

Background: Environmental factors may modulate brain function and dysfunction.
Results: Short term exposure to enriched environment produces antidepressant-like effects via a vascular endothelial growth factor-mediated increase in spinogenesis.
Conclusion: Enriched environment may act synergistically with other antidepressant treatments to rapidly provide beneficial antidepressant effects.
Significance: These findings provide new insights into the molecular mechanisms underlying the therapeutic effects of the enriched environment.
Keywords: Brain-derived Neurotrophic Factor (BDNF), Depression, Hypoxia-inducible Factor (HIF), MicroRNA, Vascular Endothelial Growth Factor (VEGF), Antidepressant-like Action, Enriched Environment, Spinogenesis
Abstract
Current antidepressant treatments remain limited by poor efficacy and a slow onset of action. Increasing evidence demonstrates that enriched environment (EE) treatment can promote structural and behavioral plasticity in the brain and dampen stress-induced alterations of neuroplasticity. Here, we have examined whether short term exposure to EE is able to produce antidepressant-like effects. Our results show that housing adult mice in an EE cage for 7 days led to antidepressant-like behavioral profiles and a significant increase in the number of dendritic spines in hippocampal CA1 pyramidal neurons. These EE-induced antidepressant-like effects are primarily attributed to increased vascular endothelial growth factor (VEGF) expression through a hypoxia-inducible factor-1α (HIF-1α)-mediated transcriptional mechanism. Blockade of HIF-1α synthesis by lentiviral infection with HIF-1α small hairpin RNAs completely blocked the increase in expression of VEGF and the antidepressant-like effects induced by EE. Moreover, no significant antidepressant-like effects were observed with EE treatment in VEGF receptor 2 (Flk-1) knock-out mice. The increase in HIF-1α expression in the hippocampus induced by EE was associated with a decrease in endogenous levels of microRNA-107 (miR-107). Overexpression of miR-107 in the hippocampus completely blocked EE-induced HIF-1α expression and the antidepressant-like effects. These results support a model in which the down-regulation of miR-107, acting through HIF-1α, mediates VEGF-dependent spinogenesis to underlie the EE-induced antidepressant-like effects.
Introduction
Major depressive disorder (MDD)2 is a common, chronic, and recurrent mental illness that affects about 17% of the population and is a substantial social and economic burden worldwide (1, 2). There are a wide variety of different medicines available to alleviate depressive symptoms, but serious limitations still exist. Most notably, the available treatments usually take weeks to months to achieve their antidepressant effects, and only about one-third of depressed patients respond to the first medication prescribed (3). There is an urgent need to develop more effective and faster acting antidepressant treatments. Interestingly, it has been reported recently that the N-methyl-d-aspartate receptor antagonist ketamine can elicit rapid and effective antidepressant-like effects through the activation of the mammalian target of rapamycin pathway in the prefrontal cortex, leading to a sustained elevation of synaptic signaling proteins and dendritic spine number (4, 5). Ketamine also significantly improved depressive symptoms in patients with treatment-resistant MDD (6, 7). However, the widespread clinical use of ketamine is limited due to the potential for abuse and toxicity (8).
Evidence for hippocampal atrophy in MDD patients (9, 10), as well as derailment of many hippocampus-related functions in depression (11, 12), indicates a critical role of the hippocampus in the disease. Although most recent studies have focused on establishing the causal relationship between impaired adult neurogenesis in the hippocampus and the development of MDD (13), there is evidence in the rodent model that the increase in depressive-like behaviors was associated with a decrease in the number of dendritic spines in hippocampal principal neurons (14–16). Moreover, chronic antidepressant administration reverses the dendritic spine loss and ameliorates depressive-like behaviors (16, 17). These observations have raised the possibility that novel strategies for producing a rapid induction of spinogenesis in the hippocampus might provide more effective therapeutic interventions for MDD.
Environmental factors may modulate brain function and dysfunction. Exposure to enriched environment (EE) has been shown to ameliorate the deleterious effects of stress on neurobiological and endocrine systems and promote adaptability to stress in both humans (18, 19) and rodents (20, 21). There is evidence that adult hippocampal neurogenesis, dendritic complexity of hippocampal neurons, and some learning abilities can be improved by housing animals in EE (22–24). Moreover, long term exposure to EE has been shown to produce antidepressant-like effects in rodent models of depression (25, 26). However, the molecular mechanisms underlying the effects of EE and the question of whether short term exposure to EE might also produce antidepressant-like effects remain unexplored.
Vascular endothelial growth factor (VEGF) was originally thought to be a potent and selective endothelial cell mitogen implicated in vascularization and angiogenesis (27). Recent studies have revealed that VEGF can act as a neurotrophic factor that regulates neurogenesis and mediates the effects of EE and antidepressants on hippocampal plasticity (28, 29). In this study, we have investigated the effects of short term EE treatment on mouse models of depressive-like behaviors. Our results demonstrate that housing adult mice in an EE cage for 7 days produces antidepressant-like effects and suggests a mechanism by which down-regulation of miR-107, acting through hypoxia-inducible factor-1α (HIF-1α), mediates VEGF-dependent hippocampal spinogenesis to underlie the antidepressant-like effects of EE.
EXPERIMENTAL PROCEDURES
Animals
Adult (8–12 weeks old) male C57BL/6J, heterozygous VEGF receptor 2 (Flk-1)-deficient (B6.129-Kdrtm1Jrt/J), homozygous brain-derived neurotrophic factor (BDNF)-floxed (Bdnftm3Jae/J), and calmodulin kinase II-cre transgenic mice were originally obtained from The Jackson Laboratory (Bar Harbor, ME) and bred within our animal facility. Bdnftm3Jae/J mice were crossed to calmodulin kinase II-cre mice to generate BDNF conditional knock-out mice. Bdnftm3Jae/J mice were used as the control littermates for comparison with BDNF conditional knock-out mice. Mice were genotyped by a polymerase chain reaction (PCR)-based method using genomic DNA isolated from tail samples. The knock-out mice were backcrossed to C57BL/6J mice for five generations to obtain a homogeneous genetic background. All experimental procedures were performed according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of National Cheng Kung University. Four mice were housed per cage in a temperature-controlled room under a 12-h light/dark cycle with ad libitum access to food and water. All behavioral tests were done during the light cycle of the day (10:00 a.m. to 4:00 p.m.). All efforts were made to minimize animal suffering and to reduce the number of mice used.
Housing Conditions
Mice were housed in groups of four in standard laboratory cages sized 28 × 15 × 15 cm3 (length × wide × height) until the beginning of the experiments. After 1 week, mice were then randomly assigned to standard (SE) or enriched rearing environment (EE) groups. SE used the same type of cage during the habituation period. The EE consisted a large cage (34 × 25 × 29 cm3) provided with different devices such as plastic tubes, ropes, two running wheels, and 3–4 toys. In some experiments, mice were housed in the enriched cage with locked running wheels. The devices were rearranged and renewed every 3 days to stimulate animal's exploratory behavior.
Tail Suspension Test
The mice were tested on a modified version of the tail suspension test (TST) described by Steru et al. (30). On the day of testing, experimental mice were transferred to the experiment room and allowed to acclimatize for 3 h. Mice were individually suspended by the tail to a horizontal ring-stand bar (distance from floor = 25 cm) using adhesive tape wrapped around its tail (1 cm from tip). Typically, mice displayed multiple escape-oriented behaviors interspersed with bouts of immobility as the session progressed. A 3-min test session was used that was recorded by a digital video camera positioned in front of the TST apparatus. The total duration of immobility for individual animal was recorded. Mice were considered immobile when they hung passively and motionless. Because of a propensity of C57BL/6J mice to climb up their tails during the testing session (31), mice that climbed their tail or fell off the hanger were excluded from the final analysis (less than 20% of the total population). We also noted that mice with relatively lower body weight or younger than 8 weeks old may display a high propensity to climb up their tails during the test. Thus, 8-week-old male mice, 20–24 g body weight, were used for TST. The behavioral performance was scored by a trained observer blind to the experimental conditions.
Sucrose Preference Test
Sucrose preference test (SPT) was performed on a modified version as described previously (32). Mice were first habituated to consume water in the two-bottle-choice paradigm for 3 days. At the beginning of the experiment, mice were housed individually in the test chamber identical to their home cage, and water was deprived for 1 h to increase drinking behavior. In the test session, mice were provided access to two bottles with 1% sucrose solution and water, respectively, for 3 h. The sucrose preference score was determined dividing the volume of sucrose consumed by the total liquid consumption.
Chronic Mild Stress
To induce chronic mild stress (CMS), mice were exposed to a variable sequence of mild and unpredictable stressors according to the procedure adapted from Willner et al. (33) and Moreau et al. (34). The procedure included the following: one overnight period of food and water deprivation for 16 h; two 5-min periods of social defeat; one period of continuous overnight illumination for 16 h; one 3-h 4 °C cold exposure; two 0.5-h periods of restraint stress; one overnight period of confinement to small cages (14 × 14 × 15 cm3, length × wide × height) for 16 h; one 3-h 40° cage tilt; and one overnight period in a soiled cage for 16 h (by adding 100 ml of water to the home cage). The CMS procedure was carried out in a randomized fashion for a 1 week period and repeated over 3 weeks. After CMS, mice were subjected to SE or EE for 7 days before behavioral tests.
Plasma Corticosterone Assay
Blood samples were obtained from the tail vein just before and after CMS and after exposure to SE or EE for 7 days and were centrifuged at 1000 × g, and plasma was separated and stored at −20 °C. Because behavioral tests were performed between 10:00 a.m. and 4:00 p.m., blood samples were collected at 10:00 a.m. (2 h after the start of the light cycle of experimental animals). Plasma corticosterone levels were determined using a commercially available enzyme immunoassay kit (Cayman Chemical, Ann Arbor, MI) according to the manufacturer's instructions.
Hippocampal Neuronal Cultures
Primary cultures of hippocampal neurons were prepared from newborn C57BL/6J mouse pups on postnatal day 0 as described elsewhere (35, 36). Mouse pups were decapitated, and hippocampi were dissected out in ice-cold Hanks' balanced salt solution (Invitrogen). Tissues were enzymatically digested with 0.25% trypsin (Sigma) for 15 min at 37 °C. Cells were disaggregated by trituration and plated on poly-l-lysine-coated Petri dishes or glass coverslips in Neurobasal-A medium containing B27 serum-free supplement (Invitrogen), 0.5 mm l-glutamine, and antibiotics (50 units/ml penicillin and 50 μg/ml streptomycin). Half of the growth medium was replaced every 3 days thereafter.
Quantitative Real Time PCR (qPCR)
Total RNA was isolated from hippocampal CA1 tissue lysates using a TriReagent kit (Molecular Research Center, Cincinnati, OH) and treated with RNase-free DNase (RQ1; Promega) to remove potential contamination by genomic DNA. Total RNA (1 μg) from samples was reverse-transcribed using a SuperScript cDNA synthesis kit (Invitrogen). qPCR was performed on the Roche LightCycler instrument (Roche Diagnostics) using the FastStart DNA Master SYBR Green I kit (Roche Diagnostics) according to the manufacturer's instructions. The primers used in this experiment were as follows: HIF-1α, 5′-TGCTTGGTGCTGATTTGTGA-3′ (forward) and 5′-GGTCAGATGATCAGAGTCCA-3′ (reverse); VEGF, 5′-GCTCTCTTGGGTGCACTGGA-3′ (forward) and 5′-CACCGCCTTGGCTTGTCACA-3′ (reverse); and 18 S rRNA, 5′-CAACTTTCGATGGTAGTCGC-3′ (forward) and 5′-CGCTATTGGAGCTGGAATTAC-3′ (reverse). Samples were amplified for 40 cycles consisting of 95 °C (20 s), 60 °C (20 s), and 72 °C (40 s), and PCR amplifications were repeated in duplicate. After amplification, equal volumes of PCR products were subjected to electrophoresis on 1% (w/v) agarose gels and visualized with ethidium bromide. A melting curve was created at the end of the PCR cycle to confirm that a single product had been amplified. Data were analyzed by LightCycler quantification software to determine the threshold cycle above background for each reaction. The relative transcript amount of the gene of interest, which was calculated using standard curves of serial RNA dilutions, was normalized to that of 18 S rRNA.
