

Background: Group A Streptococcus (GAS) produces streptokinase (Ska), which activates plasminogen to plasmin while binding the host enzyme to the bacterial surface.
Results: Ska-activated plasmin can degrade human cathelicidin LL-37, promoting GAS resistance to the antimicrobial peptide.
Conclusion: Ska contributes to GAS innate immune evasion and virulence.
Significance: A human pathogen can co-opt the activity of a host protease to subvert peptide-based innate immune defense.
Keywords: Antimicrobial Peptides, Bacterial Pathogenesis, Innate Immunity, Plasmin, Streptococcus pyogenes, Cathelicidin, Virulence Factor
Abstract
The bacterial pathogen Group A Streptococcus (GAS) colonizes epithelial and mucosal surfaces and can cause a broad spectrum of human disease. Through the secreted plasminogen activator streptokinase (Ska), GAS activates human plasminogen into plasmin and binds it to the bacterial surface. The resulting surface plasmin protease activity has been proposed to play a role in disrupting tissue barriers, promoting invasive spread of the bacterium. We investigated whether this surface protease activity could aid the immune evasion role through degradation of the key innate antimicrobial peptide LL-37, the human cathelicidin. Cleavage products of plasmin-degraded LL-37 were analyzed by matrix-assisted laser desorption ionization mass spectrometry. Ska-deficient GAS strains were generated by targeted allelic exchange mutagenesis and confirmed to lack surface plasmin activity after growth in human plasma or media supplemented with plasminogen and fibrinogen. Loss of surface plasmin activity left GAS unable to efficiently degrade LL-37 and increased bacterial susceptibility to killing by the antimicrobial peptide. When mice infected with GAS were simultaneously treated with the plasmin inhibitor aprotinin, a significant reduction in the size of necrotic skin lesions was observed. Together these data reveal a novel immune evasion strategy of the human pathogen: co-opting the activity of a host protease to evade peptide-based innate host defenses.
Introduction
Group A Streptococcus (GAS,4 Streptococcus pyogenes) is a Gram-positive, human-specific pathogen responsible for over 500,000 deaths each year worldwide (1). Recently, mutations in the two-component regulatory system covR/S in strains of the globally disseminated GAS M1T1 serotype have been associated with invasive human infections and increased virulence in murine models of infection (2–4). This mutation arises spontaneously in vivo in the murine model of GAS M1T1 necrotizing fasciitis, leading to loss of expression of the cysteine protease SpeB and increased expression of hyaluronic acid capsule and other virulence determinants (2, 3). One of the characteristics of this hypervirulent animal-passaged (AP) variant is increased ability to acquire surface plasmin activity through the secreted GAS plasmin activator streptokinase (Ska) (3, 5).
Plasmin is a broad-spectrum host protease produced in the liver that is involved in fibrinolysis. It is expressed initially in a zymogen form, plasminogen, present in plasma at concentrations of ∼1–2 μm (6). Activation of plasminogen to plasmin is a process that is tightly controlled in the host involving plasminogen activators, tissue-type plasminogen activator and urokinase-type plasminogen activator, plasminogen activator inhibitors, PAI-1 and PAI-2, and plasmin inhibitors α2-antiplasmin and α2-macroglubulin (7). GAS, however, is able to circumvent the host regulation of plasminogen activation through the streptococcal secreted plasminogen activator, Ska (8).
Ska activates plasminogen in a manner not regulated by PAI-1 or PAI-2, nor inhibited by host factors including α2-antiplasmin (7). A Ska-plasminogen complex or Ska-fibrinogen-plasminogen complex results in an active protease that is bound to the bacterial surface through plasminogen and fibrinogen receptors (8). In the absence of the broad-spectrum secreted GAS protease SpeB, active plasmin accumulates, resulting in bacterial surface protease activity that may enable the bacteria to degrade tissue barriers, facilitating invasive spread of the pathogen (5, 9, 10).
We hypothesized that the ability of GAS to co-opt from the host broad-spectrum protease activity on its own surface may provide the pathogen additional virulence properties. A critical element of host innate defense at the initial focus of infection is the activity of cathelicidin antimicrobial peptide (AMP). Produced by epithelial cells such as keratinocytes and immune cells including neutrophils in response to injury or infection, cathelicidin exerts direct antibacterial activity and stimulates other components of the innate immune response (11, 12); loss of cathelicidin renders animals highly susceptible to invasive infection with GAS (13) and other bacterial pathogens (14, 15). In this study, we investigate the effect of surface plasmin accumulation on GAS susceptibility to cathelicidin antimicrobial peptides.
