chemother. 54, 4253–4261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Watkins T., Resch W., Irlbeck D., Swanstrom R. (2003) Selection of high-level resistance to human immunodeficiency virus type 1 protease inhibitors. Antimicrob. Agents Chemother. 47, 759–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Arribas J. R., Pulido F., Delgado R., Lorenzo A., Miralles P., Arranz A., González-García J. J., Cepeda C., Hervás R., Paño J. R., Gaya F., Carcas A., Montes M. L., Costa J. R., Peña J. M. (2005) Lopinavir/ritonavir as single-drug therapy for maintenance of HIV-1 viral suppression: 48-week results of a randomized, controlled, open-label, proof-of-concept pilot clinical trial (OK Study). J. Acquir. Immune Defic. Syndr. 40, 280–287 [DOI] [PubMed] [Google Scholar]
- 48. Katlama C., Valantin M. A., Algarte-Genin M., Duvivier C., Lambert-Niclot S., Girard P. M., Molina J. M., Hoen B., Pakianather S., Peytavin G., Marcelin A. G., Flandre P. (2010) Efficacy of darunavir/ritonavir maintenance monotherapy in patients with HIV-1 viral suppression: a randomized open-label, noninferiority trial, MONOI-ANRS 136. AIDS 24, 2365–2374 [DOI] [PubMed] [Google Scholar]
- 49. Valantin M. A., Lambert-Niclot S., Flandre P., Morand-Joubert L., Cabiè A., Meynard J. L., Ponscarme D., Ajana F., Slama L., Curjol A., Cuzin L., Schneider L., Taburet A. M., Marcelin A. G., Katlama C. (2012) Long-term efficacy of darunavir/ritonavir monotherapy in patients with HIV-1 viral suppression: week 96 results from the MONOI-ANRS 136 study. J. Antimicrob. Chemother. 67, 691–695 [DOI] [PubMed] [Google Scholar]
- 50. Gulnik S. V., Suvorov L. I., Liu B., Yu B., Anderson B., Mitsuya H., Erickson J. W. (1995) Kinetic characterization and cross-resistance patterns of HIV-1 protease mutants selected under drug pressure. Biochemistry 34, 9282–9287 [DOI] [PubMed] [Google Scholar]
- 51. Nijhuis M., Schuurman R., de Jong D., Erickson J., Gustchina E., Albert J., Schipper P., Gulnik S., Boucher C. A. (1999) Increased fitness of drug-resistant HIV-1 protease as a result of acquisition of compensatory mutations during suboptimal therapy. AIDS 13, 2349–2359 [DOI] [PubMed] [Google Scholar]
- 52. Partaledis J. A., Yamaguchi K., Tisdale M., Blair E. E., Falcione C., Maschera B., Myers R. E., Pazhanisamy S., Futer O., Cullinan A. B. (1995) In vitro selection and characterization of human immunodeficiency virus type 1 (HIV-1) isolates with reduced sensitivity to hydroxyethylamino sulfonamide inhibitors of HIV-1 aspartyl protease. J. Virol. 69, 5228–5235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pazhanisamy S., Stuver C. M., Cullinan A. B., Margolin N., Rao B. G., Livingston D. J. (1996) Kinetic characterization of human immunodeficiency virus type 1 protease-resistant variants. J. Biol. Chem. 271, 17979–17985 [DOI] [PubMed] [Google Scholar]
- 54. Schock H. B., Garsky V. M., Kuo L. C. (1996) Mutational anatomy of an HIV-1 protease variant conferring cross-resistance to protease inhibitors in clinical trials. Compensatory modulations of binding and activity. J. Biol. Chem. 271, 31957–31963 [DOI] [PubMed] [Google Scholar]
- 55. Prabu-Jeyabalan M., Nalivaika E. A., King N. M., Schiffer C. A. (2004) Structural basis for coevolution of a human immunodeficiency virus type 1 nucleocapsid-p1 cleavage site with a V82A drug-resistant mutation in viral protease. J. Virol. 78, 12446–12454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Croteau G., Doyon L., Thibeault D., McKercher G., Pilote L., Lamarre D. (1997) Impaired fitness of human immunodeficiency virus type 1 variants with high-level resistance to protease inhibitors. J. Virol. 71, 1089–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Resch W., Ziermann R., Parkin N., Gamarnik A., Swanstrom R. (2002) Nelfinavir-resistant, amprenavir-hypersusceptible strains of human immunodeficiency virus type 1 carrying an N88S mutation in protease have reduced infectivity, reduced replication capacity, and reduced fitness and process the Gag polyprotein precursor aberrantly. J. Virol. 