

Background: Thiopurine prodrugs can result in the formation of S6-methylthioguanine (S6mG) and 6-thioguanine (SG) in DNA.
Results: We examined how SG and S6mG affect DNA transcription in vitro and in human cells.
Conclusion: S6mG, but not SG, causes a strong mutagenic and inhibitory effects on transcription.
Significance: This work provides new knowledge that S6mG-mediated transcriptional alternations might contribute to thiopurine-induced cytoxicity and potential therapy-related cancers.
Keywords: Anticancer Drug, DNA Damage, DNA Mismatch Repair, DNA Repair, Transcription, Transcription Coupled Repair
Abstract
Thiopurine drugs are extensively used as chemotherapeutic agents in clinical practice, even though there is concern about the risk of therapy-related cancers. It has been previously suggested that the cytotoxicity of thiopurine drugs involves their metabolic activation, the resultant generation of 6-thioguanine (SG) and S6-methylthioguanine (S6mG) in DNA, and the futile mismatch repair triggered by replication-induced SG:T and S6mG:T mispairs. Disruption of transcription is known to be one of the major consequences of DNA damage induced by many antiviral and antitumor agents; however, it remains undefined how SG and S6mG compromise the efficiency and fidelity of transcription. Using our recently developed competitive transcription and adduct bypass assay, herein we examined the impact of SG and S6mG on transcription in vitro and in human cells. Our results revealed that, when situated on the transcribed strand, S6mG exhibited both inhibitory and mutagenic effects during transcription mediated by single-subunit T7 RNA polymerase or multisubunit human RNA polymerase II in vitro and in human cells. Moreover, we found that the impact of S6mG on transcriptional efficiency and fidelity is modulated by the transcription-coupled nucleotide excision repair capacity. In contrast, SG did not considerably compromise the efficiency or fidelity of transcription, and it was a poor substrate for NER. We propose that S6mG might contribute, at least in part, to thiopurine-mediated cytotoxicity through inhibition of transcription and to potential therapy-related carcinogenesis via transcriptional mutagenesis.
Introduction
FDA-approved thiopurine drugs such as azathioprine, 6-mercaptopurine, and 6-thioguanine (SG)2 are among the most effective chemotherapeutic drugs in clinical practice. They are widely used as anti-inflammatory, anticancer, and immunosuppressive agents in the treatment of a variety of human diseases including childhood acute leukemia and inflammatory disorders, even though there is concern about therapy-induced malignancies resulting from long term thiopurine use (1–4). Despite their successful clinical applications for over half a century, the exact molecular mechanisms by which thiopurines exert their cytotoxic effects remain incompletely understood.
Thiopurines are prodrugs, and to achieve their efficacy and cytotoxicity, they need to be converted to active metabolites, i.e., SG nucleotides, and subsequently incorporated into DNA (2, 3, 5). The incorporation of SG nucleotides into DNA has been detected in several different types of mammalian cells and tissues after treatment with thiopurines, although the levels of DNA SG vary widely among different biological samples (6–10). SG in DNA has been implicated in the mitochondrial pathway of apoptosis by suppressing the activation of Rac1 GTPase through its binding to SGTP instead of GTP (11). In addition, DNA SG is known to perturb some other cellular pathways that involve global cytosine demethylation or the formation of oxidative damage products such as DNA interstrand cross-links and DNA-protein cross-links (6, 7, 12–15).
Postreplicative mismatch repair (MMR) system is also thought to play an important role in thiopurine-mediated cytotoxicity (3). MMR-deficient cells in culture are more resistant to thiopurine treatment than MMR-proficient cells (16–19). Some earlier studies have suggested that the toxicity of thiopurines involves methylation of the S6 position of SG in DNA by S-adenosyl-l-methionine to form S6-methylthioguanine (S6mG) (Fig. 1A), misincorporation of thymidine opposite the S6mG during DNA replication, and recognition of the resultant mispairs by the MMR system (8, 20). Our recent studies further revealed that S6mG is capable of inducing G → A mutations at frequencies of 94 and 39% in Escherichia coli and mammalian cells, respectively (8, 21–23). Additionally, replicative bypass of SG is also mildly mutagenic in these cells, and the resulting SG:T base pairs can be efficiently recognized by MMR proteins; thus, SG may also trigger the postreplicative MMR system and contribute to thiopurine toxicity (21, 22, 24, 25).
FIGURE 1.

Experimental outline. A, chemical structures of guanine (G), SG, and S6mG. B, strategy for assessing the impact of SG and S6mG on DNA transcription. X indicates an SG, S6mG, or guanine that was located on the transcribed strand of TurboGFP gene downstream of the CMV and T7 promoters. The +1 transcription start sites are indicated as arrows. Lesion-bearing or control plasmids were mixed with the competitor genome as DNA templates for in vitro or in vivo transcription. Although truncated RNA may be produced when transcription arrests at or near a lesion site, only run-off RNA is shown and used for RT-PCR. Among the RT-PCR products, only the wild-type sequence arising from the lesion-containing genome is shown. The arrows indicate the cleavage sites of SacI and FspI, which are used to digest the RT-PCR products for subsequent PAGE and LC-MS/MS analyses.
Disruption of transcription and related processes is considered as one of the major factors contributing to the cytotoxicity of potent antiviral and anticancer agents currently used in the clinic or in clinical trials (26–30). Many of these drugs interfere with transcription through chemical modifications of cellular DNA (26, 31). In this context, some drug-induced DNA damage may inhibit the initiation of RNA synthesis by altering the binding of some transcription factors to DNA, whereas others may act as a physical impediment to transcription elongation by RNA polymerase (RNAP) (26, 27, 31). The latter type of DNA lesions usually trigger transcription-coupled nucleotide excision repair (TC-NER) that preferentially removes lesions on the transcribed strand of DNA (32). On the other hand, transcriptional bypass of some DNA lesions may lead to the generation of mutant transcripts in a process called transcriptional mutagenesis (33, 34). A recent study revealed that SG in DNA is only marginally inhibitory to transcription in vitro (10). However, it remains undefined whether SG compromises transcription fidelity and how S6mG affects DNA transcription.