Western Blotting Analysis
Hippocampal tissue samples were lysed in ice-cold Tris-HCl buffer solution (TBS; pH 7.4) containing a mixture of protein phosphatase and proteinase inhibitors (50 mm Tris-HCl, 100 mm NaCl, 15 mm sodium pyrophosphate, 50 mm sodium fluoride, 1 mm sodium orthovanadate, 5 mm EGTA, 5 mm EDTA, 1 mm phenylmethylsulfonyl fluoride, 1 μm microcystin-LR, 1 μm okadaic acid, 0.5% Triton X-100, 2 mm benzamidine, 60 μg/ml aprotinin, and 60 μg/ml leupeptin) to avoid dephosphorylation and degradation of proteins and ground with a pellet pestle (Kontes Glassware). Samples were sonicated and spun down at 15,000 × g at 4 °C for 15 min. The supernatant was then assayed for total protein concentration using the Bradford protein assay kit (Bio-Rad). Each homogenate tissue sample was separated in a 7% SDS-polyacrylamide gel. Following transfer onto nitrocellulose membranes, blots were blocked in TBS containing 3% bovine serum albumin (BSA) and 0.1% Tween 20 for 1 h and then blotted overnight at 4 °C with antibodies for HIF-1α (1:1000; Novus, Littleton, CO), HIF-1β (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA), synapsin I (1:2000; Cell Signaling, Danvers, MA), PSD-95 (1:5000; Millipore, Temecula, CA), or β-actin (1:2000; Sigma). Each blot was probed with horseradish peroxidase (HRP)-conjugated secondary antibody for 1 h and developed using the ECL immunoblotting detection system (GE Healthcare). Immunoblots were analyzed by densitometry using Bio-profil BioLight PC software. Only film exposures that were not saturated were used for quantification analysis. Expression of HIF-1α or HIF-1β was evaluated relative to that of β-actin. Background correction values were subtracted from each lane to minimize variability across membranes.
Preparation of Synaptoneurosomes
Hippocampal CA1 tissues were minced in ice-cold homogenization buffer (0.32 m sucrose, 20 mm HEPES, 1 mm EDTA, 1× protease inhibitor mixture, 5 mm sodium fluoride, and 1 mm sodium vanadate) and homogenized using a glass tissue grinder with a Teflon pestle. The homogenate was centrifuged at 1000 × g for 10 min to remove nuclei and cell debris, and the supernatant was transferred to a new tube and centrifuged at 10,000 × g for 30 min to generate a crude synaptoneurosomal fraction. After centrifugation, the pellet was resuspended and sonicated in protein lysis buffer (50 mm Tris-HCl, 150 mm NaCl, 1% Triton X-100, 0.1% SDS, 2 mm EDTA, 1 mm sodium vanadate, 5 mm sodium fluoride, and 1× protease inhibitor mixture). All procedures were performed at 4 °C.
Construction and Production of Engineered Lentiviruses
Engineered self-inactivating recombinant lentiviruses were used for stably overexpressing or silencing the HIF-1α gene in mouse hippocampus in vivo or primary culture of mouse hippocampal neurons in vitro described by Huang et al. (36). Viruses were produced by co-transfection of lentiviral DNA with two helper plasmids in HEK293T cells as follows: vesicular stomatitis virus envelope glycoprotein and Δ8.9 (37). Medium containing recombinant lentiviruses was harvested 36–48 h after transfection and ultracentrifuged to obtain concentrated lentiviral particles. Pellets were resuspended by phosphate buffer solution with titers of 108 to 109 units/ml. For HIF-1α knockdown experiments, small hairpin RNAs (shRNAs) were expressed under a human H1 promoter and EGFP (as a marker for infection efficacy) under the CAG promoter (Addgene plasmid 12247). A set of shRNAs targeted at the coding region of mice HIF-1α (shRNA-HIF-1α) and one control shRNA directed against DsRed (shRNA-DsRed) were designed. Generation of short hairpin sequences were cloned into lentiviral vector using a PCR-based strategy, GTGATGAAAGAATTACTGAGT (shRNA-HIF-1α) and AGTTCCAGTACGGCTCCAA (shRNA-DsRed). The shRNAs were injected into the CA1 region of the dorsal hippocampus, and the specificity and efficiency of the shRNAs were confirmed by collecting virus-infected hippocampal CA1 tissues for Western blotting analysis. For overexpression of constitutively active HIF-1α (CA-HIF-1α), a lentiviral vector (UXIE) with bicistronic expression of transgenes and EGFP was constructed under the control of ubiquitin promoter and separated by an internal ribosomal entry site. CA-HIF-1α was cloned by internal deletion of the oxygen regulation degradation domain (amino acids 392–520), which resulted in a sustained increase in expression levels of HIF-1α under normoxic conditions (38), and the efficiency of CA-HIF-1α was proved by luciferase activity in our previous study (36).
For Flk-1 receptor knockdown experiments, the short hairpin sequences of Flk-1 receptors were cloned into lentiviral vector based on a PCR strategy, CCCTGTGAAGTATCTCAGTTA (shRNA-Flk-1). For experiments requiring overexpression of the dominant negative form of Flk-1 receptors (DN-Flk-1), the transmembrane domain sequence and 23 residues of the cytoplasmic part of full-length Flk-1 receptors were retained but truncated the intracellular kinase domain by deletion of the 561 C-terminal amino acids (39). The DN-Flk-1 was further constructed fusing with EGFP under the control of CAG promoter. For analysis of dendritic spines, hippocampal cultured neurons were grown on coverslips that transiently co-transfected with constructs encoding β-actin-targeted EGFP (β-actin-EGFP) with shRNA-DsRed, shRNA-Flk-1, or DN-Flk-1 plasmids using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
Construction of Lentiviral Vectors Expressing miRNAs and Antagomirs
To generate miRNA expression constructs, a 200-bp DNA fragment surrounding the individual miRNA sequence was amplified by PCR from the mouse genome library. The PCR products were then cloned into the lentiviral expression vectors under the control of H1 promoter. The EGFP-expressing cassette with its EF1α promoter was cloned into a lentivirus expression vector to visualize the infected cells. The following primers were used: miR-107, 5′-ACGCGTCGCGCCATTACTGCCGTAGACCA-3′ and 5′-ATCGATGAACTTAGCAATCTTTCAAA-3′; miR-20b, 5′-ACGCGTCGCATTTGAACGGCACAATTTC-3′ and 5′-ATCGATCAGCCATATATTCACATATA-3′; miR-134, 5′-ACGCGTCGCCAACCTTGGTGAGGCAGCTG-3′ and 5′-ATCGATATCCTGGTCCACTGAGCAGG-3′; and miR-138, 5′-ACGCGTCGCGGGTTCTGGGCTTTCTACCT-3′, and 5′-ATCGATTGAACATCACCTTTCACAGT-3′. All constructs were generated by using standard protocols in molecular biology and confirmed by DNA sequencing.
For stable interference with endogenous miRNA expression and function, we used antagomiR treatment (40, 41). The individual antagomiR was cloned into the lentiviral vector under the control of H1 promoter. Short hairpin sequences targeting different endogenous miRNAs were cloned into the lentiviral vector based on a PCR strategy as follows: miR-107, AGCAGCATTGTACAGGGCTATCA; miR-20b, CAAAGTGCTCATAGTGCAGGTA; miR-134, TGTGACTGGTTGACCAGAGGG; and miR-138, AGCTGGTGTTGTGAATCAGGCCG.
Stereotaxic Viral Injection
Recombinant lentiviruses that express control shRNA-DsRed, shRNA-HIF-1α, miR-107, or antagomiR-107 were injected bilaterally into the CA1 region of the dorsal hippocampus using standard stereotaxic procedures. Under pentobarbital anesthesia, concentrated virus stock solution was injected into the targeted sites (0.5 μl per site at 0.25 μl/min) by using a Hamilton syringe with a 34-gauge blunted tip needle. All mice received four sites of viral injection targeting the CA1 region of the dorsal hippocampus (coordinates: anteroposterior = −2 mm from bregma; lateral = ±1.8 mm; ventral = 1.7 mm; anteroposterior = −2.5 mm from bregma; lateral = ±2.3 mm; ventral = 1.9 mm) in accordance with the description by Franklin and Paxinos (42). Animals were allowed to recover for 10 days before starting the experiments.
Golgi Impregnation
Mice were deeply anesthetized with sodium pentobarbital (50 mg/kg intraperitoneally) and transcardially perfused with phosphate-buffered solution (PBS), and brains were then removed and immersed in Golgi-Cox solution at room temperature for 2 weeks. After transferring to a 30% sucrose solution for 48 h, 200-μm thick coronal sections were prepared by use of a vibrating microtome (VT1200S; Leica, Nussloch, Germany). The slices were subsequently alkalinized in 18.7% ammonia, fixed in Kodak Rapid Fix solution, dehydrated in alcohol, cleared with xylene, mounted onto gelatinized slides, and coverslipped under Permount (Fisher). Spine number was determined blind to the treatment condition. At least 10 pyramidal neurons from each animal (three animals per group) were chosen for analysis. Images of dendritic spines were taken from the secondary and tertiary branches of basilar dendrites (30–100 μm from the soma) of hippocampal CA1 pyramidal neurons by using the Olympus microscope equipped with a 100× 1.25 NA oil immersion objective. Multiple Z-stack images of neurons from CA1 regions were collected with the aid of a computer-assisted neuron tracing system (NeuroLucida, Microbrightfield, Inc.) and further reconstructed by using ImageJ software. Spines were defined as dendritic protrusions ≤2 μm in length, with an obvious head structure. The number of spines was counted with 20-μm dendrite segments and presented as the number of dendritic spines in 20 μm.
Quantification of VEGF-A Protein by Enzyme-linked Immunosorbent Assay
The amount of VEGF-A in hippocampal tissues or the release from cultured hippocampal neurons was measured by ELISA using a commercially available kit specific for mice VEGF-A (Bender MedSystems, Vienna, Austria) according to the manufacturer's instructions.
Quantification of BDNF by Enzyme-linked Immunosorbent Assay (ELISA)
The BDNF levels were measured with a conventional two-site enzyme-linked immunosorbent assay kit according to the manufacturer's protocol (Promega, Charbonnière, France). Briefly, hippocampal tissues were homogenized with ice-cold lysis TBS and centrifuged for 20 min at 15,000 × g at 4 °C. BDNF concentrations were calculated from regression analysis of human recombinant BDNF standard curve run in each assay and expressed as normalized per μg of tissue weight.
Plasmid Transfection and VEGF Treatment
For quantitative analysis of dendritic spine number in vitro, hippocampal neuronal cultures (3 × 105 cells) were grown on glass coverslips (10-mm diameter) inserted in Petri dishes. The mature neuronal cultures at 12 days in vitro (DIV) were co-transfected with 0.5 μg of β-actin-EGFP plasmid and 1 μg of shRNA-DsRed, shRNA-Flk-1, or DN-Flk-1 plasmid by Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
To determine the effect of VEGF on the number of dendritic spines, mature neuronal cultures at 12 DIV were transfected with the indicated plasmids for 3 days, followed by stimulation with VEGF (100 ng/ml) for 24 h. VEGF (Sigma) was first dissolved in distilled water as stock solution and further diluted into culture medium to achieve final concentrations of 1–100 ng/ml. The stock solutions of VEGF were aliquots stored at −20 °C.