EXPERIMENTAL PROCEDURES
Bacterial Strains, Media, and Growth Conditions
The well characterized AP SpeB-negative, covS mutant M1T1 strain 5448AP and its parental strain 5448 were used in this study (3). Escherichia coli strains MC1061 and TOP10 were used for cloning. GAS strains were grown in Todd-Hewitt broth (THB) or on Todd-Hewitt agar plates, and E. coli was grown in Luria-Bertani broth (LB) or on Luria-Bertani agar plates. For antibiotic selection, erythromycin was used at 5 μg/ml for GAS or 500 μg/ml for E. coli. Kanamycin was used at 50 μg/ml. Bacteria were routinely grown at 37 °C unless noted otherwise.
Allelic Exchange Mutagenesis
A precise, in-frame allelic replacement of the ska gene with cat encoding chloramphenicol transferase was created in GAS wild-type (WT) strain 5448 using established methods (16). Briefly, an 854-bp fragment upstream of ska was PCR-amplified with primers ska-upF (5′-TGTACCCGCAGTTACCTGATACC-3′) and ska-upRcat (5′-AGAAACCTCCTACTAAAAGTTAAG-3′), the latter containing a 30-bp 5′ overhang homologous to the 5′ end of cat, and an 853-bp fragment downstream of ska was amplified by primers ska-downFcat (5′-CCACGATCTTCTAAAATGATG-3′), containing a 30-bp 5′ overhang homologous to the 3′ end of cat, and ska-downR (5′-TGGCTACCAAGAACGCTTGATTG-3′). The upstream and downstream fragments were combined with the 650-bp cat gene in a fusion PCR reaction using primers ska-upF and ska-downR, creating an amplicon in which ska had been precisely replaced with cat. The resulting triple fragment was TA-cloned into pCR-2.1-TOPO (Invitrogen) and subsequently digested with restriction enzymes BamHI and XbaI, ligated into the temperature-sensitive vector pHY304 to generate the knock-out plasmid pskaKO, and then transformed into GAS strain 5448 by electroporation and transformants identified at the permissive temperature of 30 °C under erythromycin selection. Transformants were then grown at the nonpermissive temperature of 37 °C with erythromycin present to select for homologous recombination and integration of the plasmid into the genome. Single crossovers were confirmed by PCR analysis. Relaxation of the plasmid was carried out at 30 °C with no antibiotic. Screening for erythromycin-sensitive colonies was used to identify double crossover events, allelic replacement mutants were confirmed by PCR, and the strain was denoted 5448Δska.
To create an AP Ska knock-out, equivalent to the animal-passaged covR/S mutant 5448AP, the knock-out strain 5448Δska was passaged in mice. 10-week-old female CD1 mice were injected subcutaneously with 1 × 108 colony-forming units (cfu) of 5448Δska. At 72 h after infection, mice were sacrificed, lesions were homogenized, and bacteria were plated on Todd-Hewitt agar. Individual colonies were screened for cysteine protease activity to detect SpeB using the azocasein assay described previously (17). SpeB-negative colonies were used for covR/S PCR and sequencing as described previously (18). A covR/S mutant was identified containing a duplication of nucleotides 1056–1078 resulting in a premature stop codon after amino acid 369. This strain was subsequently used in this study and designated 5448APΔska. Although additional mutations during animal passage cannot be ruled out entirely, we ensured that the key phenotypic changes under examination were reversed upon plasmid-based complementation. Complementation was performed by transforming 5448APΔska with the plasmid pDCerm containing the ska gene, creating the resultant strain, 5448APΔska[pSka]. All experiments involving the complemented strain were performed using 5448AP and 5448APΔska transformed with the empty vector pDCerm.
Plasminogen Activation and Cathelicidin Cleavage
Plasminogen was activated by combining 2 μg of group C streptococcal Ska (Sigma) with 2 μg of human plasminogen (Sigma) in 20 μl of PBS; 2 μg of human plasmin (Sigma) without streptokinase was also incubated with LL-37. For cathelicidin degradation, 32 μm human LL-37 or CRAMP (American Peptide Co.) was added, and the reaction was incubated at 37 °C for the specified time. Reactions were stopped by the addition of 20 μg/ml aprotinin. Samples were run on 12% Bis-Tris NuPAGE gels (Invitrogen) with MES buffer as per the manufacturer's instructions or processed for mass spectrometry analysis.
MALDI Analysis of LL-37 Cleavage by Plasmin
Cleavage reaction samples at differential time points were prepared for MALDI-TOF analysis by mixing a 1:1 ratio with Sigma-Aldrich universal MALDI matrix resuspended in 78% acetonitrile and 0.1% trifluoroacetic acid. 1 μl of each mixed sample was plated on a Bruker MALDI target plate for analysis. The Bruker microflex MALDI-TOF was calibrated at 25 ppm mass error with the Bruker peptide calibration standard. After calibration, each spot was analyzed using reflectron positive ionization and a laser power of 60%. Masses that corresponded to truncated forms of LL-37 and were absent in the control samples were noted. Select LL-37 cleavage ions were further confirmed by tandem mass spectrometry for identification purposes.