To address these questions, here we have examined quantitatively the impact of SG and S6mG on the transcriptional efficiency and accuracy in vitro and in vivo by using our recently developed competitive transcription and adduct bypass assay (35). We also investigated whether TC-NER is involved in the removal of these lesions in human cells.
EXPERIMENTAL PROCEDURES
Materials and Cell Culture Conditions
Unmodified oligodeoxyribonucleotides (ODNs), [γ-32P]ATP, and shrimp alkaline phosphatase were purchased from Integrated DNA Technologies, PerkinElmer Life Technologies, and USB Corporation, respectively. Chemicals and enzymes unless otherwise specified were obtained from Sigma-Aldrich and New England Biolabs, respectively. The 293T human embryonic kidney epithelial cells were purchased from ATCC. The human skin fibroblast cell lines GM00637 and GM04429 were kindly provided by Prof. Gerd P. Pfeifer (City of Hope). The cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (Invitrogen), 100 units/ml penicillin, and 100 μg/ml streptomycin (ATCC) and incubated at 37 °C in 5% CO2 atmosphere.
Transcription Template Preparation
To construct lesion-free control vector for SG and S6mG, a 52-mer ODN with the sequence of 5′-CTAGCGCTACCGGACTCAGATCTCGAGCTCTAGCTTTGCGCAAGCGACTCCG-3′ was annealed with the complementary strand and ligated to an NheI-EcoRI restriction fragment from the nonreplicating pTGFP-T7-Hha10 plasmids (35). We next treated the control plasmids with the nicking enzyme Nt.BstNBI and subsequently produced a gapped vector by removing a 33-mer single-stranded ODN. The gapped plasmid was subsequently annealed with a 5′-phosphorylated SG- or S6mG-bearing ODN and ligated with T4 DNA ligase. The ligation mixture was incubated with ethidium bromide, and the resulting supercoiled lesion-bearing plasmid was isolated by agarose gel electrophoresis, following recently published procedures (21, 36, 37). The competitor vector used for this study was previously constructed for studying the impact of N2-(1-carboxyethyl)-2′-deoxyguanosine on transcription (35). Purified control or lesion-bearing genome was mixed with a competitor genome and used as transcription templates, with a molar ratio of competitor vector to control or lesion-bearing genome being 1:6 for all experiments in this study.
In Vitro Transcription Assay
Multiple rounds of transcription reactions using T7 RNAP or HeLa nuclear extract (Promega) were performed as described elsewhere (35). Briefly, T7 RNAP-mediated reaction was incubated at 37 °C for 1 h in a 20-μl mixture containing 20 units of T7 RNAP, 10 units of RNase inhibitor, 0.5 mm each ATP, CTP, GTP, and UTP, and 50 ng of NotI-linearized DNA template. The hRNAPII-mediated reaction was incubated at 30 °C for 1 h in a 25-μl mixture containing 8 units of HeLa nuclear extract, 10 units of RNase inhibitor, 0.4 mm each of all four ribonucleotides, and 50 ng of NotI-linearized DNA template.
In Vivo Transcription Assay
Human skin fibroblast cells (GM00637 and GM04429) were grown to ∼70% confluence in 24-well plates and co-transfected with 50 ng of mixed genome and 450 ng of carrier plasmid (self-ligated pGEM-T; Promega) with Lipofectamine 2000 (Invitrogen), according to the manufacturer's protocol. To prepare the carrier plasmid, we first treated the linearized pGEM-T vector (Promega) containing 3′-T overhangs with T4 DNA polymerase, followed by self-ligating the resultant plasmid DNA, which contains blunt ends. Highly efficient depletion of cockayne syndrome group B (CSB) and xeroderma pigmentosum group C (XPC) genes was achieved by siRNA treatments as previously described (35), and all siRNAs were obtained from Dharmacon: CSB SMARTpool (L-004888), XPC SMARTpool (L-016040), and siGENOME NonTargeting siRNA (D-001210). Briefly, for siRNA experiments, 293T cells were grown to 40–60% confluence in 24-well plates and transfected with either or both XPC and CSB siRNAs (25 pmol each). After a 48-h incubation, 50 ng of mixed genome, 450 ng of carrier DNA, and another aliquot of siRNAs were co-transfected into the cells with Lipofectamine 2000 (Invitrogen). For all in vivo transcription assays, the cells were harvested for RNA extraction 24 h after transfection with the mixed genome.
RNA Extraction and RT-PCR
RNA transcripts arising from in vitro or in vivo transcription were isolated using the Total RNA Kit I (Omega) and subjected to two rounds of DNase I treatment with the DNA-free kit (Ambion) to eliminate the DNA contamination. cDNA was generated by using M-MLV reverse transcriptase (Promega) and a mixture of an oligo(dT)16 primer and a gene-specific primer (5′-TCGGTGTTGCTGTGAT-3′). RT-PCR amplification for PAGE and LC-MS/MS analyses were then performed by using Phusion high fidelity DNA polymerase and a pair of primers spanning the lesion site as described in a recent study (35). Real time quantitative RT-PCR for evaluating the extent of siRNA knockdown was performed by using the iQ SYBR Green Supermix kit (Bio-Rad) and gene-specific primers for CSB, XPC, or the control gene GAPDH as described elsewhere (35).