76, 8659–8666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zennou V., Mammano F., Paulous S., Mathez D., Clavel F. (1998) Loss of viral fitness associated with multiple Gag and Gag-Pol processing defects in human immunodeficiency virus type 1 variants selected for resistance to protease inhibitors in vivo. J. Virol. 72, 3300–3306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chen Z., Li Y., Schock H. B., Hall D., Chen E., Kuo L. C. (1995) Three-dimensional structure of a mutant HIV-1 protease displaying cross-resistance to all protease inhibitors in clinical trials. J. Biol. Chem. 270, 21433–21436 [DOI] [PubMed] [Google Scholar]
- 60. Ho D. D., Toyoshima T., Mo H., Kempf D. J., Norbeck D., Chen C. M., Wideburg N. E., Burt S. K., Erickson J. W., Singh M. K. (1994) Characterization of human immunodeficiency virus type 1 variants with increased resistance to a C2-symmetric protease inhibitor. J. Virol. 68, 2016–2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kaplan A. H., Michael S. F., Wehbie R. S., Knigge M. F., Paul D. A., Everitt L., Kempf D. J., Norbeck D. W., Erickson J. W., Swanstrom R. (1994) Selection of multiple human immunodeficiency virus type 1 variants that encode viral proteases with decreased sensitivity to an inhibitor of the viral protease. Proc. Natl. Acad. Sci. U.S.A. 91, 5597–5601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mammano F., Trouplin V., Zennou V., Clavel F. (2000) Retracing the evolutionary pathways of human immunodeficiency virus type 1 resistance to protease inhibitors: virus fitness in the absence and in the presence of drug. J. Virol. 74, 8524–8531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Markowitz M., Mo H., Kempf D. J., Norbeck D. W., Bhat T. N., Erickson J. W., Ho D. D. (1995) Selection and analysis of human immunodeficiency virus type 1 variants with increased resistance to ABT-538, a novel protease inhibitor. J. Virol. 69, 701–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Martinez-Picado J., Savara A. V., Sutton L., D'Aquila R. T. (1999) Replicative fitness of protease inhibitor-resistant mutants of human immunodeficiency virus type 1. J. Virol. 73, 3744–3752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chang M. W., Torbett B. E. (2011) Accessory mutations maintain stability in drug-resistant HIV-1 protease. J. Mol. Biol. 410, 756–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bally F., Martinez R., Peters S., Sudre P., Telenti A. (2000) Polymorphism of HIV type 1 Gag p7/p1 and p1/p6 cleavage sites: clinical significance and implications for resistance to protease inhibitors. AIDS Res. Hum. Retroviruses 16, 1209–1213 [DOI] [PubMed] [Google Scholar]
- 67. Doyon L., Croteau G., Thibeault D., Poulin F., Pilote L., Lamarre D. (1996) Second locus involved in human immunodeficiency virus type 1 resistance to protease inhibitors. J. Virol. 70, 3763–3769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Koch N., Yahi N., Fantini J., Tamalet C. (2001) Mutations in HIV-1 Gag cleavage sites and their association with protease mutations. AIDS 15, 526–528 [DOI] [PubMed] [Google Scholar]
- 69. Mammano F., Petit C., Clavel F. (1998) Resistance-associated loss of viral fitness in human immunodeficiency virus type 1: phenotypic analysis of protease and Gag coevolution in protease inhibitor-treated patients. J. Virol. 72, 7632–7637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zhang Y. M., Imamichi H., Imamichi T., Lane H. C., Falloon J., Vasudevachari M. B., Salzman N. P. (1997) Drug resistance during indinavir therapy is caused by mutations in the protease gene and in its Gag substrate cleavage sites. J. Virol. 71, 6662–6670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Parry C. M., Kohli A., Boinett C. J., Towers G. J., McCormick A. L., Pillay D. (2009) Gag determinants of fitness and drug susceptibility in protease inhibitor-resistant human immunodeficiency virus type 1. J. Virol. 83, 9094–9101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Deeks S. G., Hoh R., Grant R. M., Wrin T., Barbour J. D., Narvaez A., Cesar D., Abe K., Hanley M. B., Hellmann N. S., Petropoulos C. J., McCune J. M., Hellerstein M. K. (2002) CD4+ T cell kinetics and activation in human immunodeficiency virus-infected patients who remain viremic despite long-term treatment with protease inhibitor-based therapy. J. Infect. Dis. 185, 315–323 [DOI] [PubMed] [Google Scholar]