Immunofluorescence Staining
Cultured hippocampal neurons were fixed with 4% paraformaldehyde in PBS for 15 min at room temperature. After fixation, neurons were washed three times with PBS at room temperature and then permeabilized and blocked simultaneously in a solution containing 2% goat serum, 3% BSA, and 0.2% Triton X-100 in PBS for 1 h at room temperature. The primary antibody against GFP (1:5000; Abcam) was added in blocking solution and incubated overnight at 4 °C to enhance the EGFP-expressed signal. After rinses with PBS, coverslips with neurons were incubated for 1 h at room temperature with appropriate Alexa Fluor 488-conjugated secondary antibodies diluted in 3% BSA, counterstained with nucleic marker 4,6-diamidino-2-phenylindole (1:2000; Santa Cruz Biotechnology), rinsed extensively in PBS, and mounted with ProLong Gold Antifade Reagent (Invitrogen).
Immunohistochemistry
Mice were deeply anesthetized with sodium pentobarbital (50 mg/kg, intraperitoneally) and perfused transcardially with PBS and 4% paraformaldehyde. After the perfusion, brains were removed and continued to fix in 4% paraformaldehyde for 48 h at 4 °C and then transferred to the solution containing 30% sucrose that was immersed at 4 °C for at least 48 h before slicing. Coronal slices were sectioned to a 40-μm thickness, washed with 0.3% Triton X-100, and then incubated for blocking with solution containing 3% goat serum in PBS. After blocking, floating tissue slices were washed with wash buffer (TBS-T, 10 mm Tris-HCl, 150 mm NaCl, and 0.025% Tween 20; pH 7.4), incubated with 1% H2O2 for 30 min to block endogenous peroxidase, and incubated in PBS. The sections were incubated in the primary antibody against GFP (1:1000; Abcam) overnight at 4 °C in PBS with 0.1% Triton X-100. Finally, sections were washed with TBS-T and then incubated with the secondary Alexa Fluor 488 anti-rabbit antibodies for 1 h at room temperature. The immunostained sections were collected on separate gelatin-coated glass slides, washed, dehydrated, and air-dried, and the slices were coverslipped with Permount (Fisher).
Image Acquisition and Quantification
Fluorescence microscopic images of neurons were obtained using an Olympus BX51 microscope (Olympus, Tokyo, Japan) or an Olympus FluoView FV1000 confocal microscope with sequential acquisition setting at a resolution of 1024 × 1024 pixels and a sampling of six consecutive optical sections in the Z-stack. The high magnification images were recorded with an Olympus Plan Apochromat 60× oil immersion objective (1.42 numerical aperture and 0.15 working distance). At least 20 neurons (∼3–5 dendrites per neuron) from three independent experiments were used to determine spine density for each experimental condition by using MetaMorph image analysis software (version 7.0, Molecular Devices, Downingtown, PA). The number of dendrites used for quantification is indicated in the figures. To quantify dendritic spine number, dendritic segments 50–100 μm from the soma were used, and each individual spine present on the dendrites was manually traced. The number of dendritic spines was measured at 50 μm distant from the soma and a 50-μm length of dendritic segment. Dendritic spines were defined as any protrusions of 0.5–5 μm in length that had a clear neck and a relatively large head structure (43, 44). Blind conditions were used for the morphometric quantification of acquired images.
Statistical Analysis
Data for each experiment were normalized relative to controls and presented as means ± S.E. The significance of any difference between means was calculated by using analysis of variance with Bonferroni's post hoc analyses in comparison between multiple groups or the unpaired Student's t test between two groups. Probability values (p) less than 0.05 were considered to be statistically significant. The number of experiments performed was indicated by n.
RESULTS
Short Term Exposure to EE Produces Antidepressant-like Effects
To determine whether EE has antidepressant-like effects in behavioral models of depression, we used the TST (30), a variable that is considered to reflect “behavioral despair,” to assess depressive-like behaviors in adult male mice previously housed in SE or EE for a period of 3 or 7 days. As shown in Fig. 1A, mice previously housed in EE for 7 days spent significantly less time immobile compared with mice previously housed in SE as indicated by a main effect of housing (F(1,42) = 6.8; p < 0.05) and a significant housing × treatment duration interaction (F(2,42) = 5.7; p < 0.05). In addition, previous EE treatment also had antidepressant-like effects in both the SPT and forced swimming test (data not shown), two other classical experimental models used to evaluate behavioral despair. However, no significant differences in total distance traveled and percentage of time spent in the central zone in the open field were observed in EE-housed mice when compared with SE-housed mice, indicating that previous exposure to EE did not alter the anxiety state of the animals (data not shown). Moreover, the antidepressant-like effects of enriched housing in the TST were still evident 1 week following EE treatment and were able to restore upon re-exposure to EE (Fig. 1B).
FIGURE 1.

Short term exposure to EE produced antidepressant-like effects. A, depressive-like behaviors were measured by the TST. The immobility time during a 3-min TST was measured in mice after housing in an SE or EE for a period of 3 or 7 days. B, schematic representation of the experimental designs for examining the lasting antidepressant-like effects by EE. The antidepressant-like effects in the TST were still evident 7 days following EE treatment and were able to be restored upon re-exposure to EE. C, levels of VEGF-A proteins in the hippocampus of mice subjected to SE or EE for 7 days. D, levels of BDNF proteins in the hippocampus of mice subjected to SE or EE for 7 days. E, correlation analysis of VEGF-A levels in the hippocampus of mice and the immobility time in the TST. F, correlation analysis of BDNF levels in the hippocampus of mice and the immobility time in the TST. G, immobility time in the TST for wild type (WT) or heterozygous Flk-1 knock-out (Flk-1+/−) mice subjected to SE or EE for 7 days. H, immobility time in the TST for homozygous BDNF-floxed or BDNF conditional knock-out (BDNF−/−) mice subjected to SE or EE for 7 days. The total number of animals examined is indicated by n in parentheses. Data are presented as means ± S.E. *, p < 0.05 compared with SE group.
EE Increases Hippocampal VEGF Expression Associated with the Antidepressant-like Effects
Because mice previously housed in EE for 7 days consistently showed antidepressant efficacy in the TST, we chose this assessment for all subsequent experiments designed to elucidate the mechanisms underlying the antidepressant-like effects of EE. We next evaluated potential mediators underlying the EE-induced antidepressant-like effects. Although EE may affect a wide range of signaling events (45), we chose to focus on the growth factor signaling pathways for the following two reasons: 1) increased hippocampal expression of VEGF and BDNF are observed after long term EE treatment (46–48), and 2) the effectiveness of antidepressant treatments are correlated with the expression levels of VEGF and BDNF in the hippocampus (49–52). When we measured VEGF-A and BDNF protein expression in the hippocampal tissue lysates of EE- or SE-housed mice at the end of the 7-day treatment period by ELISA, both hippocampal VEGF-A and BDNF levels in EE-housed mice (VEGF-, 35.3 ± 0.8 pg/mg tissue; BDNF, 254.2 ± 25 pg/mg tissue) were significantly higher than in SE-housed mice (VEGF-A, 29.8 ± 1.8 pg/mg tissue; BDNF, 165.5 ± 21 pg/mg tissue) (Fig. 1, C and D). To further characterize the relationship between growth factors and EE-induced antidepressant-like effects, we correlated the TST performance with the expression levels of VEGF-A or BDNF protein in the hippocampus. A significant inverse correlation between the immobility time in the TST and the levels of VEGF-A protein was observed (r = −0.64; p < 0.01; Fig. 1E), whereby the higher the levels of VEGF-A protein, the lower the immobility time obtained in the TST. However, the immobility time and BDNF protein levels were not correlated with each other (r = −0.08; p = 0.77; Fig. 1F).
VEGF exerts its biological functions via activation of the protein-tyrosine kinase receptors, VEGF receptor 1 (VEGFR1/Flt-1) or VEGF receptor 2 (VEGFR2/Flk-1), which differ considerably in their individual signaling properties (27). Because pharmacological blockade of the Flk-1 inhibited the action of chemical antidepressants (28, 29), we investigated whether VEGF signaling through the Flk-1 is required for EE-induced antidepressant-like effects. For this, we assessed the antidepressant-like effects of EE in heterozygous Flk-1 knock-out mice. There was no obvious difference in the immobility time between the heterozygous Flk-1 receptor knock-out mice previously housed in EE and SE for 7 days (Flk+/−-SE, 89.3 ± 5.4 s; Flk+/−-EE, 97.1 ± 7.1 s; p = 0.39; Fig. 1G). However, immobility time in the TST was significantly reduced in EE-housed wild type mice when compared with SE-housed mice (WT-SE, 91.2 ± 4.1 s; WT-EE, 64.6 ± 6.7 s; p < 0.01). To examine further the relationship between BDNF and EE-induced antidepressant-like effects, we measured the antidepressant-like effects of EE in BDNF conditional knock-out mice. As shown in Fig. 1H, EE-housed BDNF conditional knock-out mice (BDNF−/−-SE, 81.38 ± 3.8 s; BDNF−/−-EE, 56.6 ± 2.6 s; p < 0.01) and littermate controls (homozygous BDNF-floxed) (floxed-SE, 79.9 ± 5.2 s; floxed-EE, 50.8 ± 6.1 s; p < 0.01) showed a similarly decreased immobility time in the TST when compared with SE-housed mice. These results underscore an important role of VEGF/Flk-1 signaling in mediating EE-induced antidepressant-like actions.
We also determined whether EE has antidepressant-like effects in the CMS model of depression. The experimental design is shown in Fig. 2A. As expected, CMS exposure exhibited a significant reduction in the SPT (F(3,32) = 12.04; p < 0.01; Fig. 2B) and an increase in immobility time in the TST (F(3,32) = 15.8; p < 0.01; Fig. 2C), indications of elevated levels of depression. Exposure to EE for 7 days significantly reversed the CMS-induced behavioral deficits in both the SPT (CMS-SE, 61.3 ± 3.7%; CMS-EE, 75.2 ± 3.2%; p < 0.02) and TST (CMS-SE, 113.2 ± 3.9 s; CMS-EE, 91.8 ± 4.8 s; p < 0.01), when compared with SE-housed CMS mice. In addition, a significant increase in basal plasma corticosterone levels was noted in CMS mice before being housed in SE or EE (F(1,34) = 5.1; p < 0.001; Fig. 2D). However, we found no significant effect of housing treatment on basal plasma corticosterone levels in CMS mice (CMS-SE, 56.4 ± 2.7 ng/ml; CMS-EE, 58.8 ± 4.6 ng/ml; p = 0.65), indicating that the antidepressant-like effects of enriched housing is not due to the reversal of CMS-induced increases in plasma corticosterone levels.
FIGURE 2.