PAGE Analysis
PAGE analysis of transcription products of SG and S6mG was performed as described elsewhere (35). Briefly, a portion of the above RT-PCR fragments was incubated in a 10-μl reaction containing 1× NEB buffer 4, 10 units of SacI, and 1 unit of shrimp alkaline phosphatase at 37 °C for 1 h and then at 80 °C for 20 min. The resulting dephosphorylated DNA was incubated in a 15-μl solution containing 1× NEB buffer 4, ATP (50 pmol of cold, premixed with 1.66 pmol of [γ-32P]ATP), and 10 units of T4 polynucleotide kinase. The mixture was then incubated at 37 °C for 30 min and at 65 °C for 20 min, after which 10 units of FspI was added and incubated at 37 °C for 1 h. The resulting 32P-labeled restriction fragments were resolved by using 30% polyacrylamide gel (acrylamide:bis-acrylamide = 19:1) with 8 m urea and quantified by phosphorimaging analysis (21, 22). The relative mutation frequency (RMF) was determined by the ratio of the amount of detectable mutant restriction product to the total amounts of restriction fragments arising from the lesion-bearing genome. The relative bypass efficiency (RBE) was calculated using the following formula, RBE = (lesion signal/competitor signal)/(nonlesion control signal/competitor signal) (22, 35, 38).
LC-MS/MS Analysis
To identify the transcription products of SG and S6mG using LC-MS/MS, RT-PCR products were treated with 50 units of SacI, 50 units of FspI, and 20 units of shrimp alkaline phosphatase in 250 μl of NEB buffer 4 at 37 °C for 4 h, followed by heating at 80 °C for 20 min. The resulting solution was extracted with phenol/chloroform/isoamyl alcohol (25:24:1, v/v/v), and the aqueous portion was dried with a SpeedVac, desalted with HPLC, and dissolved in water. The resultant ODN mixture was subjected to LC-MS/MS analysis following previously described conditions (21, 22, 35). Briefly, a 0.5 × 150-mm Zorbax SB-C18 column (Agilent Technologies) was used. The flow rate was 8.0 μl/min, and a 5-min linear gradient of 5–35% methanol followed by a 15 min of 35–95% methanol in 400 mm 1,1,1,3,3,3-hexafluoro-2-propanol buffer (pH was adjusted to 7.0 by the addition of triethylamine) was employed for the separation. The LTQ linear ion trap mass spectrometer was set up for monitoring the fragmentation of the [M-3H]3− ions of the 14-mer (d(GCAAAMCTAGAGCT), where M designates A, T, C, or G) ODNs.
RESULTS
Construction of Plasmids Containing a Site-specifically Inserted SG or S6mG
To investigate how SG or S6mG perturbs the efficiency and fidelity of transcription, we first constructed nonreplicating double-stranded plasmids with a single SG or S6mG at a defined site on the transcribed strand (Fig. 1B). The lesions were positioned 63 and 44 nucleotides downstream of the transcription start sites of the cytomegalovirus and T7 promoters, respectively. In this context, we chose T7 RNA polymerase (T7 RNAP) as a model for assessing the potential impact of these lesions on transcription of mitochondrial genome, because there is a high degree of homology between T7 RNAP and eukaryotic mitochondrial RNAPs (39).
Effects of SG and S6mG on Transcription in Vitro
To examine the impact of SG or S6mG on transcription in vitro, the lesion-bearing or nonlesion control plasmids were individually premixed with a competitor genome and used as DNA templates for multiple rounds of transcription with T7 RNAP or HeLa cell nuclear extract. The latter served as a source of human RNA polymerase II (hRNAPII) transcription machinery. Relative to the control plasmids containing an unmodified guanine in lieu of a lesion, the competitor genome has three additional nucleotides near the relevant site (Fig. 1B). After in vitro transcription, the RNA products of interest were identified and quantified by restriction digestion of RT-PCR products, and PAGE and LC-MS/MS analyses of appropriate restriction fragments (Figs. 1B and 2A). The effects of SG and S6mG on transcriptional efficiency and fidelity were determined, as previously described (35), by the RBE and RMF, respectively (see “Experimental Procedures”).
FIGURE 2.

Transcriptional alternations induced by SG and S6mG in in vitro transcription systems using T7 RNAP or HeLa nuclear extract (hRNAPII). A, sample processing for PAGE analysis (p* indicates 32P-labeled phosphate group). The RT-PCR products were first treated with SacI, and the 5′-phosphate groups in the resulting products were removed with shrimp alkaline phosphatase. The dephosphorylated DNA was radiolabeled on the 5′ end with [γ-32P]ATP and T4 polynucleotide kinase (T4 PNK), after which the second enzyme (FspI) was added to produce the 32P-labeled restriction fragments for PAGE analysis. Only the RT-PCR product arising from the lesion-containing genome is shown. B, representative gel images showing the restriction fragments released from the RT-PCR products arising from transcription templates containing SG (lanes 1 and 4), S6mG (lanes 2 and 5), or normal guanine (G, lanes 3 and 6). 10mer-TA, 10mer-TG, 10mer-TT, and 10mer-TC represent standard ODNs d(CTAGNTTTGC), where N is A, G, T, and C, respectively (lanes 7–10, respectively). 13mer-Comp represents standard ODN d(CATCAAGCTTTGC) (lane 11), which corresponds to the restriction fragment arising from the competitor genome. C and D, the RBE values of SG and S6mG (C) and RMF values of S6mG (D) in in vitro transcription systems using T7 RNAP or HeLa nuclear extract (hRNAPII). The data represent the means and standard error of results from three independent experiments.
PAGE analysis showed that the 10-mer DNA fragments (d(CTAGNTTTGC), where N designates A, T, C, or G) with a single-nucleotide difference opposite the lesion site can be resolved from each other (Fig. 2B, lanes 7–10). Except for the 10-mer 32P-labeled product with the wild-type sequence (i.e., d(CTAGCTTTGC)), no mutated products were detectable for SG-bearing plasmid by T7 RNAP and hRNAPII in vitro (Fig. 2B, lanes 1 and 4). By using LC-MS and MS/MS analyses, we monitored the fragmentation of the [M-3H]3− ions of the complementary 14-mer fragments (d(GCAAAMCTAGAGCT), where M designates A, T, C, or G). Again, we found that only the wild-type sequence (d(GCAAAGCTAGAGCT)) could be detected in the restriction mixtures arising from the in vitro transcription of SG-containing substrates (data not shown). In addition, a single SG on the transcribed strand did not considerably affect the transcriptional bypass efficiencies of T7 RNAP or hRNAPII in vitro (Fig. 2C).