Ska Western Blot
Bacterial cultures were grown to mid-log phase (A600 = 0.4), and bacteria were pelleted by centrifugation for 10 min at 3200 × g. Supernatants were run on 10% Bis-Tris NuPAGE gels (Invitrogen) in MOPS buffer as per the manufacturer's instructions. Gels were transferred to nitrocellulose membranes (Invitrogen) using a Trans-Blot SD semi-dry transfer cell (Bio-Rad) as per the manufacturer's instructions. Membranes were blocked overnight with 5% skim milk in PBS + 0.05% Tween 20 (PBST). Membranes were incubated with polyclonal rabbit-anti-Ska antibody (5) at 1:1000 in PBST + 0.5% skim milk for 1.5 h, washed with PBST, and incubated with goat-anti-rabbit-HRP conjugate (Bio-Rad) at 1:10,000 in PBST + 0.5% skim milk for 1 h. The membrane was subsequently washed and developed using SuperSignal West Pico chemiluminescent substrate (Thermo Scientific).
Secreted Streptokinase Activity Assay
Bacterial cultures were grown to mid-log phase (A600 = 0.4), pelleted as described above, and filter-sterilized through 0.22 μm syringe-driven filters. 90 μl of sterile supernatant was mixed 1:1 with 1 unit/ml human plasminogen (Calbiochem) and 7 μm human fibrinogen (Calbiochem) in THB in flat bottom 96-well plates (Costar). 20 μl of 3 μm S-2251 (Chromogenix) was added, and the plates were incubated for 1 h at 37 °C. Absorbance was then read at 405 nm. 1 μg of group C Ska (Sigma) was used as a positive control. Assays were performed in triplicate, and the data are presented as mean ± S.D.
Blood Collection and Plasma Preparation
Blood was collected from healthy volunteers under informed consent by venipuncture with heparin used as the anticoagulant. Plasma was obtained by centrifuged for 5 min at 1000 × g. The supernatant was subsequently centrifuged for 5 min at 3000 × g.
Surface Plasmin Activity Assay
Bacterial cultures were grown to mid-log phase (A600 = 0.4) in THB + 50% fresh human plasma, THB supplemented with 1 unit/ml human plasminogen (Calbiochem) and 7 μm human fibrinogen (Calbiochem), or THB only. Bacteria were pelleted by centrifugation for 5 min at 7000 × g. Bacterial pellets were washed with PBS and resuspended in 0.1 volume of sterile PBS. 20 μl of S-2251 (Chromogenix) was added to 180 μl of bacteria in flat bottom 96-well plates (Costar) and incubated for 4 h at 37 °C. Absorbance was read at 405 nm. Assays were performed in triplicate, and the data are presented as mean ± S.D.
Degradation of LL-37 in Bacterial Supernatants
Bacteria were grown to mid-log phase (A600 = 0.4) in THB supplemented with plasminogen and fibrinogen as described above. The presence of plasmin activity at the bacterial surface was confirmed as described above. Bacteria were incubated with 10 μg of LL-37 at 37 °C for 1 h. Bacteria were then pelleted as above, and the supernatant was collected to screen for LL-37. Prior to SDS-PAGE and Western blot analysis, supernatants were concentrated by trichloroacetic acid (TCA) precipitation. An equal volume of 10% TCA was added to supernatants and incubated on ice for 20 min. Following centrifugation at 7000 × g, the protein pellet was washed with 100% ethanol and allowed to air dry. The protein pellet was then resuspended in 40 μl of 100 mm Tris-HCl (pH 7.6) and electrophoresed as described above. Proteins were transferred to nitrocellulose membrane using a Bio-Rad Trans-Blot apparatus (Bio-Rad). Following protein transfer, membranes were blocked for 1 h at room temperature with PBS, 1% skim milk. Membranes were then incubated with goat-anti-LL-37 (Eurogenetics, Tessenderlo, Belgium) diluted 1 in 500 in PBS, 1% skim milk) followed by rabbit anti-goat IgG HRP diluted 1 in 1000 in PBS, 1% skim milk. Excess secondary antibody was removed by three PBS washes for 10 min each, and all blots were developed in a solution of 100 mm Tris-HCl (pH 7.6) containing 1.4 mm diaminobenzidine and 0.06% (v/v) hydrogen peroxide.