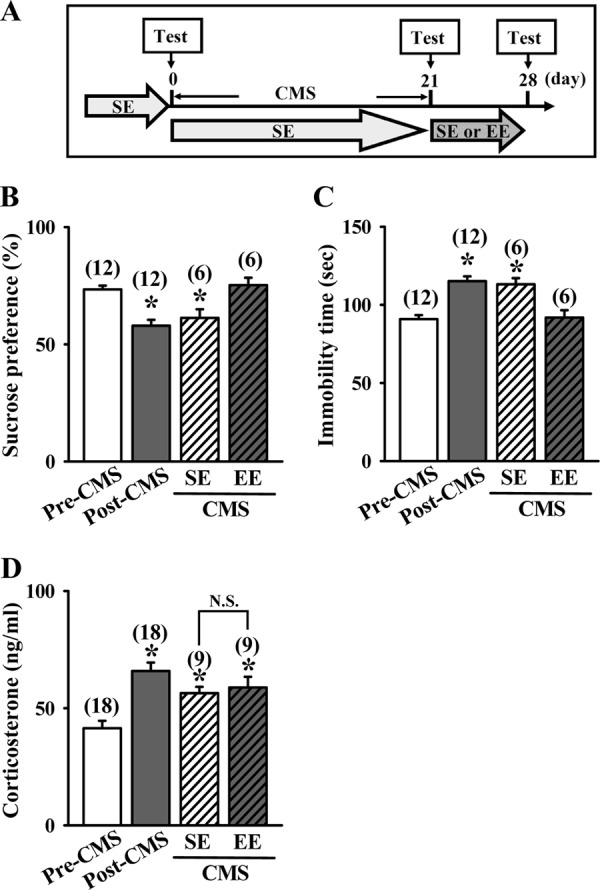
EE treatment leads to antidepressant-like effects in chronic mild stress model. A, schematic representation of the experimental designs for examining the EE-induced antidepressant-like effects in mice subjected to a 21-day CMS before SE or EE treatment. B, depressive-like behaviors were measured by the SPT. Preference ratios for sucrose solution over water were measured in CMS mice after housing in an SE or EE for a period of 7 days. C, depressive-like behaviors were measured by the TST. The immobility time during a 3-min TST was measured in CMS mice after housing in an SE or EE for a period of 7 days. D, plasma corticosterone levels were measured in CMS mice before and after housing in an SE or EE for a period of 7 days. The total number of animals examined is indicated by n in parentheses. Data are presented as means ± S.E. *, p < 0.05 compared with pre-exposed CMS (Pre-CMS) group.
EE Increases Dendritic Spine Number in Hippocampal CA1 Pyramidal Neurons
Because alterations in the morphology and number of dendritic spines are closely associated with depressive-like behaviors and their treatments (4, 14–16), we therefore examined the influence of EE on spine number of hippocampal CA1 pyramidal neurons based on Golgi-Cox staining. Golgi impregnation demonstrated that the spine number of CA1 pyramidal neurons was significantly increased in the hippocampus of EE-housed mice when compared with SE-housed mice (Fig. 3A). The number of dendritic spines was significantly elevated after housing mice in EE for 3 days and reached maximal level for 5 days (SE, 21.7 ± 0.6; EE-1 day, 22.5 ± 0.5; EE-3 days, 27.2 ± 0.5; EE-5 days, 30.9 ± 0.6; EE-7 days, 32.1 ± 0.6; F(4,96) = 61.4; p < 0.05; Fig. 3B). In addition, we also observed that the levels of presynaptic protein synapsin I and postsynaptic protein PSD-95 were significantly increased in the synaptoneurosomal fractions prepared from hippocampal CA1 tissues of EE-housed mice when compared with SE-housed mice (Fig. 3, C and D). Levels of synapsin I and PSD-95 were elevated after housing mice in EE for 5 days and reached maximal level for 7 days.
FIGURE 3.
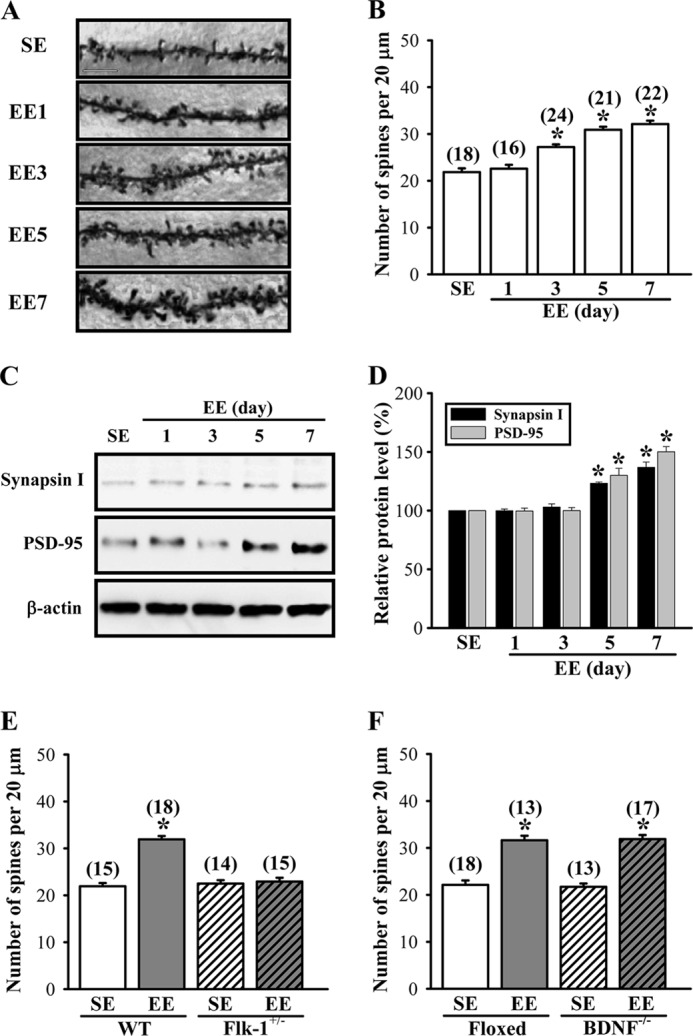
EE increases the number of dendritic spines in hippocampal CA1 pyramidal neurons. A and B, representative images and summary graph showing the number of Golgi-impregnated hippocampal CA1 neuronal dendritic spines in mice subjected to SE or EE for 1–7 days. Scale, 10 μm. C and D, representative immunoblot and corresponding densitometric analysis showing the expression levels of synapsin I and PSD-95 in the synaptoneurosomal fractions prepared from hippocampal CA1 tissues of mice subjected to SE or EE for 1–7 days. E, summary graph showing the number of dendritic spines of hippocampal CA1 pyramidal neurons in WT or Flk-1+/− mice subjected to SE or EE for 7 days. F, summary graph showing the number of dendritic spines of hippocampal CA1 pyramidal neurons in BDNF-floxed or BDNF−/− mice subjected to SE or EE for 7 days. The total number of dendrites examined is indicated by n in parentheses. Data are presented as means ± S.E. *, p < 0.05 compared with SE group.
To test whether VEGF/Flk-1 signaling is required for EE-induced increase in spine number, we measured the effect of EE on dendritic spine number in heterozygous Flk-1 knock-out mice (F(3,58) = 57.4; p < 0.01). As shown in Fig. 3E, no significant difference was observed in the spine number in hippocampal CA1 pyramidal neurons between the heterozygous Flk-1 knock-out mice previously housed in EE and SE for 7 days (Flk+/−-SE, 22.5 ± 0.5; Flk+/−-EE, 22.9 ± 0.8; p = 0.66). However, a significant increase in spine number was observed in EE-housed wild type mice when compared with SE-housed mice (WT-SE, 21.9 ± 0.7; WT-EE, 31.9 ± 0.6; p < 0.01). We also performed similar experiments in BDNF conditional knock-out mice (F(3,57) = 58.6; p < 0.01). EE-housed BDNF conditional knock-out mice (BDNF−/−-SE, 21.7 ± 0.5; BDNF−/−-EE, 31.9 ± 0.8; p < 0.01) and littermate controls (floxed-SE, 22.1 ± 0.5; floxed-EE, 31.6 ± 1.0; p < 0.01) showed a similar increase in spine number when compared with SE-housed mice (Fig. 3F). These results strongly support a role for VEGF/Flk-1 signaling in mediating an EE-induced increase in dendritic spine number in hippocampal CA1 pyramidal neurons.
To further confirm that VEGF can promote dendritic spine formation, we took advantage of an in vitro neuronal culture system to examine the role of VEGF/Flk-1 signaling in the regulation of spinogenesis. For this, hippocampal neuronal cultures were transfected with a DNA construct encoding β-actin-EGFP at 12 DIV. Three days after transfection, neurons were treated with different doses of VEGF (1–100 ng/ml) for 24 h, followed by the morphological analysis. We found that VEGF produced a significant and dose-dependent increase in the number of dendritic spines (F(3,159) = 25.2, p < 0.001; Fig. 4, A and B). In addition, the increase in spine number by VEGF was time-dependent, which became apparent as soon as 12 h and remained elevated at 24 and 48 h following VEGF (100 ng/ml) treatment (F(4,291) = 81.6, p < 0.001; Fig. 4, C and D). To determine whether the increase in spine number by VEGF was mediated through the activation of Flk-1, we co-transfected hippocampal neurons with β-actin-EGFP DNA construct and a lentiviral vector expressing shRNA specifically targeted against Flk-1 (shRNA-Flk-1) or a control vector expressing an shRNA sequence targeted against DsRed (shRNA-DsRed) at 12 DIV and then examined at 16 DIV after treatment with VEGF for 24 h. As shown in Fig. 5, A and C, the spine number was increased by VEGF treatment in hippocampal neurons transfected with shRNA-DsRed (control, 38.7 ± 0.9; VEGF, 47.4 ± 1.1; p < 0.01), but this increase was not observed in shRNA-Flk-1-transfected neurons (control, 37.6 ± 0.6; VEGF, 38.6 ± 0.6; p = 0.2). Neither shRNA-DsRed nor shRNA-Flk-1 transfection alone significantly altered the spine number. Similarly, overexpression of a dominant negative mutant of Flk-1 (DN-Flk-1) completely prevented the VEGF-induced increase in the spine number (control, 38.1 ± 0.6; VEGF, 38.4 ± 0.5; p = 0.78) when compared with neurons transfected with the control vector (control, 37.9 ± 0.7; VEGF, 46.5 ± 0.9; p < 0.001; Fig. 5, B and D). Neither DN-Flk-1 nor vector transfection alone significantly altered the spine number of hippocampal neuronal cultures.
FIGURE 4.

VEGF stimulates spinogenesis in primary cultures of mouse hippocampal neurons. A and B, representative images and summary graph showing the number of dendritic spines in hippocampal neuronal cultures in response to vehicle (Veh) or VEGF (1–100 ng/ml) treatment for 24 h. C and D, representative images and summary graph showing the number of dendritic spines in hippocampal neuronal cultures in response to VEGF (100 ng/ml) treatment for 6–48 h. The total number of dendrites examined is indicated by n in parentheses. Data are presented as means ± S.E. *, p < 0.05 compared with vehicle group.
FIGURE 5.

VEGF stimulates spinogenesis through the activation of Flk-1 receptors. A and C, representative images and summary graph showing the number of dendritic spines in shRNA-DsRed- or shRNA-Flk-1-transfected hippocampal neuronal cultures in response to VEGF (100 ng/ml) treatment for 24 h. B and D, representative images and summary graph showing the number of dendritic spines in vector (Vec)- or DN-Flk-1-transfected hippocampal neuronal cultures in response to VEGF (100 ng/ml) treatment for 24 h. The total number of dendrites examined is indicated by n in parentheses. Data are presented as means ± S.E. *, p < 0.05 compared with vehicle group. Con, control.
EE Stimulates VEGF Production by the Up-regulation of HIF-1α
To determine whether EE-induced elevation of VEGF protein production was due to an increase in new mRNA and protein synthesis, we examined the expression patterns of VEGF mRNA and protein in the hippocampus of EE-housed mice. We found that steady-state protein levels of VEGF were elevated after housing mice in EE for 3 days, reached maximal levels for 4 days, and remained elevated for 7 days (F(7,40) = 3.09; p < 0.05; Fig. 6A). In parallel, we observed that EE treatment caused a significant increase in VEGF mRNA levels based on qPCR analysis. The levels of VEGF mRNA were elevated after housing mice in EE for 2 days, reached maximal levels for 3 days, and remained elevated for 4 days (F(7,67) = 8.65; p < 0.01; Fig. 6B).