PAGE analysis also showed that, unlike SG, transcriptional bypass of S6mG in vitro with both T7 RNAP and hRNAPII generated at least one type of mutant transcript (Fig. 2B, lanes 2 and 5), which contains a uridine misincorporation opposite the S6mG (Fig. 3A). We further confirmed the identity of this mutation by LC-MS and MS/MS analyses. Again, we monitored the fragmentation of the [M-3H]3− ions of the complementary 14-mer fragments (d(GCAAAMCTAGAGCT), where M designates A, T, C, or G) and found that only the wild-type 14-mer sequence (i.e., d(GCAAAGCTAGAGCT)) and another 14-mer fragment with the mutated sequence (i.e., d(GCAAAACTAGAGCT)) were detectable (Fig. 3, B and C). The quantification data from PAGE analysis revealed that S6mG was highly mutagenic in in vitro transcription by T7 RNAP and hRNAPII, with RMF values of 49 and 85%, respectively (Fig. 2D). In addition, we found that S6mG acted as a modest inhibitor of transcription by T7 RNAP while posing a major impediment to transcription by hRNAPII, with the RBE values being 69 and 24%, respectively (Fig. 2C).
FIGURE 3.

LC-MS and MS/MS for monitoring the 14-mer restriction fragments of S6mG-bearing substrate from the in vitro transcription with T7 RNAP. A, the sequences of WT and mutant (Mu) transcripts are indicated above the double-stranded DNA construct. B, product ion spectrum (MS/MS) of the [M-3H]3− ion (m/z 1425.0) of the complementary 14-mer fragment of the mutant sequence d(GCAAAACTAGAGCT). Shown above the spectrum is a scheme summarizing the observed [an − Base] and wn fragment ions, and nomenclature follows that described previously (55). C, high resolution “ultra zoom-scan” ESI-MS revealed the presence of the [M-3H]3− ions of the wild-type sequence d(GCAAAGCTAGAGCT) (m/z 1430.4) and the mutant sequence d(GCAAAACTAGAGCT) (m/z 1425.0) but the absence of the [M-3H]3− ions of other single-base substitution products of d(GCAAAMCTAGAGCT) (M = C or T, with the calculated m/z being 1417.2 and 1422.2, respectively).
Effects of SG and S6mG on Transcription in Human Cells
We then performed the competitive transcription and adduct bypass assay to examine how SG and S6mG affect DNA transcription in vivo. Similar to the aforementioned in vitro experiments, we premixed either lesion-bearing or control plasmids with the competitor genome and co-transfected the mixed DNA substrates into NER-deficient (GM04429, lacking XPA) and repair-proficient (GM00637) human skin fibroblasts for in vivo transcription studies. After a 24-h incubation, the RNA products were isolated and processed as described above, and the resulting restriction fragments were subjected to PAGE and LC-MS/MS analysis (Fig. 1B).
PAGE analysis showed that transcriptional bypass of SG did not produce any detectable mutant transcripts in human skin fibroblast cells (Fig. 4A, lanes 6 and 9). We also confirmed these results with LC-MS and MS/MS analyses (data not shown). Furthermore, SG only modestly compromised transcriptional efficiency in both GM00637 and GM04429 cells at 24 h after transfection, with similar RBE values of ∼ 65% (Fig. 4B). On the other hand, ∼20% of the transcripts contained a uridine misincorporation opposite the S6mG in GM00637 cells, and a higher degree of this mutation (∼ 75%) was observed in XPA-deficient GM04429 cells (Fig. 4, A and C). In addition, the RBE value for S6mG was significantly (p < 0.01) lower in GM04429 cells (∼29%) than in the repair-proficient GM00637 cells (∼43%) (Fig. 4B). Thus, these results indicated that XPA, a core component of NER machinery (32), contributes to the removal of S6mG, but not SG, in human cells.
FIGURE 4.

Transcriptional alternations induced by SG and S6mG in NER-deficient (GM04429) or repair-proficient (GM00637) cells. A, representative gel images showing the restriction fragments released from the RT-PCR products arising from transcription templates containing SG (lanes 6 and 9), S6mG (lanes 7 and 10), or normal guanine (G, lanes 8 and 11). 10mer-TA, 10mer-TG, 10mer-TT, and 10mer-TC represent standard ODNs d(CTAGNTTTGC), where N is A, G, T, and C, respectively (lanes 1–4, respectively). 13mer-Comp represents standard ODN d(CATCAAGCTTTGC) (lane 5), which corresponds to the restriction fragment arising from the competitor genome. The band between 13mer-Comp and 10mer-TT is most likely a nonspecific digestion product and is not lesion-dependent, because it is also observed with lesion-free control templates. B and C, the RBE values of SG and S6mG (B) and RMF values of S6mG (C) based on in vivo transcription experiments using GM04429 or GM00637 cells. The data represent the means and standard error of results from three independent experiments. **, p < 0.01; ***, p < 0.001. The p values were calculated by using unpaired two-tailed Student's t test.
Because XPC and CSB are key players in global-genome repair and TC-NER subpathways, respectively (32), we used siRNAs to diminish the expression of these two genes, individually or in combination, in 293T cells and assessed their roles in the transcriptional alternations induced by SG and S6mG. Real time PCR results revealed that the siRNA knockdown was highly efficient for both CSB and XPC genes (Fig. 5). The results from PAGE analysis showed that depletion of CSB alone caused a considerable increase in transcriptional mutagenesis induced by S6mG, with the RMF values significantly (p < 0.01) increasing from 10% in control siRNA-treated cells to 28% in CSB-depleted cells (Fig. 6, A, lanes 7 and 10, and B). In contrast, knockdown of XPC alone did not change appreciably the mutagenic properties of S6mG (Fig. 6, A, lanes 7 and 13, and B), and simultaneous knockdown of CSB and XPC gave a similar RMF value for S6mG as knockdown of CSB alone (Fig. 6, A, lanes 10 and 16, and B). Likewise, although depletion of XPC had no effect (Fig. 6, A, lanes 7 and 13, and C), siRNA knockdown of CSB conferred a significant (p < 0.01) reduction in RBE value for S6mG compared with control siRNA treatment (Fig. 6, A, lanes 7 and 10, and C). In addition, we found that SG modestly decreases transcriptional efficiency without introducing a mutation in 293T cells, and neither CSB nor XPC was required for the removal of SG in human cells (Fig. 6, A and C).