Surface Degradation of LL-37
Bacteria were grown to mid-log phase (A600 = 0.4) in THB supplemented with plasminogen and fibrinogen as described above. Bacteria were centrifuged for 5 min at 7000 × g and resuspended in 0.1 volume of sterile PBS. Bacteria were incubated at 37 °C, and reactions were stopped at the desired time points by the addition of aprotinin at a final concentration of 20 μg/ml. Bacteria were then pelleted as above and resuspended in 100 mm glycine (pH 2.0) to elute proteins bound to the bacterial surface. After a 30-min incubation at room temperature, 0.1 volume of 1.0 m Tris-HCl (pH 9.0) was added to neutralize the solution. Bacteria were pelleted as above, and supernatants were run on 12% Bis-Tris NuPAGE gels (Invitrogen) in MES buffer as per the manufacturer's instructions.
LL-37 Minimum Inhibitory Concentration Testing and Killing Kinetic Assay
Bacterial cultures were grown to mid-log phase (A600 = 0.4) in PBS + 50% THB supplemented with 1 unit/ml human plasminogen (Calbiochem) and 7 μm human fibrinogen (Calbiochem). Bacteria were then diluted 1:1000 in the same media and added to LL-37 (American Peptide Co.) at concentrations of 0–8 μm in 1 μm increments in flat bottom 96-well plates (Costar) and incubated for 24 h at 37 °C. Aprotinin was added to relevant cultures at a final concentration of 2 μg/ml 10 min prior to addition of LL-37. Minimum inhibitory concentration was determined as the lowest concentration at which no growth was observed by absorbance at 600 nm. Assays were performed in triplicate, and where there was variation between replicates, the data are presented as a range. For killing kinetics, bacteria were prepared as above, and LL-37 was added at a final concentration of 4 μm in siliconized tubes. Tubes were incubated at 37 °C with rotation, and aliquots were taken at the specified time points and serially diluted for enumeration of cfu. Statistical significance was determined by Student's t test at each time point. Assays were performed in triplicate, and the data are presented as mean ± S.D.
Murine Infection Model
Bacteria were grown to mid-log phase in THB supplemented with human plasminogen and fibrinogen as described above. Bacteria were then washed with sterile PBS and resuspended to 1 × 107 cfu/ml in PBS containing 1 unit/ml human plasminogen (Calbiochem) and 7 μm human fibrinogen (Calbiochem) with or without 20 μg/ml aprotinin. 1 × 106 cfu in 100 μl volume was injected into the flank of 10-week-old female CD1 mice. Pictures of mice were taken daily, and lesion area was calculated using ImageJ software. Statistical significance was determined by two-way analysis of variance. Each data point represents one lesion, n = 9 for each group.
Ethics Approvals
Permission to obtain human blood and undertake animal experiments was obtained from the University of California, San Diego. Human volunteers provided informed consent before blood samples were obtained.
RESULTS
Ska-activated Plasmin Degrades LL-37
Ska-activated plasmin as well as purified human plasmin was observed to degrade the human cathelicidin AMP, LL-37 (Fig. 1A). No degradation of LL-37 was observed in the absence of either Ska or plasmin. A previous study of LL-37 degradation by cutaneous host proteases (e.g. kallikreins) identified a range of degradation products, some retaining and some losing antimicrobial activity (19). To elucidate the plasmin cleavage products of LL-37, we incubated Ska-activated plasmin with purified LL-37 for varying times between 0 and 120 min (Fig. 1B). Reactions were then analyzed by MALDI-TOF mass spectrometry. Degradation products of LL-37 were observed beginning at as early as 1 min of incubation time (Fig. 1C). Previously described inactive fragments LLGDFFR and IKDFLRNLVPRTES were identified, as well as a fragment with known antimicrobial activity, KSKEKIGKEFKRIVQRIKDFLRNLVPRTES, (19) which was present at 1 min of incubation time but was not detectable after 120 min. The previously undescribed fragments RIVQRIKDFLRNLVPRTES and DFLRNLVPRTES were also observed and still detectable after 120 min. Because the exact contribution of a dynamic mixture of degradation products to antimicrobial activity over time was uncertain, further analysis of the interaction between plasmin-accumulating bacteria and LL-37 was needed.
FIGURE 1.

Plasmin degradation of human cathelicidin, LL-37. A, SDS-PAGE gel showing incubation of human plasminogen (hPLG), Ska, and purified LL-37. Human plasminogen (PLN) without streptokinase is included as a control. Arrow indicates the size of full-length LL-37. B, SDS-PAGE gel showing the time course of Ska-activated plasmin incubated with LL-37. Ctrl, control. C, MS analysis of LL-37 cleavage products shown in panel B. Asterisks indicate [M+H]+ of LL-37 with an m/z of 4492 Da and [M+2H]2+ of LL-37 with an m/z 2247 Da. Numbers correspond to fragments identified in panel D. a. u., arbitrary units. D, LL-37 fragments identified by MS analysis.