FIGURE 6.
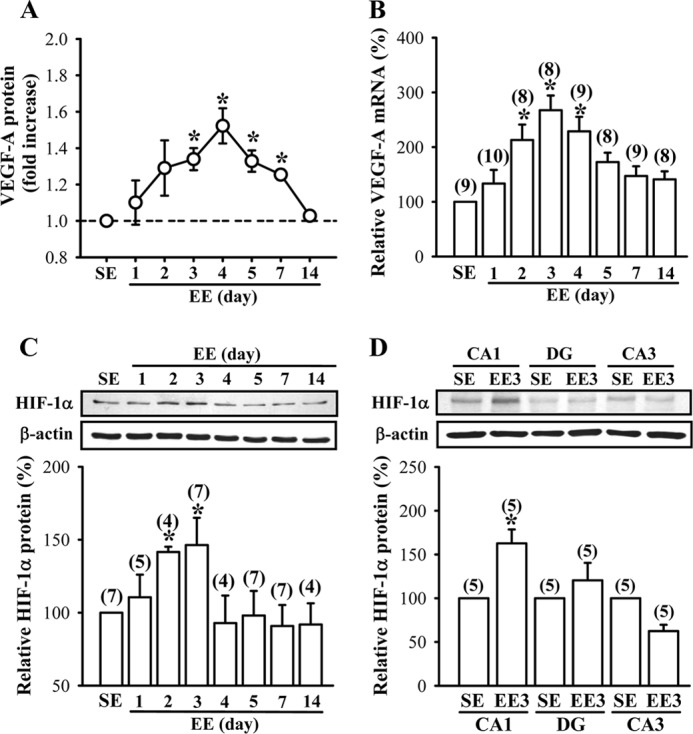
EE stimulates VEGF production by an up-regulation of HIF-1α. A, time course of EE-induced VEGF-A secretion in the hippocampus. B, time course of EE-induced VEGF-A mRNA expression in the hippocampus. The samples in A and B were collected from the same mice. C, representative immunoblots and corresponding densitometric analysis showing HIF-1α protein expression in the hippocampus of mice subjected to SE or EE for 1–14 days. D, representative immunoblots and corresponding densitometric analysis showing HIF-1α protein expression in the CA1, CA3, and dentate gyrus (DG) areas of the hippocampus of mice subjected to SE or EE for 3 days. The total number of animals examined is indicated by n in parentheses. Data are presented as means ± S.E. *, p < 0.05 compared with SE group.
Because the expression of VEGF is tightly regulated by HIF-1α (53–55), we therefore examined the influence of EE on HIF-1α protein expression in the hippocampus. Western blotting analysis revealed that HIF-1α protein in hippocampal CA1 region was significantly increased by exposure to EE for 2 or 3 days (F(7,38) = 2.3; p < 0.05; Fig. 6C). Notably, this enhancement did not persist by increasing the period of EE treatment. In addition, the increase in HIF-1α protein levels following a 3-day EE treatment was only observed in the CA1 region but not in the CA3 region or the dentate gyrus of the hippocampus (Fig. 6D). However, the expression levels of HIF-1β protein were not significantly affected by EE treatments (data not shown). These results suggest that the stimulatory effect of EE on VEGF production is mediated, at least in part, by the up-regulation of HIF-1α.
HIF-1α Is Essential for EE-induced Antidepressant-like Effects
The above results clearly indicate that HIF-1α plays a crucial role in EE-induced elevation of VEGF production, and the subsequent activation of VEGF/Flk-1 signaling is required specifically for EE-induced antidepressant-like effects. These findings prompted us to investigate whether knockdown of hippocampal CA1 HIF-1α could inhibit the EE-induced antidepressant-like effects. For this, we generated a lentiviral vector expressing shRNA specifically targeted against HIF-1α (shRNA-HIF-1α) and a control shRNA-DsRed. Measurement of HIF-1α levels by Western blotting analysis revealed significant reduction in expression of HIF-1α protein in hippocampal neuronal cultures transfected with shRNA-HIF-1α when compared with shRNA-DsRed-transfected neurons, confirming the ability of shRNAs to knock down HIF-1α (36). Having confirmed the knockdown efficiency of shRNA-HIF-1α in vitro, we then microinjected shRNA-DsRed or shRNA-HIF-1α bilaterally into the CA1 region of the dorsal hippocampus of mice. Ten days after the injection of shRNA, mice were subjected to EE or SE for 7 days, followed by the TST (Fig. 7A). Lentivirus-mediated gene delivery resulted in widespread transgene expression in the neurons, as shown by EGFP fluorescence in mice injected with shRNA-HIF-1α (Fig. 7B). In addition, a significant decrease in hippocampal HIF-1α protein expression was observed in mice transfected with shRNA-HIF-1α when compared with naive or shRNA-DsRed-transfected mice (Fig. 7C). Furthermore, we observed no significant difference in the expression of VEGF-A protein in the hippocampal tissue lysates of EE- or SE-housed shRNA-HIF-1α-transfected mice at the end of 7-day treatment period (SE, 111.6 ± 14.2%; EE, 115.3 ± 13.8%; p = 0.86; Fig. 7D). However, the protein levels of VEGF-A were significantly increased in EE-housed shRNA-DsRed-transfected mice when compared with SE-housed shRNA-DsRed-transfected mice (EE-housed mice, 164.0 ± 12.3%, versus SE-housed mice; p < 0.001). In the TST, EE-induced antidepressant-like effects were completely blocked in shRNA-HIF-1α-transfected mice (SE, 93.5 ± 7.0 s; EE, 104.1 ± 6.9 s; p = 0.31) when compared with SE-housed shRNA-HIF-1α-transfected mice (Fig. 7E). The immobility time in the TST was significantly reduced in EE-housed shRNA-DsRed-transfected mice when compared with SE-housed shRNA-DsRed-transfected mice (SE, 94.1 ± 5.4 s; EE, 67.0 ± 5.5 s; p < 0.01). Conversely, overexpression of the constitutively active form of HIF-1α (CA-HIF-1α) in the hippocampus significantly reduced the immobility time in the TST (F(3,24) = 12.4; p < 0.001) and occluded the antidepressant effect of EE (CA-HIF-1α-SE, 67.1 ± 4.2 s; CA-HIF-1α-EE, 69.6 ± 3.1 s; p = 0.64; Fig. 7F). We also investigated the role of HIF-1α in EE-induced increase in spine number of hippocampal CA1 pyramidal neurons (F(3,84) = 98.2; p < 0.01). We found no significant difference in spine number between the shRNA-HIF-1α-transfected mice housed in an EE and SE for 7 days (SE, 25.7 ± 0.5; EE, 25.1 ± 0.4; p = 0.37; Fig. 7, G and H). However, a significant increase in spine number was observed in EE-housed shRNA-DsRed-transfected mice when compared with SE-housed shRNA-DsRed-transfected mice (SE, 25.9 ± 0.4; EE, 35.5 ± 0.6; p < 0.001). These results suggest an important role for the HIF-1α-dependent induction of VEGF expression in mediating EE-induced elevation of dendritic spine number and antidepressant-like effects.
FIGURE 7.

HIF-1α is essential for EE-induced antidepressant-like effects. A, schematic representation of the experimental designs for examining the EE-induced antidepressant-like effects in mice receiving bilateral intrahippocampal injections of shRNA-DsRed or shRNA-HIF-1α before SE or EE treatment. B, representative images showing the delivery of EGFP shRNA-HIF-1α in the CA1 region of the dorsal hippocampus. Scale, 200 μm. C, representative immunoblots and corresponding densitometric analysis showing HIF-1α protein expression in hippocampal CA1 region of mice with or without (naive) intrahippocampal injections of shRNA-DsRed or shRNA-HIF-1α and subsequently subjected to EE for 7 days. D, levels of VEGF-A proteins in the hippocampus of shRNA-DsRed- or shRNA-HIF-1α-treated mice subjected to SE or EE for 7 days. E, immobility time in the TST for shRNA-DsRed- or shRNA-HIF-1α-treated mice subjected to SE or EE for 7 days. F, immobility time in the TST for GFP- or CA-HIF-1α-treated mice subjected to SE or EE for 7 days. G and H, representative images and summary graph showing the number of dendritic spines of hippocampal CA1 pyramidal neurons in shRNA-DsRed- or shRNA-HIF-1α-treated mice subjected to SE or EE for 7 days. Scale, 5 μm. The total number of animals or neurons examined is indicated by n in parentheses. Data are presented as means ± S.E. *, p < 0.05 compared with SE group.
The importance of HIF-1α-dependent induction of VEGF expression in the regulation of spinogenesis was further determined by transfection of hippocampal neuronal cultures with the lentiviral construct encoding CA-HIF-1α. Overexpression of CA-HIF-1α resulted in a significant increase in VEGF protein expression (F(2,17) = 8.6; p < 0.01) and occluded the stimulatory effect of VEGF on dendritic spines (CA-HIF-1α-Control, 48.2 ± 1.0; CA-HIF-1α-VEGF, 47.9 ± 0.7; p = 0.77; data not shown). These results demonstrate a new role for HIF-1α in controlling dendritic spine number.
Role of miR-107 in EE-induced Elevation of HIF-1α Protein Expression
We next asked if the elevated HIF-1α protein expression after EE treatment was due to an increase in HIF-1α mRNA transcription. Unexpectedly, we found that although HIF-1α protein levels were significantly increased after housing mice in EE for 2 or 3 days (F(7,38) = 2.3; p < 0.05), the mRNA levels of HIF-1α were not altered by EE treatment (F(7,70) = 0.12; p = 0.99; Fig. 8A). Recent reports have suggested that miRNAs can regulate HIF-1α expression in a broad spectrum of cancer cell lines (56–59). We therefore considered the possibility that EE may increase HIF-1α protein expression through a post-transcriptional regulation of gene expression via specific miRNAs. Because computational analysis (60, 61) and loss-of-function experiments (58, 59, 62) predicted that HIF-1α is a potential direct target of miR-107 and miR-20b, we measured the expression levels of these miRNAs in the hippocampal tissue lysates of EE- or SE-housed mice, using qPCR by LNA-based primers specific for individual mature miRNAs. We found that housing mice in EE for 1–3 days led to a significant decrease in miR-107 (F(3,30) = 4.59; p < 0.01), miR-20b (F(3,35) = 6.25; p < 0.01), and miR-134 (F(3,32) = 4.25; p < 0.05) expression (Fig. 8B). However, the expression of miR-138 was not altered by exposure to EE (F(3,37) = 0.42; p = 0.743). To directly verify the nature of HIF-1α inhibition by these miRNAs, we performed overexpression and knockdown experiments targeting different miRNAs in primary hippocampal neuronal cultures. As shown in Fig. 8C, lentiviral overexpression of miR-107, but not miR-20b, miR-134, or miR-138, in neurons resulted in reduced levels of HIF-1α protein relative to untransfected control or vector-transfected neurons. By contrast, abrogation of endogenous miR-107 function with the expression of antisense sequences specific for miR-107 (antagomiR-107) significantly increased the expression of HIF-1α protein relative to untransfected control or vector-transfected neurons (Fig. 8D). Specific inhibition of other three miRNAs by specific antagomirs had no effect on HIF-1α protein expression. Together, these results support a role for miR-107 in negatively regulating HIF-1α protein expression.
FIGURE 8.