FIGURE 5.
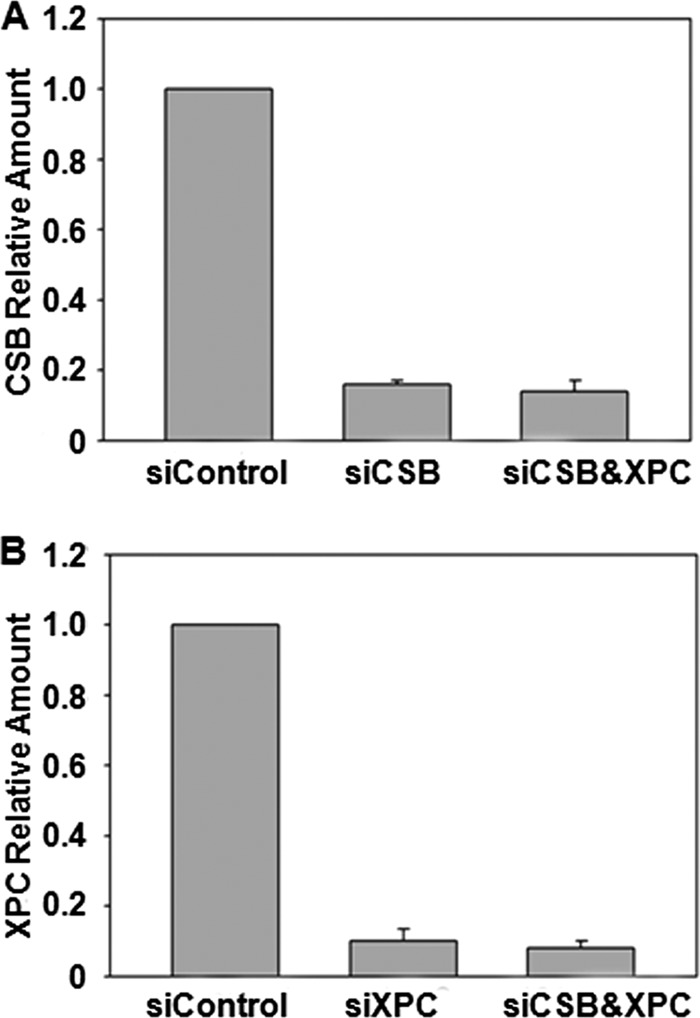
Real time quantitative RT-PCR analysis for monitoring the siRNA-induced knockdown of CSB (A) and/or XPC (B) in human 293T cells. GAPDH was used as a control for real time quantitative RT-PCR analysis. The PCR data represent the means and standard error of results from three separate experiments.
FIGURE 6.

Transcriptional alternations induced by SG and S6mG in 293T cells treated with either or both CSB and XPC siRNAs. A, representative gel images showing the restriction fragments released from the RT-PCR products arising from transcription templates containing SG (lanes 6, 9, 12, and 15), S6mG (lanes 7, 10, 13, and 16), or normal guanine (G, lanes 8, 11, 14, and 17). 10mer-TA, 10mer-TG, 10mer-TT, and 10mer-TC represent standard ODNs d(CTAGNTTTGC), where N is A, C, G, and T, respectively (lanes 1–4, respectively). 13mer-Comp represents standard ODN d(CATCAAGCTTTGC) (lane 5), which corresponds to the restriction fragment arising from the competitor genome. The band observed between 13mer-Comp and 10mer-TT is most likely a nonspecific digestion product and is not lesion-dependent, because it is also obtained with lesion-free control templates. B and C, the RMF values of S6mG (B) and the RBE values of SG and S6mG (C) in 293T cells treated with either or both CSB and XPC siRNAs. The data represent the means and standard error of results from three independent experiments. *, p < 0.05; **, p < 0.01. The p values were calculated by using unpaired two-tailed Student's t test.
DISCUSSION
It has been previously shown that a single SG on the transcribed strand weakly inhibits transcription by yeast RNAPII in vitro (10). In agreement with this finding, our results demonstrated that SG in DNA does not considerably compromise transcription elongation mediated by single-subunit T7 RNAP or multisubunit hRNAPII in vitro and in human cells. In addition, we found that depletion of NER factors, including XPA, CSB, and XPC, does not lead to a change in the transcription bypass efficiency for SG. Again, these results are consistent with previously published data indicating that DNA SG is a poor substrate for NER in human cells (10, 21). Moreover, to our knowledge, we showed for the first time that SG is not mutagenic during transcription in vitro or in vivo. In this vein, it was reported recently that SG does not substantially block DNA replication, although it can induce G → A mutations at frequencies of 8–10% in E. coli and mammalian cells (21, 22).
Unlike SG, a single S6mG in the transcribed strand was found to appreciably impede transcription by hRNAPII and induce transcriptional mutagenesis in vitro and in human cells. A similar but less pronounced effect of S6mG on transcription was also observed in T7 RNAP-mediated in vitro transcription system. More importantly, we found that the impact of S6mG on transcriptional efficiency and fidelity is modulated by XPA and CSB, but not XPC. These findings indicate that, when located on the template strand of an actively transcribed gene, S6mG is primarily repaired by TC-NER in human cells. Similar to our results, it was reported that O6-methylguanine, a structural analog of S6mG, contributes to mutagenesis and blockage of transcription and thus might invoke TC-NER (40, 41). Previous replication studies have also shown that S6mG is capable of inducing high frequencies of G → A transition in E. coli and mammalian cells (21, 22). However, S6mG is not a strong block to DNA replication, and deficiency in NER factor XPA does not considerably alter the effects of S6mG on the efficiency and fidelity of DNA replication (21, 22).