- 73. Mortuza G. B., Haire L. F., Stevens A., Smerdon S. J., Stoye J. P., Taylor I. A. (2004) High-resolution structure of a retroviral capsid hexameric amino-terminal domain. Nature 431, 481–485 [DOI] [PubMed] [Google Scholar]
- 74. von Schwedler U. K., Stemmler T. L., Klishko V. Y., Li S., Albertine K. H., Davis D. R., Sundquist W. I. (1998) Proteolytic refolding of the HIV-1 capsid protein amino terminus facilitates viral core assembly. EMBO J. 17, 1555–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Li S., Hill C. P., Sundquist W. I., Finch J. T. (2000) Image reconstructions of helical assemblies of the HIV-1 CA protein. Nature 407, 409–413 [DOI] [PubMed] [Google Scholar]
- 76. Sticht J., Humbert M., Findlow S., Bodem J., Müller B., Dietrich U., Werner J., Kräusslich H. G. (2005) A peptide inhibitor of HIV-1 assembly in vitro. Nat. Struct. Mol. Biol. 12, 671–677 [DOI] [PubMed] [Google Scholar]
- 77. Ternois F., Sticht J., Duquerroy S., Kräusslich H. G., Rey F. A. (2005) The HIV-1 capsid protein C-terminal domain in complex with a virus assembly inhibitor. Nat. Struct. Mol. Biol. 12, 678–682 [DOI] [PubMed] [Google Scholar]
- 78. Kelly B. N., Kyere S., Kinde I., Tang C., Howard B. R., Robinson H., Sundquist W. I., Summers M. F., Hill C. P. (2007) Structure of the antiviral assembly inhibitor CAP-1 complex with the HIV-1 CA protein. J. Mol. Biol. 373, 355–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Tang C., Loeliger E., Kinde I., Kyere S., Mayo K., Barklis E., Sun Y., Huang M., Summers M. F. (2003) Antiviral inhibition of the HIV-1 capsid protein. J. Mol. Biol. 327, 1013–1020 [DOI] [PubMed] [Google Scholar]
- 80. Blair W. S., Pickford C., Irving S. L., Brown D. G., Anderson M., Bazin R., Cao J., Ciaramella G., Isaacson J., Jackson L., Hunt R., Kjerrstrom A., Nieman J. A., Patick A. K., Perros M., Scott A. D., Whitby K., Wu H., Butler S. L. (2010) HIV capsid is a tractable target for small molecule therapeutic intervention. PLoS Pathog. 6, e1001220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Shi J., Zhou J., Shah V. B., Aiken C., Whitby K. (2011) Small-molecule inhibition of human immunodeficiency virus type 1 infection by virus capsid destabilization. J. Virol. 85, 542–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lemke C. T., Titolo S., von Schwedler U., Goudreau N., Mercier J. F., Wardrop E., Faucher A. M., Coulombe R., Banik S. S., Fader L., Gagnon A., Kawai S. H., Rancourt J., Tremblay M., Yoakim C., Simoneau B., Archambault J., Sundquist W. I., Mason S. W. (2012) Distinct effects of two HIV-1 capsid assembly inhibitor families that bind the same site within the N-terminal domain of the viral CA protein. J. Virol. 86, 6643–6655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Pettit S. C., Henderson G. J., Schiffer C. A., Swanstrom R. (2002) Replacement of the P1 amino acid of human immunodeficiency virus type 1 Gag processing sites can inhibit or enhance the rate of cleavage by the viral protease. J. Virol. 76, 10226–10233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Coren L. V., Thomas J. A., Chertova E., Sowder R. C., 2nd, Gagliardi T. D., Gorelick R. J., Ott D. E. (2007) Mutational analysis of the C-terminal Gag cleavage sites in human immunodeficiency virus type 1. J. Virol. 81, 10047–10054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Li F., Goila-Gaur R., Salzwedel K., Kilgore N. R., Reddick M., Matallana C., Castillo A., Zoumplis D., Martin D. E., Orenstein J. M., Allaway G. P., Freed E. O., Wild C. T. (2003) PA-457: a potent HIV inhibitor that disrupts core condensation by targeting a late step in Gag processing. Proc. Natl. Acad. Sci. U.S.A. 100, 13555–13560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Zhou J., Yuan X., Dismuke D., Forshey B. M., Lundquist C., Lee K. H., Aiken C., Chen C. H. (2004) Small-molecule inhibition of human immunodeficiency virus type 1 replication by specific targeting of the final step of virion maturation. J. Virol. 