miR-107 negatively regulates EE-induced HIF-1α protein expression. A, time course of changes in the expression of HIF-1α mRNAs and proteins in the hippocampus of mice subjected to SE or EE for 1–14 days. B, expression levels of miR-107, miR-20b, miR-134, and miR-138 in the hippocampus of mice subjected to SE or EE for 1–3 days. C, representative immunoblots and corresponding densitometric analysis showing that HIF-1α protein expression in hippocampal neuronal cultures was specifically decreased by overexpression (OE) of miR-107. D, representative immunoblots and corresponding densitometric analysis showing that HIF-1α protein expression in hippocampal neuronal cultures was specifically increased after transfection with antagomiR-107. The total number of animals examined or experiments performed is indicated by n in parentheses. Data are presented as means ± S.E. *, p < 0.05 compared with SE or control (Con) group. Vec, vector.
miR-107 Negatively Regulates Dendritic Spine Formation by Inhibiting the HIF-1α/VEGF/Flk-1 Signaling
To investigate a possible regulatory mechanism between miR-107 and dendritic spine formation, we knocked down endogenous miR-107 in hippocampal neuronal cultures obtained from wild type and heterozygous Flk-1 knock-out mice and examined dendritic spine number and VEGF-A protein expression. Transfection of wild type but not Flk-1 knock-out neurons with antagomiR-107 led to a significant increase in spine number when compared with control vector (wild type, F(1,137) = 21.9, p < 0.01; Flk-1 knock-out, F(1,119) = 2.11; p = 0.15; Fig. 9, A and B). Similar effects were observed in wild type, and Flk-1 knock-out neurons received VEGF (100 ng/ml) treatment for 24 h (wild type, F(1,126) = 32.9, p < 0.01; Flk-1 knock-out, F(1,96) = 0.64; p = 0.43). In addition, VEGF treatment did not cause a further increase in spine number in neurons transfected with antagomiR-107 (F(1,107) = 0.71; p = 0.4; Fig. 9, C and D). Co-transfection of antagomiR-107 with shRNA-HIF-1α suppressed antagomiR-107-induced increase in spine number (F(1,91) = 0.013; p = 0.91). Moreover, the expression of VEGF-A protein appears significantly higher in neurons transfected with antagomiR-107 when compared with control vector (wild type, F(1,12) = 9.28, p < 0.01; Flk-1 knock-out, F(1,12) = 2.94; p = 0.11; Fig. 9E). These results suggest that miR-107 negatively regulates dendritic spine formation by an inhibition of HIF-1α/VEGF/Flk-1 signaling in neurons.
FIGURE 9.

Inhibition of miR-107 increases dendritic spine number through the activation of VEGF/Flk-1 signaling. A and B, representative images and summary graph showing the number of dendritic spines in hippocampal neuronal cultures from WT or Flk-1+/− mice subjected to VEGF (100 ng/ml) treatment for 24 h or transfection with antagomiR-107. Con, control; Vec, vector. C and D, representative images and summary graph showing the number of dendritic spines in antagomiR-107-transfected neuronal cultures subjected to VEGF treatment or shRNA-HIF-1α co-transfection. E, levels of VEGF-A protein secretion in hippocampal neuronal cultures from WT or Flk-1+/− mice in response to Vec or antagomiR-107 treatments. The total number of dendrites examined or experiments performed is indicated by n in parentheses. Data are presented as means ± S.E. *, p < 0.05. N.S., not significant.
Role of miR-107 in EE-induced Antidepressant-like Effects
The above results clearly indicate that miR-107 plays a crucial role in the regulation of HIF-1α and VEGF expression as well as dendritic spine formation. We next examined whether miR-107 is involved in EE-induced antidepressant-like effects using an overexpression strategy. We microinjected lentiviral vectors expressing miR-107 bilaterally into the CA1 region of the dorsal hippocampus of mice. Ten days after the overexpression of miR-107, mice were subjected to EE or SE housing for 7 days, followed by the TST (Fig. 10A). Lentivirus-mediated gene delivery resulted in widespread transgene expression in the neurons, as shown by EGFP fluorescence in mice injected with miR-107 (Fig. 10B). Mice that received miR-107 exhibited no significant change in hippocampal HIF-1α protein expression after EE housing (SE, 90.1 ± 8.4%; EE, 111.5 ± 11.6%; p = 0.14) when compared with control vector (EE, 175.9 ± 10.9% versus SE-housed mice; p < 0.01; Fig. 10C). In addition, we observed no significant difference in expression of VEGF-A protein in the hippocampal tissue lysates of EE- or SE-housed miR-107-transfected mice at the end of the 7-day treatment period (SE, 120.3 ± 15.5%; EE, 108 ± 13.7%; p = 0.56; Fig. 10D). However, the protein levels of VEGF-A were significantly increased in EE-housed control vector-transfected mice when compared with SE-housed control vector-transfected mice (EE, 161.1 ± 16.3% versus SE-housed mice; p < 0.05). In the TST, EE-induced antidepressant activity was completely blocked in miR-107-transfected mice when compared with SE-housed miR-107-transfected mice (SE, 89.7 ± 2.5 s; EE, 108 ± 13.7 s; p = 0.36; Fig. 10E). Immobility time in the TST was significantly reduced in EE-housed control vector-transfected mice when compared with SE-housed control vector-transfected mice (SE, 93.7 ± 6.3 s; EE, 65.7 ± 6.5 s; p < 0.01). We also examined the influence of miR-107 overexpression on EE-induced increase in dendritic spine number in hippocampal CA1 pyramidal neurons. We found no significant difference in spine number between the miR-107-transfected mice housed in EE and SE for 7 days (SE, 25.1 ± 0.4; EE, 24.9 ± 0.5; p = 0.82; Fig 10, F and G). However, a significant increase in spine number was observed in EE-housed control vector-transfected mice when compared with SE-housed control vector-transfected mice (SE, 25.8 ± 0.4; EE, 35.6 ± 0.5; p = 0.82; p < 0.001). These findings strongly support a negative regulatory role of miR-107 in EE-induced elevation in spine number and antidepressant-like effects.
FIGURE 10.

Role of miR-107 in EE-induced antidepressant-like effects. A, schematic representation of the experimental designs for examining the EE-induced antidepressant-like effects in mice receiving bilateral hippocampal overexpression (OE) of miR-107 before SE or EE treatment. B, representative image showing the delivery of EGFP miR-107 in the CA1 region of the dorsal hippocampus. Scale bar, 200 μm. C, representative immunoblots and corresponding densitometric analysis showing HIF-1α protein expression in hippocampal CA1 region of mice with hippocampal overexpression of vector (Vec) or miR-107 and subsequently subjected to EE for 7 days. D, levels of VEGF-A proteins in the hippocampus of vector- or miR-107-overexpressing mice subjected to SE or EE for 7 days. E, immobility time in the TST for vector- or miR-107-overexpressing mice subjected to SE or EE for 7 days. F and G, representative images and summary graph showing the number of dendritic spines of hippocampal CA1 pyramidal neurons in vector- or miR-107-overexpressing mice subjected to SE or EE for 7 days. Scale bar, 5 μm. The total number of animals or neurons examined is indicated by n in parentheses. Data are presented as means ± S.E. *, p < 0.05 compared with SE group.
We finally evaluated whether a correlation exists between changes in the number of dendritic spines in hippocampal CA1 pyramidal neurons and antidepressant-like effects in mice. We pooled different treatment group data and found a significant inverse correlation between spine number and the immobility time in the TST (r = −0.75; p < 0.01; Fig. 11), confirming the strong relationship of dendritic spine number and depressive-like behaviors.
FIGURE 11.
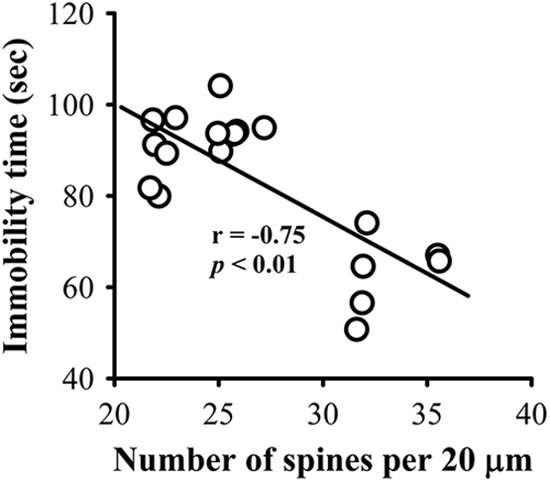
Correlation between the number of dendritic spines and antidepressant-like effects. Correlation analysis of the spine numbers of hippocampal CA1 pyramidal neurons and the immobility time in the TST (n = 19).
DISCUSSION
In this study, we highlight a novel role for HIF-1α-mediated VEGF expression in EE-induced increase in dendritic spine number in hippocampal CA1 neurons and behavioral features of an antidepressant performance. We confirm that HIF-1α is a specific target of miR-107 and suggest that EE enhances HIF-1α expression by a post-transcriptional regulation of HIF-1α gene through a mechanism that involves the down-regulation of miR-107. VEGF promotes dendritic spine formation and thereby exerts its antidepressant-like effects through the activation of Flk-1.
Exposure to EE has previously been shown to exhibit antidepressant-like effects in different animal models of depression (25, 26, 63). Although an EE may exert its antidepressant-like effects through a number of mechanisms (45), most of previous studies have focused on examining the regulation of hippocampal neurogenesis following long term exposure (3–8 weeks) to EE. Here, we extend these findings and demonstrate for the first time that previous short term exposure (3–7 days) to EE is able to produce antidepressant-like effects. Our results highlight the existence of a positive correlation between EE-induced increase in spine number in hippocampal CA1 pyramidal neurons and antidepressant-like effects. It is noteworthy that although EE-induced antidepressant-like effects are not permanent and progressively decay over a period of days, these effects are able to be restored upon re-exposure to EE. Given that increased environmental complexity and novelty may lead to greater levels of mental stimulation and physical activity (45), it will therefore be interesting to determine whether a more complex rearing environment may enhance and prolong the antidepressant-like effects of EE.
Recent studies have demonstrated that EE may alter the hippocampal expression of VEGF (48, 64), but no consensus has thus far emerged. Cao et al. (48) have shown that rats housed in EE for 4 weeks resulted in a prominent increase in hippocampal VEGF mRNA and protein levels. However, no significant change in VEGF mRNA levels in the hippocampus has been reported in mice housed in EE for 4 weeks (65). Therefore, it cannot be determined from these inconsistent results whether altered hippocampal VEGF expression may represent a molecular basis for mediating the effects of EE on brain function. Our data demonstrate a positive correlation between EE-induced increase in hippocampal VEGF protein levels and the antidepressant-like effects. In accordance with previous findings in the mouse model (65), our data also indicate that the initial increase in VEGF levels returns to near basal levels when the duration of exposure to EE is extended beyond 14 days. In addition, by using the heterozygous Flk-1 knock-out mice, we demonstrate that VEGF, acting through Flk-1 receptors, mediates the effects of EE on dendritic spine formation and depressive-like behaviors. Moreover, our results confirm and extend previous findings that, in addition to VEGF, exposure to EE also enhances BDNF expression in the hippocampus (46, 47). Surprisingly, we found no significant correlation between hippocampal BDNF levels and EE-induced antidepressant-like effects. This finding seems to contradict the previous hypothesis illustrating that depression is associated with reduced BDNF levels in the hippocampus, and BDNF-induced neuroplasticity contributes to the actions of antidepressants (13, 66). One possible interpretation of our finding is that VEGF production or signaling may underlie rapid antidepressant effects by regulation of hippocampal dendritic spine morphogenesis, whereas the more long term antidepressant effects are potentially mediated by BDNF signaling in regulating neurogenesis. Further studies are required to test this possibility.