Characterization of Ska Knock-out GAS
To study the role of plasmin in GAS resistance to cathelicidin antimicrobial peptides, we created a precise, in-frame allelic replacement knock-out of Ska in hypervirulent, SpeB-negative, covS mutant GAS generated by animal passage and characteristic of the human invasive GAS M1T1 genotype/phenotype (2–4). Western blot analysis showed the presence of Ska in the supernatants from WT strain 5448AP, but not from the knock-out strain (5448APΔska), from here on referred to as WT and Δska, respectively (Fig. 2A). Empty vector controls, containing the plasmid pDCerm, showed similar levels of expression to their parental strains. Ska expression was restored in the plasmid-complemented strain, Δska[pSka]. Ska activity was observed in the supernatants of WT GAS (Fig. 2B), and the WT strain accumulated surface plasmin activity when grown in fresh human plasma or in THB supplemented with human plasminogen and fibrinogen (Fig. 2C). To avoid the possible confounding influence of other plasma proteins, subsequent experiments were performed in THB supplemented with human plasminogen and fibrinogen.
FIGURE 2.

LL-37 degradation by plasmin on the bacterial surface. A, Western blot for Ska in culture supernatants. B, Ska activity assay from culture supernatants. Values shown are mean ± S.D. hPLG, human plasminogen. C, plasmin activity assay on washed bacteria after growth in 50% human plasma, THB supplemented with human plasminogen and fibrinogen (+Plg/Fn), or THB without supplementation (−Plg/Fn). Values shown are mean ± S.D. D, Western blot showing degradation of LL-37 in supernatants after incubation with bacteria grown in THB supplemented with human plasminogen and fibrinogen. Ctrl, control. E, SDS-PAGE gel showing LL-37 eluted from the surface of bacteria grown in THB supplemented with human plasminogen and fibrinogen.
Surface-associated Plasmin Degrades LL-37
After allowing bacteria to accumulate surface plasmin activity by growth in the presence of human plasminogen and fibrinogen, we demonstrated that these bacteria are able to degrade LL-37 in a manner dependent on the expression of Ska (Fig. 2D). Because LL-37 targets the bacterial cell wall to exert its antimicrobial activity, we further investigated whether surface plasmin could degrade LL-37 peptide associated with the bacterial surface. Bacteria allowed to accumulate surface plasmin activity were incubated with LL-37, and after washing away unbound LL-37, a low pH buffer was used to elute surface-bound LL-37. Although Ska-expressing WT GAS showed reduced levels of intact LL-37 on their surface over time, the level of LL-37 bound to Δska mutant bacteria remained relatively constant (Fig. 2E). Although it is possible that other factors may play a role in degradation of LL-37 over this time period, it is apparent from the data presented in Fig. 2E that surface-acquired plasmin is the major contributor in this assay.
Surface-associated Plasmin Increases Resistance to LL-37
To determine the physiological relevance of GAS surface plasmin cleavage of cathelicidin, we conducted LL-37 sensitivity testing. After growth in medium supplemented with plasminogen and fibrinogen to allow accumulation of surface plasmin activity, Ska-expressing WT GAS exhibited increased minimum inhibitory concentrations for LL-37 when compared with the Δska mutant (Fig. 3A). Ska-expressing WT bacteria also had significantly increased survival versus the isogenic Δska mutant in an LL-37 kinetic killing assay (Fig. 3B). These findings show that accumulation of surface plasmin, activated by Ska, provides GAS with increased resistance to LL-37 cathelicidin antimicrobial peptides.
FIGURE 3.

Surface plasmin activity increases resistance to LL-37. A, LL-37 minimum inhibitory concentration (MIC) assay on bacteria grown in media supplemented with human plasminogen and fibrinogen. B, LL-37 killing kinetics assay on bacteria grown in THB supplemented with human plasminogen and fibrinogen. Values in all panels represent mean ± S.D.; * indicates p < 0.05.
Plasmin Inhibition Reduces Virulence in Vivo
To study the effect of plasmin inhibition in vivo, we utilized a murine model of necrotizing fasciitis in which cathelicidin antimicrobial peptides have been shown to be abundantly expressed (20). Ska-activated plasmin was confirmed to degrade the murine cathelicidin, CRAMP, in vitro (Fig. 4A). Because Ska activity is specific to human plasminogen and not murine plasminogen, GAS strains were allowed to accumulate surface plasmin activity during growth, and bacteria were co-administered with human plasminogen and fibrinogen, with or without the addition of the plasmin inhibitor aprotinin. Significantly smaller lesions were observed when plasmin acquisition was absent, either in the Ska-knock-out strain or with the addition of aprotinin (Fig. 4B), demonstrating a potential therapeutic benefit of plasmin inhibition in restricting GAS proliferation.
FIGURE 4.