Transcriptional inhibition has been suggested to be a potential strategy for cancer therapy (29–31). It has been shown that the survival of tumor cells requires the expression of antiapoptotic factors, and tumor cells appear to be more sensitive to inhibition of transcription than normal cells (42). In this context, the half-lives of mRNAs encoding antiapoptosis proteins are generally shorter than those of apoptosis-promoting factors. In addition, many of these antiapoptosis genes are on average larger in size than the apoptosis-promoting genes. Thus, transcription inhibitors are thought to selectively impede the expression of these antiapoptosis factors and induce the apoptosis of cancer cells (29, 43, 44). Many transcriptional inhibitors have been clinically tested as anticancer agents, including cisplatin and other platinum-based drugs (31, 45–47). Given that S6mG, but not SG, exhibits a strong inhibitory effect on transcription in our experimental systems, it is therefore possible that SG in human genomic DNA has little or no direct role, at the level of transcription, in the cytotoxic effects of thiopurine drugs; however, S6mG, a methylation damage derived from SG in DNA, may contribute, at least in part, to the cytotoxicity of thiopurine drugs through serving as a transcriptional inhibitor. On the other hand, it has been previously suggested that both S6mG and SG can induce replication errors and subsequently trigger futile cycles of MMR, which is widely regarded as a major contributor to thiopurine toxicity (8, 20–22, 24, 25).
The mechanisms through which S6mG interferes with transcription may be similar to those of platinum drug-induced DNA damage (31). It has been shown that DNA damage induced by platinum-based agents may block transcription at both the initiation and elongation stages (31, 45, 46). Thus, aside from its effect on transcription elongation observed in this study, it would be interesting to investigate, in the future, whether S6mG also affects transcription initiation by altering the binding of transcription factors to DNA.
Long term use of thiopurines in patients is associated with an elevated incidence of certain iatrogenic cancers (48–51). Previous studies have provided some clues that the mutations induced by S6mG and SG during replication might have a role in the carcinogenicity of thiopurine drugs (21, 22). In this vein, transcriptional mutagenesis has also been proposed as one of the principal inducers of cancer and other human diseases, despite the lack of direct evidence linking transcriptional mutations to cancer (33, 40, 52, 53). Notably, a previous study has provided important implications for the role of transcriptional mutagenesis in tumorigenesis in that mutagenic transcriptional bypass of 8-oxoguanine could lead to activation of an oncogenic pathway (53). Despite the very low frequency of formation after thiopurine treatment, S6mG in cellular DNA appears to be at a similar level as O6-methylguanine and abundant oxidative DNA lesions such as 8-oxoguanine (7, 8, 54). The observation that S6mG, but not SG, can lead to the generation of mutant transcripts at high frequency, suggests that S6mG-induced transcriptional mutagenesis may also contribute to the development of thiopurine therapy-related cancers and other complications.
Taken together, the present study demonstrates that, when located on the transcribed DNA strand, S6mG, but not SG, can cause strong mutagenic and inhibitory effects on transcription and act as an efficient substrate for TC-NER. These findings suggest that, at the level of transcription, SG has little or no direct impact on the activities of thiopurine drugs, but its methylation product (i.e., S6mG) might have two opposite effects: one contributes to thiopurine-induced cytotoxicity through inhibition of transcription, and the other leads to thiopurine therapy-related cancers by inducing mutant transcripts. Therefore, the work reported here improves our understanding of the mechanisms by which thiopurine drugs exert their cytotoxic and potential carcinogenic effects.
Acknowledgments
We thank Prof. Gerd P. Pfeifer for kindly providing the GM00637 and GM04429 human skin fibroblast cells and Prof. Timothy R. O'Connor for providing the initial pTGFP-Hha10 vector.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 DK082779.
- SG
- 6-thioguanine
- S6mG
- S6-methylthioguanine
- MMR
- mismatch repair
- TC-NER
- transcription-coupled nucleotide excision repair
- RMF
- relative mutation frequency
- RBE
- relative bypass efficiency
- T7 RNAP
- T7 RNA polymerase
- hRNAPII
- human RNA polymerase II
- XPA
- xeroderma pigmentosum group A
- XPC
- xeroderma pigmentosum group C
- CSB
- cockayne syndrome group B
- ODN
- oligodeoxyribonucleotide.