A pressing question that follows these observations is how EE enhances VEGF gene expression. Our results suggest that HIF-1α is the principal regulator for governing the stimulatory effect of EE on VEGF production. We found that the increase in HIF-1α protein levels by EE is transient, consistent with our data showing no persistent increase in VEGF-A mRNA levels by EE treatment. Knockdown of HIF-1α by RNA interference completely blocked the EE-induced hippocampal VEGF production. Surprisingly, the levels of HIF-1α mRNA remained unchanged following exposure to an EE, suggesting that EE increases HIF-1α protein expression by a post-transcriptional mechanism. This finding is in contrast with observations made in a previous study demonstrating that the HIF-1α mRNA levels increased 6- and 7-fold after 3 and 6 h of EE treatment (67). These apparently disparate results could be attributed to the different sampling times. This interpretation is supported by previous observation that housing rats in EE for 4 weeks only induced a transient increase in HIF-1α mRNA expression in the hippocampus (48). Thus, it is reasonable to speculate the participation of other gene regulatory mechanisms in mediating the EE-induced increase in HIF-1α protein expression. Furthermore, under hypoxic conditions, lack of hydroxylation prevents HIF-1α degradation by the ubiquitin-proteasome system and leads to rapid HIF-1α accumulation (68, 69). Thus, we could not exclude the possibility that an inhibition of HIF-1α degradation may play some role in EE-induced increased HIF-1α protein expression.
Hundreds of miRNAs have been characterized, many of which are enriched in the brain (70, 71), but few studies have been conducted to determine the influence of EE on brain miRNA expression profile. We observed a significant down-regulation of miR107, miR20b, and miR134 following housing mice in EE. More importantly, our data indicate that expression of miR-107 is inversely correlated with HIF-1α protein levels in the hippocampus, suggesting an important role for miR-107 in controlling EE-induced HIF-1α expression and subsequent VEGF production. These results are in agreement with a previous computational approach that revealed the potential binding site for miR-107 in the 3′-untranslated region of HIF-1α mRNA (61). Our results also confirmed that expression levels of HIF-1β protein remain unchanged following housing mice in EE (data not shown). This contrasts with observations made in a recent study, which showed that knockdown of miR-107 enhances HIF-1β expression and hypoxic signaling in human colon cancer cells (72). This discrepancy could be due to the use of different cell types that may vary in their mode of action.
The accumulation of a specific miRNA is dependent on the rates of transcription, processing, and also decay. Thus, the decrease in miR-107 abundance in response to EE treatment could be associated with a decrease in gene transcription, an increase in miR-107 turnover rate, or both. Although the true molecular basis of how EE reduces the expression of miR-107 in the hippocampus remains unclear, a number of potential mechanisms may be involved. First, EE may act indirectly by reducing expression of endogenous p53 and subsequently decreasing miR-107 expression. Indeed, recent research indicates that p53 can stimulate miR-107 expression in human colon cancer cells (72). Second, EE may act on general miRNA processing machinery (i.e. diminished Dicer or Argonaute levels) to reduce miR-107 expression (73, 74). Although we cannot completely exclude this possibility, there was no significant difference in the Dicer protein levels between EE- and SE-housed mice (data not shown). Third, EE may promote miR-107 degradation by activation of small RNA-degrading nuclease (e.g. 5′–3′ exoribonuclease 2), resulting in a rapid reduction of miR-107 levels. In this regard, a previous study has revealed that housing rats in an EE cage for 7–10 days can lead to a significant increase in 5′–3′-exoribonuclease 2 expression in the hippocampus (75). Nevertheless, additional studies are needed to clarify all these possibilities.
A leading hypothesis of depression supports a role for hippocampal dendritic spine loss in the development of depressive-like behaviors (14–16). This hypothesis is supported by the observations that chronic antidepressant administration reverses the dendritic spine loss and ameliorates depressive-like behaviors in a rat learned helplessness model of depression (16, 17). Furthermore, Li et al. (4) demonstrate that the rapid antidepressant-like response following ketamine treatment is mediated by an increase in dendritic spine density in the prefrontal cortex. In accordance with these findings, these results show a strong correlation between the EE-induced increase in the number of dendritic spines in hippocampal CA1 pyramidal neurons and antidepressant activity and suggest a new pathway in regulating these processes via the down-regulation of miR-107. Although a recent study indicates a functional relevance of miR-134 in dendritic spine formation (76), our results do not support a role for miR-134 in mediating the effect of EE on dendritic spine formation.
These observations beg the question of how could the remodeling of hippocampal dendritic spines contribute to change depressive-like behaviors. The hippocampus has been firmly established in playing a critical role in learning and memory (77), regulating the brain's response to stress (78) and modulating the mesolimbic and mesocortical dopamine reward and motivation circuitries (79, 80), which all may make contribution to the mechanisms of depressive behaviors (16, 81). Because dendritic spines represent the major postsynaptic compartment for excitatory synaptic inputs, changes in the structure and number of dendritic spines may alter the functional activity of hippocampal neurons. Deterioration of hippocampal neuronal activity might contribute to the development of cognitive impairment, dysregulation of the stress response, and loss of motivation, common traits of depression in humans and animals (66).
Exercise has also been shown to increase the expression of a variety of neurotrophic factors, including BDNF, VEGF, and insulin-like growth factor-1 (IGF-1) (82–85), and to enhance dendritic length and numbers of spines in the hippocampus (86). As our enriched environments contained running wheels to enhance motor activity, it is thus possible that increased motor activity might contribute to some of antidepressant-like effects of EE. Although we cannot exclude this possibility, it should be noted that previous work on exercise-induced antidepressant-like effects were done using long term exercise regimens (3–4 weeks) (51, 87). It is noteworthy that similar antidepressant-like effects were seen in mice housed in the identical enriched cage with the locked running wheels for 7 days (data not shown), partially excluding the contribution of exercise in EE-induced antidepressant-like effects observed in our short term EE exposure protocol.
Recently, significant progress has been made at uncovering the role of VEGF in the actions of chemical antidepressants. Multiple classes of antidepressants, including selective serotonin and noradrenalin uptake inhibitors, have been reported to increase the expression levels of VEGF mRNA and protein in the hippocampus (28, 29). Likewise, intracerebroventricular infusion of VEGF mimics the behavioral actions produced by antidepressants (28). Because the induction of VEGF requires chronic administration of chemical antidepressants for at least 2 weeks and the inconvenience of the direct infusion of VEGF, a new strategy is needed, directed at a faster increase in hippocampal VEGF. We show here that short term exposure to EE is able to induce a rapid increase in hippocampal VEGF that ultimately produces antidepressive-like behaviors in both naive and CMS mice. It is noteworthy that although the results obtained from animal experiments cannot be fully translated into human experiments, our findings suggest that EE may provide a reservoir of protection against the potential detrimental effect of ensuring depression-inducing situations and have the ability to reduce or reverse a previously established depressive disorder. In addition, it is unclear whether EE may act synergistically with other antidepressant treatments to rapidly provide beneficial antidepressant effects. Furthermore, because activation of the HIF-1 transcriptional complex may regulate numerous downstream targets, some of these molecules may work together with VEGF to underlie the EE-induced antidepressant-like effects. Further studies are required to explore these issues in humans and animals.
In conclusion, we provide compelling evidence that previous short term exposure to EE can be highly effective to provide beneficial antidepressant-like effects. By using molecular genetic approaches, we show that these effects are mediated through the down-regulation of miR-107, leading to an increase in HIF-1α expression, which in turn enhances VEGF production in the hippocampus. Moreover, we observed that EE-induced antidepressant-like effects are significantly correlated with a VEGF/Flk-1-mediated increase in dendritic spine number in hippocampal CA1 pyramidal neurons. These data also suggest that VEGF may be a key mediator linking the environment to depressive-like behaviors (88).
Acknowledgments
We thank members of the Hsu laboratory and Dr. Ming-Hong Tai for helpful discussions and suggestions.
This work was supported by National Health Research Institute Research Grant NHRI-EX101-9618NI and by National Science Council Grant NSC99-2321-B-006-001, Taiwan.
- MDD
- major depressive disorder
- BDNF
- brain-derived neurotrophic factor
- CMS
- chronic mild stress
- DIV
- days in vitro
- EE
- enriched environment
- Flk-1
- vascular endothelial growth factor receptor 2
- HIF-1α
- hypoxia-inducible factor-1α
- IGF-1
- insulin-like growth factor-1
- miR
- microRNA
- qPCR
- quantitative real time PCR
- SE
- standard environment
- SPT
- sucrose preference test
- TST
- tail suspension test
- EGFP
- enhanced GFP.