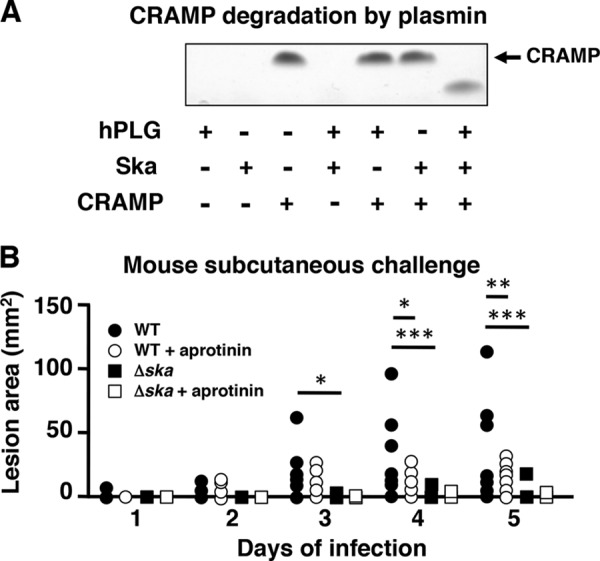
Inhibition of plasmin reduces virulence in vivo. A, SDS-PAGE gel showing incubation of human plasminogen (hPLG), Ska, and purified murine cathelicidin, CRAMP. Arrow indicates the size of full-length CRAMP. B, lesion size of mice following subcutaneous infection with GAS strains grown in media supplemented with human plasminogen and fibrinogen, co-administered with or without the addition of aprotinin. Values represent mean ± S.D.; * indicates p < 0.05, ** indicates p < 0.01, *** indicates p < 0.001.
DISCUSSION
The ability of GAS to activate plasmin through Ska and bind the active protease to the bacterial surface has been known for some time (8, 9), with potential contributions to tissue barrier disruption and invasive spread of the bacterium (5, 9). In this study, we demonstrate the ability of surface-acquired plasmin activity to degrade host cathelicidin antimicrobial peptides.
Cathelicidin peptides serve a critical front line role in mammalian innate immunity against invasive bacterial infection as first demonstrated in a murine model of GAS infection; cathelicidin-deficient mice are more susceptible to invasive GAS infection (13), and overexpression of an additional (porcine) cathelicidin in mice provides increased resistance (21). The invasive M1T1 clone of GAS demonstrates increased resistance to cathelicidin AMPs when compared with other GAS strains (22), which may in part be attributable to cathelicidin binding properties of the M1 protein hypervariable domain (22) or protein SIC (streptococcal inhibitor of complement) uniquely expressed only in M1 and M57 strains (23). Upon selection of the covS mutations associated with the shift to invasive infection (2, 4), GAS increases expression of the hyaluronic acid capsule, which can impair cathelicidin killing, (17) and accumulates more plasmin activity on its surface (5). Here we show that the co-optation of this host protease activity represents another important cathelicidin resistance phenotype of the globally disseminated hyperinvasive M1T1 GAS clone.
Plasmin is an important host factor involved in fibrinolysis and tissue remodeling following injury (24). Because of its potent proteolytic activity, plasmin activation is tightly regulated under normal conditions (6). GAS-secreted Ska is able to activate plasmin independent of host plasmin activators (8), creating the opportunity for active plasmin to function in environments that are not present in an uninfected host.
Cathelicidin has previously been shown to influence the expression of Ska through the two-component regulatory system covR/S (25); however, no relationship between Ska expression and resistance to cathelicidin has been shown. The genetically unrelated plasmin activator of Staphylococcus aureus, staphylokinase, can increase resistance to human defensins through direct binding of the AMP by staphylokinase, independent of its plasmin-activating capacity (26). In contrast to these findings, here we showed that Ska-activated plasmin increases resistance to cathelicidin antimicrobial peptides by virtue of its plasmin protease activity. Interestingly, cathelicidin has been shown to increase staphylokinase-dependent fibrinolysis by plasmin (27). It is not known whether staphylokinase activation of plasmin results in increased resistance of S. aureus to cathelicidin or whether cathelicidin can influence the fibrinolytic capacity of Ska-activated plasmin.
We demonstrated the therapeutic potential of plasmin inhibition for reducing the severity of GAS subcutaneous infections. Blocking the ability of GAS to acquire Ska-activated plasmin activity on its surface, either by genetic manipulation of Ska or by inhibition of plasmin activity with aprotinin, significantly reduced virulence in a murine infection model. Pharmacological inhibition of Ska in combination with other virulence determinants has recently been shown to reduce GAS virulence in mice (28). Our data have shown that inhibition of plasmin protease activity (the critical effector mechanism of Ska) is sufficient to reduce GAS virulence in a murine skin infection model.
In summary, these data present a new look at a GAS virulence factor. In addition to providing GAS with an ability to spread through tissue barriers, Ska also provides the bacterium with an immune evasion tool. Although there is no current inhibitor of Ska in clinical development, plasmin protease activity itself provides a valid target for novel GAS therapeutics.