REFERENCES
- 1. Pui C. H., Jeha S. (2007) New therapeutic strategies for the treatment of acute lymphoblastic leukemia. Nat. Rev. Drug Discov. 6, 149–165 [DOI] [PubMed] [Google Scholar]
- 2. Elion G. B. (1989) The purine path to chemotherapy. Science 244, 41–47 [DOI] [PubMed] [Google Scholar]
- 3. Karran P., Attard N. (2008) Thiopurines in current medical practice. Molecular mechanisms and contributions to therapy-related cancer. Nat. Rev. Cancer 8, 24–36 [DOI] [PubMed] [Google Scholar]
- 4. Chouchana L., Narjoz C., Beaune P., Loriot M. A., Roblin X. (2012) Review article. The benefits of pharmacogenetics for improving thiopurine therapy in inflammatory bowel disease. Aliment Pharmacol. Ther. 35, 15–36 [DOI] [PubMed] [Google Scholar]
- 5. LePage G. A. (1963) Basic biochemical effects and mechanism of action of 6-thioguanine. Cancer Res. 23, 1202–1206 [PubMed] [Google Scholar]
- 6. O'Donovan P., Perrett C. M., Zhang X., Montaner B., Xu Y. Z., Harwood C. A., McGregor J. M., Walker S. L., Hanaoka F., Karran P. (2005) Azathioprine and UVA light generate mutagenic oxidative DNA damage. Science 309, 1871–1874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang H., Wang Y. (2010) LC-MS/MS coupled with stable isotope dilution method for the quantification of 6-thioguanine and S6-methylthioguanine in genomic DNA of human cancer cells treated with 6-thioguanine. Anal. Chem. 82, 5797–5803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Swann P. F., Waters T. R., Moulton D. C., Xu Y. Z., Zheng Q., Edwards M., Mace R. (1996) Role of postreplicative DNA mismatch repair in the cytotoxic action of thioguanine. Science 273, 1109–1111 [DOI] [PubMed] [Google Scholar]
- 9. Warren D. J., Andersen A., Slørdal L. (1995) Quantitation of 6-thioguanine residues in peripheral blood leukocyte DNA obtained from patients receiving 6-mercaptopurine-based maintenance therapy. Cancer Res. 55, 1670–1674 [PubMed] [Google Scholar]
- 10. Brem R., Li F., Karran P. (2009) Reactive oxygen species generated by thiopurine/UVA cause irreparable transcription-blocking DNA lesions. Nucleic Acids Res. 37, 1951–1961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tiede I., Fritz G., Strand S., Poppe D., Dvorsky R., Strand D., Lehr H. A., Wirtz S., Becker C., Atreya R., Mudter J., Hildner K., Bartsch B., Holtmann M., Blumberg R., Walczak H., Iven H., Galle P. R., Ahmadian M. R., Neurath M. F. (2003) CD28-dependent Rac1 activation is the molecular target of azathioprine in primary human CD4+ T lymphocytes. J. Clin. Invest. 111, 1133–1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brem R., Karran P. (2012) Oxidation-mediated DNA crosslinking contributes to the toxicity of 6-thioguanine in human Cells. Cancer Res. 72, 4787–4795 [DOI] [PubMed] [Google Scholar]
- 13. Yuan B., Zhang J., Wang H., Xiong L., Cai Q., Wang T., Jacobsen S., Pradhan S., Wang Y. (2011) 6-Thioguanine reactivates epigenetically silenced genes in acute lymphoblastic leukemia cells by facilitating proteasome-mediated degradation of DNMT1. Cancer Res. 71, 1904–1911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gueranger Q., Kia A., Frith D., Karran P. (2011) Crosslinking of DNA repair and replication proteins to DNA in cells treated with 6-thioguanine and UVA. Nucleic Acids Res. 39, 5057–5066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ren X., Li F., Jeffs G., Zhang X., Xu Y. Z., Karran P. (2010) Guanine sulphinate is a major stable product of photochemical oxidation of DNA 6-thioguanine by UVA irradiation. Nucleic Acids Res. 38, 1832–1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yan T., Berry S. E., Desai A. B., Kinsella T. J. (2003) DNA mismatch repair (MMR) mediates 6-thioguanine genotoxicity by introducing single-strand breaks to signal a G2-M arrest in MMR-proficient RKO cells. Clin. Cancer Res. 9, 2327–2334 [PubMed] [Google Scholar]
- 17. Branch P., Aquilina G., Bignami M., Karran P. (1993) Defective mismatch binding and a mutator phenotype in cells tolerant to DNA damage. Nature 362, 652–654 [DOI] [PubMed] [Google Scholar]
- 18. Hawn M. T., Umar A., Carethers J. M., Marra G., Kunkel T. A., Boland C. R., Koi M. (1995) Evidence for a connection between the mismatch repair system and the G2 cell cycle checkpoint. Cancer Res. 55, 3721–3725 [PubMed] [Google Scholar]
- 19. Glaab W. E., Risinger J. I., Umar A., Barrett J. C., Kunkel T. A., Tindall K. R. (1998) Resistance to 6-thioguanine in mismatch repair-deficient human cancer cell lines correlates with an increase in induced mutations at the HPRT locus. Carcinogenesis 19, 1931–1937 [DOI] [PubMed] [Google Scholar]
- 20. Waters T. R., Swann P. F. (1997) Cytotoxic mechanism of 6-thioguanine. hMutSα, the human mismatch binding heterodimer, binds to DNA containing S6-methylthioguanine. Biochemistry 36, 2501–2506 [DOI] [PubMed] [Google Scholar]
- 21. Yuan B., O'Connor T. R., Wang Y. (2010) 6-Thioguanine and S6-methylthioguanine are mutagenic in human cells. ACS Chem. Biol. 5, 1021–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yuan B., Wang Y. (2008) Mutagenic and cytotoxic properties of 6-thioguanine, S6-methylthioguanine, and guanine-S6-sulfonic acid. J. Biol. Chem. 283, 23665–23670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gu C., Wang Y. (2007) In vitro replication and thermodynamic studies of methylation and oxidation modifications of 6-thioguanine. Nucleic Acids Res. 35, 3693–3704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Griffin S., Branch P., Xu Y. Z., Karran P. (1994) DNA mismatch binding and incision at modified guanine bases by extracts of mammalian cells. Implications for tolerance to DNA methylation damage. Biochemistry 33, 4787–4793 [DOI] [PubMed] [Google Scholar]
- 25. Krynetski E. Y., Krynetskaia N. F., Gallo A. E., Murti K. G., Evans W. E. (2001) A novel protein complex distinct from mismatch repair binds thioguanylated DNA. Mol. Pharmacol. 59, 367–374 [DOI] [PubMed] [Google Scholar]
- 26. Chifotides H. T., Fu P. K., Dunbar K. R., Turro C. (2004) Effect of equatorial ligands of dirhodium(II,II) complexes on the efficiency and mechanism of transcription inhibition in vitro. Inorg. Chem. 43, 1175–1183 [DOI] [PubMed] [Google Scholar]