78, 922–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Keller P. W., Adamson C. S., Heymann J. B., Freed E. O., Steven A. C. (2011) HIV-1 maturation inhibitor bevirimat stabilizes the immature Gag lattice. J. Virol. 85, 1420–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Zhou J., Huang L., Hachey D. L., Chen C. H., Aiken C. (2005) Inhibition of HIV-1 maturation via drug association with the viral Gag protein in immature HIV-1 particles. J. Biol. Chem. 280, 42149–42155 [DOI] [PubMed] [Google Scholar]
- 89. Nguyen A. T., Feasley C. L., Jackson K. W., Nitz T. J., Salzwedel K., Air G. M., Sakalian M. (2011) The prototype HIV-1 maturation inhibitor, bevirimat, binds to the CA-SP1 cleavage site in immature Gag particles. Retrovirology 8, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Checkley M. A., Luttge B. G., Soheilian F., Nagashima K., Freed E. O. (2010) The capsid-spacer peptide 1 Gag processing intermediate is a dominant-negative inhibitor of HIV-1 maturation. Virology 400, 137–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Rulli S. J., Jr., Muriaux D., Nagashima K., Mirro J., Oshima M., Baumann J. G., Rein A. (2006) Mutant murine leukemia virus Gag proteins lacking proline at the N terminus of the capsid domain block infectivity in virions containing wild-type Gag. Virology 347, 364–371 [DOI] [PubMed] [Google Scholar]
- 92. Fu W., Gorelick R. J., Rein A. (1994) Characterization of human immunodeficiency virus type 1 dimeric RNA from wild-type and protease-defective virions. J. Virol. 68, 5013–5018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Shehu-Xhilaga M., Kraeusslich H. G., Pettit S., Swanstrom R., Lee J. Y., Marshall J. A., Crowe S. M., Mak J. (2001) Proteolytic processing of the p2/nucleocapsid cleavage site is critical for human immunodeficiency virus type 1 RNA dimer maturation. J. Virol. 75, 9156–9164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Thomas J. A., Gorelick R. J. (2008) Nucleocapsid protein function in early infection processes. Virus Res. 134, 39–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Sheng N., Erickson-Viitanen S. (1994) Cleavage of p15 protein in vitro by human immunodeficiency virus type 1 protease is RNA-dependent. J. Virol. 68, 6207–6214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Sheng N., Pettit S. C., Tritch R. J., Ozturk D. H., Rayner M. M., Swanstrom R., Erickson-Viitanen S. (1997) Determinants of the human immunodeficiency virus type 1 p15NC-RNA interaction that affect enhanced cleavage by the viral protease. J. Virol. 71, 5723–5732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Breuer S., Chang M. W., Yuan J., Torbett B. E. (2012) Identification of HIV-1 inhibitors targeting the nucleocapsid protein. J. Med. Chem. 55, 4968–4977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Breuer S., Sepulveda H., Chen Y., Trotter J., Torbett B. E. (2011) A cleavage enzyme-cytometric bead array provides biochemical profiling of resistance mutations in HIV-1 Gag and protease. Biochemistry 50, 4371–4381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Hermle J., Anders M., Heuser A. M., Müller B. (2010) A simple fluorescence based assay for quantification of human immunodeficiency virus particle release. BMC Biotechnol. 10, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Vogt V. M., Eisenman R. (1973) Identification of a large polypeptide precursor of avian oncornavirus proteins. Proc. Natl. Acad. Sci. U.S.A. 70, 1734–1738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. King N. M., Prabu-Jeyabalan M., Nalivaika E. A., Schiffer C. (2004) Combating susceptibility to drug resistance: lessons from HIV-1 protease. Chem. Biol. 11, 1333–1338 [DOI] [PubMed] [Google Scholar]