This work was supported, in whole or in part, by National Institutes of Health Grants AI077780 (to V. N.), AI052453 (to R. L. G. and V. N.), and AR052728 (to R. L. G. and V. N.). This work was also supported by National Health and Medical Research Council of Australia (NHMRC) Grant 635218 (to M. S.).
- GAS
- Group A Streptococcus
- AP
- animal-passaged
- Ska
- streptokinase
- AMP
- antimicrobial peptide
- CRAMP
- cathelicidin-related AMP
- PAI
- plasminogen activator inhibitor
- THB
- Todd-Hewitt broth
- Bis-Tris
- 2-(bis(2-hydroxyethyl)amino)-2-(hydroxymethyl)propane-1,3-diol.
REFERENCES
- 1. Carapetis J. R., Steer A. C., Mulholland E. K., Weber M. (2005) The global burden of group A streptococcal diseases. Lancet Infect. Dis. 5, 685–694 [DOI] [PubMed] [Google Scholar]
- 2. Sumby P., Whitney A. R., Graviss E. A., DeLeo F. R., Musser J. M. (2006) Genome-wide analysis of group A streptococci reveals a mutation that modulates global phenotype and disease specificity. PLoS Pathog. 2, e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Walker M. J., Hollands A., Sanderson-Smith M. L., Cole J. N., Kirk J. K., Henningham A., McArthur J. D., Dinkla K., Aziz R. K., Kansal R. G., Simpson A. J., Buchanan J. T., Chhatwal G. S., Kotb M., Nizet V. (2007) DNase Sda1 provides selection pressure for a switch to invasive group A streptococcal infection. Nat. Med. 13, 981–985 [DOI] [PubMed] [Google Scholar]
- 4. Cole J. N., Barnett T. C., Nizet V., Walker M. J. (2011) Molecular insight into invasive group A streptococcal disease. Nat. Rev. Microbiol. 9, 724–736 [DOI] [PubMed] [Google Scholar]
- 5. Cole J. N., McArthur J. D., McKay F. C., Sanderson-Smith M. L., Cork A. J., Ranson M., Rohde M., Itzek A., Sun H., Ginsburg D., Kotb M., Nizet V., Chhatwal G. S., Walker M. J. (2006) Trigger for group A streptococcal M1T1 invasive disease. FASEB J. 20, 1745–1747 [DOI] [PubMed] [Google Scholar]
- 6. Ponting C. P., Marshall J. M., Cederholm-Williams S. A. (1992) Plasminogen: a structural review. Blood Coagul. Fibrinolysis 3, 605–614 [PubMed] [Google Scholar]
- 7. Parry M. A., Zhang X. C., Bode I. (2000) Molecular mechanisms of plasminogen activation: bacterial cofactors provide clues. Trends Biochem. Sci. 25, 53–59 [DOI] [PubMed] [Google Scholar]
- 8. Walker M. J., McArthur J. D., McKay F., Ranson M. (2005) Is plasminogen deployed as a Streptococcus pyogenes virulence factor? Trends Microbiol. 13, 308–313 [DOI] [PubMed] [Google Scholar]
- 9. Lottenberg R., Minning-Wenz D., Boyle M. D. (1994) Capturing host plasmin(ogen): a common mechanism for invasive pathogens? Trends Microbiol. 2, 20–24 [DOI] [PubMed] [Google Scholar]
- 10. Sanderson-Smith M. L., Dinkla K., Cole J. N., Cork A. J., Maamary P. G., McArthur J. D., Chhatwal G. S., Walker M. J. (2008) M protein-mediated plasminogen binding is essential for the virulence of an invasive Streptococcus pyogenes isolate. FASEB J. 22, 2715–2722 [DOI] [PubMed] [Google Scholar]
- 11. Dürr U. H., Sudheendra U. S., Ramamoorthy A. (2006) LL-37, the only human member of the cathelicidin family of antimicrobial peptides. Biochim. Biophys. Acta 1758, 1408–1425 [DOI] [PubMed] [Google Scholar]
- 12. Zanetti M. (2004) Cathelicidins, multifunctional peptides of the innate immunity. J. Leukoc. Biol. 75, 39–48 [DOI] [PubMed] [Google Scholar]
- 13. Nizet V., Ohtake T., Lauth X., Trowbridge J., Rudisill J., Dorschner R. A., Pestonjamasp V., Piraino J., Huttner K., Gallo R. L. (2001) Innate antimicrobial peptide protects the skin from invasive bacterial infection. Nature 414, 454–457 [DOI] [PubMed] [Google Scholar]
- 14. Bergman P., Johansson L., Wan H., Jones A., Gallo R. L., Gudmundsson G. H., Hökfelt T., Jonsson A. B., Agerberth B. (2006) Induction of the antimicrobial peptide CRAMP in the blood-brain barrier and meninges after meningococcal infection. Infect. Immun. 74, 6982–6991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chromek M., Slamová Z., Bergman P., Kovács L., Podracká L., Ehrén I., Hökfelt T., Gudmundsson G. H., Gallo R. L., Agerberth B., Brauner A. (2006) The antimicrobial peptide cathelicidin protects the urinary tract against invasive bacterial infection. Nat. Med. 12, 636–641 [DOI] [PubMed] [Google Scholar]