- 27. Darnell J. E., Jr. (2002) Transcription factors as targets for cancer therapy. Nat. Rev. Cancer 2, 740–749 [DOI] [PubMed] [Google Scholar]
- 28. Sobell H. M. (1985) Actinomycin and DNA transcription. Proc. Natl. Acad. Sci. U.S.A. 82, 5328–5331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Derheimer F. A., Chang C. W., Ljungman M. (2005) Transcription inhibition. A potential strategy for cancer therapeutics. Eur. J. Cancer 41, 2569–2576 [DOI] [PubMed] [Google Scholar]
- 30. Stellrecht C. M., Chen L. S. (2011) Transcription inhibition as a therapeutic target for cancer. Cancers 3, 4170–4190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Todd R. C., Lippard S. J. (2009) Inhibition of transcription by platinum antitumor compounds. Metallomics 1, 280–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hanawalt P. C., Spivak G. (2008) Transcription-coupled DNA repair. Two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 9, 958–970 [DOI] [PubMed] [Google Scholar]
- 33. Brégeon D., Doetsch P. W. (2011) Transcriptional mutagenesis. Causes and involvement in tumour development. Nat. Rev. Cancer 11, 218–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kuraoka I., Tanaka K. (2006) Assays for transcription elongation by RNA polymerase II using oligo(dC)-tailed template with single DNA damage. Methods Enzymol. 408, 214–223 [DOI] [PubMed] [Google Scholar]
- 35. You C., Dai X., Yuan B., Wang J., Brooks P. J., Niedernhofer L. J., Wang Y. (2012) A quantitative assay for assessing the effects of DNA lesions on transcription. Nat. Chem. Biol. 8, 817–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yuan B., You C., Andersen N., Jiang Y., Moriya M., O'Connor T. R., Wang Y. (2011) The roles of DNA polymerases κ and ι in the error-free bypass of N2-carboxyalkyl-2′-deoxyguanosine lesions in mammalian cells. J. Biol. Chem. 286, 17503–17511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Baker D. J., Wuenschell G., Xia L., Termini J., Bates S. E., Riggs A. D., O'Connor T. R. (2007) Nucleotide excision repair eliminates unique DNA-protein cross-links from mammalian cells. J. Biol. Chem. 282, 22592–22604 [DOI] [PubMed] [Google Scholar]
- 38. Delaney J. C., Essigmann J. M. (2004) Mutagenesis, genotoxicity, and repair of 1-methyladenine, 3-alkylcytosines, 1-methylguanine, and 3-methylthymine in alkB Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 101, 14051–14056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Masters B. S., Stohl L. L., Clayton D. A. (1987) Yeast mitochondrial RNA polymerase is homologous to those encoded by bacteriophages T3 and T7. Cell 51, 89–99 [DOI] [PubMed] [Google Scholar]
- 40. Burns J. A., Dreij K., Cartularo L., Scicchitano D. A. (2010) O6-methylguanine induces altered proteins at the level of transcription in human cells. Nucleic Acids Res. 38, 8178–8187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dimitri A., Burns J. A., Broyde S., Scicchitano D. A. (2008) Transcription elongation past O6-methylguanine by human RNA polymerase II and bacteriophage T7 RNA polymerase. Nucleic Acids Res. 36, 6459–6471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Koumenis C., Giaccia A. (1997) Transformed cells require continuous activity of RNA polymerase II to resist oncogene-induced apoptosis. Mol. Cell. Biol. 17, 7306–7316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ljungman M., Lane D. P. (2004) Transcription. Guarding the genome by sensing DNA damage. Nat. Rev. Cancer 4, 727–737 [DOI] [PubMed] [Google Scholar]
- 44. Lam L. T., Pickeral O. K., Peng A. C., Rosenwald A., Hurt E. M., Giltnane J. M., Averett L. M., Zhao H., Davis R. E., Sathyamoorthy M., Wahl L. M., Harris E. D., Mikovits J. A., Monks A. P., Hollingshead M. G., Sausville E. A., Staudt L. M. (2001) Genomic-scale measurement of mRNA turnover and the mechanisms of action of the anti-cancer drug flavopiridol. Genome Biol. 2, RESEARCH0041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhu G., Myint M., Ang W. H., Song L., Lippard S. J. (2012) Monofunctional platinum-DNA adducts are strong inhibitors of transcription and substrates for nucleotide excision repair in live mammalian cells. Cancer Res. 72, 790–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ang W. H., Myint M., Lippard S. J. (2010) Transcription inhibition by platinum-DNA cross-links in live mammalian cells. J. Am. Chem. Soc. 132, 7429–7435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang D., Lippard S. J. (2005) Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 4, 307–320 [DOI] [PubMed] [Google Scholar]
- 48. Karran P. (2006) Thiopurines, DNA damage, DNA repair and therapy-related cancer. Br. Med. Bull. 79–80, 153–170 [DOI] [PubMed] [Google Scholar]
- 49. Lennard L., Thomas S., Harrington C. I., Maddocks J. L. (1985) Skin cancer in renal transplant recipients is associated with increased concentrations of 6-thioguanine nucleotide in red blood cells. Br. J. Dermatol. 113, 723–729 [DOI] [PubMed] [Google Scholar]
- 50. Relling M. V., Rubnitz J. E., Rivera G. K., Boyett J. M., Hancock M. L., Felix C. A., Kun L. E., Walter A. W., Evans W. E., Pui C. H. (1999) High incidence of secondary brain tumours after radiotherapy and antimetabolites. Lancet. 354, 34–39 [DOI] [PubMed] [Google Scholar]
- 51. Penn I. (2000) Post-transplant malignancy. The role of immunosuppression. Drug Saf. 23, 101–113 [DOI] [PubMed] [Google Scholar]
- 52. Brégeon D., Peignon P. A., Sarasin A. (2009) Transcriptional mutagenesis induced by 8-oxoguanine in mammalian cells. PLoS Genet. 5, e1000577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Saxowsky T. T., Meadows K. L., Klungland A., Doetsch P. W. (2008) 8-Oxoguanine-mediated transcriptional mutagenesis causes Ras activation in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 105, 18877–18882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cadet J., Douki T., Gasparutto D., Ravanat J. L. (2003) Oxidative damage to DNA. Formation, measurement and biochemical features. Mutat. Res. 531, 5–23 [DOI] [PubMed] [Google Scholar]
- 55. McLuckey S. A., Van Berkel G. J., Glish G. L. (1992) Tandem mass spectrometry of small, multiply charged oligonucleotides. J. Am. Soc. Mass Spectrom. 3, 60–70 [DOI] [PubMed] [Google Scholar]