- 16. Buchanan J. T., Simpson A. J., Aziz R. K., Liu G. Y., Kristian S. A., Kotb M., Feramisco J., Nizet V. (2006) DNase expression allows the pathogen group A Streptococcus to escape killing in neutrophil extracellular traps. Curr. Biol. 16, 396–400 [DOI] [PubMed] [Google Scholar]
- 17. Cole J. N., Pence M. A., von Köckritz-Blickwede M., Hollands A., Gallo R. L., Walker M. J., Nizet V. (2010) M protein and hyaluronic acid capsule are essential for in vivo selection of covRS mutations characteristic of invasive serotype M1T1 group A Streptococcus. MBio 1, e00191–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hollands A., Aziz R. K., Kansal R., Kotb M., Nizet V., Walker M. J. (2008) A naturally occurring mutation in ropB suppresses SpeB expression and reduces M1T1 group A streptococcal systemic virulence. PloS One 3, e4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yamasaki K., Schauber J., Coda A., Lin H., Dorschner R. A., Schechter N. M., Bonnart C., Descargues P., Hovnanian A., Gallo R. L. (2006) Kallikrein-mediated proteolysis regulates the antimicrobial effects of cathelicidins in skin. FASEB J. 20, 2068–2080 [DOI] [PubMed] [Google Scholar]
- 20. Dorschner R. A., Pestonjamasp V. K., Tamakuwala S., Ohtake T., Rudisill J., Nizet V., Agerberth B., Gudmundsson G. H., Gallo R. L. (2001) Cutaneous injury induces the release of cathelicidin anti-microbial peptides active against group A Streptococcus. J. Invest. Dermatol. 117, 91–97 [DOI] [PubMed] [Google Scholar]
- 21. Lee P. H., Ohtake T., Zaiou M., Murakami M., Rudisill J. A., Lin K. H., Gallo R. L. (2005) Expression of an additional cathelicidin antimicrobial peptide protects against bacterial skin infection. Proc. Natl. Acad. Sci. U.S.A. 102, 3750–3755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lauth X., von Köckritz-Blickwede M., McNamara C. W., Myskowski S., Zinkernagel A. S., Beall B., Ghosh P., Gallo R. L., Nizet V. (2009) M1 protein allows Group A streptococcal survival in phagocyte extracellular traps through cathelicidin inhibition. J. Innate Immun. 1, 202–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pence M. A., Rooijakkers S. H., Cogen A. L., Cole J. N., Hollands A., Gallo R. L., Nizet V. (2010) Streptococcal inhibitor of complement promotes innate immune resistance phenotypes of invasive M1T1 group A Streptococcus. J. Innate Immun. 2, 587–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Collen D. (1999) The plasminogen (fibrinolytic) system. Thromb. Haemost. 82, 259–270 [PubMed] [Google Scholar]
- 25. Gryllos I., Tran-Winkler H. J., Cheng M. F., Chung H., Bolcome R., 3rd, Lu W., Lehrer R. I., Wessels M. R. (2008) Induction of group A Streptococcus virulence by a human antimicrobial peptide. Proc. Natl. Acad. Sci. U.S.A. 105, 16755–16760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jin T., Bokarewa M., Foster T., Mitchell J., Higgins J., Tarkowski A. (2004) Staphylococcus aureus resists human defensins by production of staphylokinase, a novel bacterial evasion mechanism. J. Immunol. 172, 1169–1176 [DOI] [PubMed] [Google Scholar]
- 27. Braff M. H., Jones A. L., Skerrett S. J., Rubens C. E. (2007) Staphylococcus aureus exploits cathelicidin antimicrobial peptides produced during early pneumonia to promote staphylokinase-dependent fibrinolysis. J. Infect Dis. 195, 1365–1372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sun H., Xu Y., Sitkiewicz I., Ma Y., Wang X., Yestrepsky B. D., Huang Y., Lapadatescu M. C., Larsen M. J., Larsen S. D., Musser J. M., Ginsburg D. (2012) Inhibitor of streptokinase gene expression improves survival after group A Streptococcus infection in mice. Proc. Natl. Acad. Sci. U.S.A. 109, 3469–3474 [DOI] [PMC free article] [PubMed] [Google Scholar]