REFERENCES
- 1. Wong M. L., Licinio J. (2001) Research and treatment approaches to depression. Nat. Rev. Neurosci. 2, 343–351 [DOI] [PubMed] [Google Scholar]
- 2. Kessler R. C., Berglund P., Demler O., Jin R., Koretz D., Merikangas K. R., Rush A. J., Walters E. E., Wang P. S. (2003) The epidemiology of major depressive disorder. Results from the National Comorbidity Survey Replication (NCS-R). JAMA 289, 3095–3105 [DOI] [PubMed] [Google Scholar]
- 3. Trivedi M. H., Greer T. L., Grannemann B. D., Church T. S., Galper D. I., Sunderajan P., Wisniewski S. R., Chambliss H. O., Jordan A. N., Finley C., Carmody T. I. (2006) TREAD. Treatment with exercise augmentation for depression. Study rationale and design. Clin. Trials 3, 291–305 [DOI] [PubMed] [Google Scholar]
- 4. Li N., Lee B., Liu R. J., Banasr M., Dwyer J. M., Iwata M., Li X. Y., Aghajanian G., Duman R. S. (2010) mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329, 959–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li N., Liu R. J., Dwyer J. M., Banasr M., Lee B., Son H., Li X. Y., Aghajanian G., Duman R. S. (2011) Glutamate N-methyl-d-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol. Psychiatry 69, 754–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zarate C. A., Jr., Singh J. B., Carlson P. J., Brutsche N. E., Ameli R., Luckenbaugh D. A., Charney D. S., Manji H. K. (2006) A randomized trial of an N-methyl-d-aspartate antagonist in treatment-resistant major depression. Arch. Gen. Psychiatry 63, 856–864 [DOI] [PubMed] [Google Scholar]
- 7. Diazgranados N., Ibrahim L., Brutsche N. E., Newberg A., Kronstein P., Khalife S., Kammerer W. A., Quezado Z., Luckenbaugh D. A., Salvadore G., Machado-Vieira R., Manji H. K., Zarate C. A., Jr. (2010) A randomized add-on trial of an N-methyl-d-aspartate antagonist in treatment-resistant bipolar depression. Arch. Gen. Psychiatry 67, 793–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cryan J. F., O'Leary O. F. (2010) Neuroscience. A glutamate pathway to faster-acting antidepressants? Science 329, 913–914 [DOI] [PubMed] [Google Scholar]
- 9. Frodl T., Meisenzahl E. M., Zetzsche T., Höhne T., Banac S., Schorr C., Jäger M., Leinsinger G., Bottlender R., Reiser M., Möller H. J. (2004) Hippocampal and amygdala changes in patients with major depressive disorder and healthy controls during a 1-year follow-up. J. Clin. Psychiatry 65, 492–499 [DOI] [PubMed] [Google Scholar]
- 10. Malykhin N. V., Carter R., Seres P., Coupland N. J. (2010) Structural changes in the hippocampus in major depressive disorder. Contributions of disease and treatment. J. Psychiatry Neurosci. 35, 337–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. O'Brien J. T., Lloyd A., McKeith I., Gholkar A., Ferrier N. (2004) A longitudinal study of hippocampal volume, cortisol levels, and cognition in older depressed subjects. Am. J. Psychiatry 161, 2081–2090 [DOI] [PubMed] [Google Scholar]
- 12. Gallassi R., Di Sarro R., Morreale A., Amore M. (2006) Memory impairment in patients with late-onset major depression: the effect of antidepressant therapy. J. Affect. Disord. 91, 243–250 [DOI] [PubMed] [Google Scholar]
- 13. Pittenger C., Duman R. S. (2008) Stress, depression, and neuroplasticity. A convergence of mechanisms. Neuropsychopharmacology 33, 88–109 [DOI] [PubMed] [Google Scholar]
- 14. Sousa N., Lukoyanov N. V., Madeira M. D., Almeida O. F., Paula-Barbosa M. M. (2000) Reorganization of the morphology of hippocampal neurites and synapses after stress-induced damage correlates with behavioral improvement. Neuroscience 97, 253–266 [DOI] [PubMed] [Google Scholar]
- 15. Law A. J., Weickert C. S., Hyde T. M., Kleinman J. E., Harrison P. J. (2004) Reduced spinophilin but not microtubule-associated protein 2 expression in the hippocampal formation in schizophrenia and mood disorders. Molecular evidence for a pathology of dendritic spines. Am. J. Psychiatry 161, 1848–1855 [DOI] [PubMed] [Google Scholar]
- 16. Hajszan T., Dow A., Warner-Schmidt J. L., Szigeti-Buck K., Sallam N. L., Parducz A., Leranth C., Duman R. S. (2009) Remodeling of hippocampal spine synapses in the rat learned helplessness model of depression. Biol. Psychiatry 65, 392–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hajszan T., Szigeti-Buck K., Sallam N. L., Bober J., Parducz A., Maclusky N. J., Leranth C., Duman R. S. (2010) Effects of estradiol on learned helplessness and associated remodeling of hippocampal spine synapses in female rats. Biol. Psychiatry 67, 168–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Salmon P. (2001) Effects of physical exercise on anxiety, depression, and sensitivity to stress: a unifying theory. Clin. Psychol. Rev. 21, 33–61 [DOI] [PubMed] [Google Scholar]
- 19. Southwick S. M., Vythilingam M., Charney D. S. (2005) The psychobiology of depression and resilience to stress: implications for prevention and treatment. Annu. Rev. Clin. Psychol. 1, 255–291 [DOI] [PubMed] [Google Scholar]
- 20. Greenwood B. N., Fleshner M. (2008) Exercise, learned helplessness, and the stress-resistant brain. Neuromolecular Med. 10, 81–98 [DOI] [PubMed] [Google Scholar]
- 21. Schloesser R. J., Lehmann M., Martinowich K., Manji H. K., Herkenham M. (2010) Environmental enrichment requires adult neurogenesis to facilitate the recovery from psychosocial stress. Mol. Psychiatry 15, 1152–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kempermann G., Gast D., Gage F. H. (2002) Neuroplasticity in old age. Sustained fivefold induction of hippocampal neurogenesis by long term environmental enrichment. Ann. Neurol. 52, 135–143 [DOI] [PubMed] [Google Scholar]
- 23. Bindu B., Alladi P. A., Mansooralikhan B. M., Srikumar B. N., Raju T. R., Kutty B. M. (2007) Short term exposure to an enriched environment enhances dendritic branching but not brain-derived neurotrophic factor expression in the hippocampus of rats with ventral subicular lesions. Neuroscience 144, 412–423 [DOI] [PubMed] [Google Scholar]
- 24. Wright R. L., Conrad C. D. (2008) Enriched environment prevents chronic stress-induced spatial learning and memory deficits. Behav. Brain Res. 187, 41–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Llorens-Martín M. V., Rueda N., Martínez-Cué C., Torres-Alemán I., Flórez J., Trejo J. L. (2007) Both increases in immature dentate neuron number and decreases of immobility time in the forced swim test occurred in parallel after environmental enrichment of mice. Neuroscience 147, 631–638 [DOI] [PubMed] [Google Scholar]
- 26. Veena J., Srikumar B. N., Mahati K., Bhagya V., Raju T. R., Shankaranarayana Rao B. S. (2009) Enriched environment restores hippocampal cell proliferation and ameliorates cognitive deficits in chronically stressed rats. J. Neurosci. Res. 87, 831–843 [DOI] [PubMed] [Google Scholar]
- 27. Ferrara N., Gerber H. P., LeCouter J. (2003) The biology of VEGF and its receptors. Nat. Med. 9, 669–676 [DOI] [PubMed] [Google Scholar]
- 28. Warner-Schmidt J. L., Duman R. S. (2007) VEGF is an essential mediator of the neurogenic and behavioral actions of antidepressants. Proc. Natl. Acad. Sci. U.S.A. 104, 4647–4652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Greene J., Banasr M., Lee B., Warner-Schmidt J., Duman R. S. (2009) Vascular endothelial growth factor signaling is required for the behavioral actions of antidepressant treatment. Pharmacological and cellular characterization. Neuropsychopharmacology 34, 2459–2468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Steru L., Chermat R., Thierry B., Simon P. (1985) The tail suspension test. A new method for screening antidepressants in mice. Psychopharmacology 85, 367–370 [DOI] [PubMed] [Google Scholar]
- 31. Dalvi A., Lucki I. (1999) Murine models of depression. Psychopharmacology 147, 14–16 [DOI] [PubMed] [Google Scholar]
- 32. Banasr M., Duman R. S. (2008) Glial loss in the prefrontal cortex is sufficient to induce depressive-like behaviors. Biol. Psychiatry 64, 863–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Willner P., Towell A., Sampson D., Sophokleous S., Muscat R. (1987) Reduction of sucrose preference by chronic unpredictable mild stress, and its restoration by a tricyclic antidepressant. Psychopharmacology 93, 358–364 [DOI] [PubMed] [Google Scholar]
- 34. Moreau J. L., Jenck F., Martin J. R., Mortas P., Haefely W. E. (1992) Antidepressant treatment prevents chronic unpredictable mild stress-induced anhedonia as assessed by ventral tegmentum self-stimulation behavior in rats. Eur. Neuropsychopharmacol. 2, 43–49 [DOI] [PubMed] [Google Scholar]
- 35. Balkowiec A., Katz D. M. (2002) Cellular mechanisms regulating activity-dependent release of native brain-derived neurotrophic factor from hippocampal neurons. J. Neurosci. 22, 10399–10407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huang Y. F., Yang C. H., Huang C. C., Tai M. H., Hsu K. S. (2010) Pharmacological and genetic accumulation of hypoxia-inducible factor-1α enhances excitatory synaptic transmission in hippocampal neurons through the production of vascular endothelial growth factor. J. Neurosci. 30, 6080–6093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kutner R. H., Zhang X. Y., Reiser J. (2009) Production, concentration, and titration of pseudotyped HIV-1-based lentiviral vectors. Nat. Protoc. 4, 495–505 [DOI] [PubMed] [Google Scholar]
- 38. Sutter C. H., Laughner E., Semenza G. L. (2000) Hypoxia-inducible factor 1α protein expression is controlled by oxygen-regulated ubiquitination that is disrupted by deletions and missense mutations. Proc. Natl. Acad. Sci. U.S.A. 97, 4748–4753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Millauer B., Shawver L. K., Plate K. H., Risau W., Ullrich A. (1994) Glioblastoma growth inhibited in vivo by a dominant-negative Flk-1 mutant. Nature 367, 576–579 [DOI] [PubMed] [Google Scholar]
- 40. Scherr M., Venturini L., Battmer K., Schaller-Schoenitz M., Schaefer D., Dallmann I., Ganser A., Eder M. (2007) Lentivirus-mediated antagomir expression for specific inhibition of miRNA function. Nucleic Acids Res. 35, e149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Surdziel E., Eder M., Scherr M. (2010) Lentivirus-mediated antagomir expression. Methods Mol. Biol. 667, 237–248 [DOI] [PubMed] [Google Scholar]
- 42. Franklin B. J., Paxinos G. (eds) (2008) The Mouse Brain in Stereotaxic Coordinates, 3rd Ed., Academic Press, New York [Google Scholar]
- 43. Li Z., Okamoto K., Hayashi Y., Sheng M. (2004) The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 119, 873–887 [DOI] [PubMed] [Google Scholar]
- 44. Abu-Elneel K., Ochiishi T., Medina M., Remedi M., Gastaldi L., Caceres A., Kosik K. S. (2008) A δ-catenin signaling pathway leading to dendritic protrusions. J. Biol. Chem. 283, 32781–32791 [DOI] [PubMed] [Google Scholar]
- 45. Nithianantharajah J., Hannan A. J. (2006) Enriched environments, experience-dependent plasticity, and disorders of the nervous system. Nat. Rev. Neurosci. 7, 697–709 [DOI] [PubMed] [Google Scholar]
- 46. Young D., Lawlor P. A., Leone P., Dragunow M., During M. J. (1999) Environmental enrichment inhibits spontaneous apoptosis, prevents seizures, and is neuroprotective. Nat. Med. 5, 448–453 [DOI] [PubMed] [Google Scholar]
- 47. Ickes B. R., Pham T. M., Sanders L. A., Albeck D. S., Mohammed A. H., Granholm A. C. (2000) Long term environmental enrichment leads to regional increases in neurotrophin levels in rat brain. Exp. Neurol. 164, 45–52 [DOI] [PubMed] [Google Scholar]
- 48. Cao L., Jiao X., Zuzga D. S., Liu Y., Fong D. M., Young D., During M. J. (2004) VEGF links hippocampal activity with neurogenesis, learning, and memory. Nat. Genet. 36, 827–835 [DOI] [PubMed] [Google Scholar]
- 49. Nibuya M., Morinobu S., Duman R. S. (1995) Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J. Neurosci. 15, 7539–7547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. De Foubert G., Carney S. L., Robinson C. S., Destexhe E. J., Tomlinson R., Hicks C. A., Murray T. K., Gaillard J. P., Deville C., Xhenseval V., Thomas C. E., O'Neill M. J., Zetterström T. S. (2004) Fluoxetine-induced change in rat brain expression of brain-derived neurotrophic factor varies depending on length of treatment. Neuroscience 128, 597–604 [DOI] [PubMed] [Google Scholar]
- 51. Greenwood B. N., Foley T. E., Day H. E., Campisi J., Hammack S. H., Campeau S., Maier S. F., Fleshner M. (2003) Freewheel running prevents learned helplessness/behavioral depression. Role of dorsal raphe serotonergic neurons. J. Neurosci. 23, 2889–2898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lee J. S., Jang D. J., Lee N., Ko H. G., Kim H., Kim Y. S., Kim B., Son J., Kim S. H., Chung H., Lee M. Y., Kim W. R., Sun W., Zhuo M., Abel T., Kaang B. K., Son H. (2009) Induction of neuronal vascular endothelial growth factor expression by cAMP in the dentate gyrus of the hippocampus is required for antidepressant-like behaviors. J. Neurosci. 29, 8493–8505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Semenza G. L. (1998) Hypoxia-inducible factor 1. Master regulator of O2 homeostasis. Curr. Opin. Genet. Dev. 8, 588–594 [DOI] [PubMed] [Google Scholar]
- 54. Sharp F. R., Bernaudin M. (2004) HIF-1 and oxygen sensing in the brain. Nat. Rev. Neurosci. 5, 437–448 [DOI] [PubMed] [Google Scholar]
- 55. Ke Q., Costa M. (2006) Hypoxia-inducible factor-1 (HIF-1). Mol. Pharmacol. 70, 1469–1480 [DOI] [PubMed] [Google Scholar]
- 56. Camps C., Buffa F. M., Colella S., Moore J., Sotiriou C., Sheldon H., Harris A. L., Gleadle J. M., Ragoussis J. (2008) Hsa-miR-210 is induced by hypoxia and is an independent prognostic factor in breast cancer. Clin. Cancer Res. 14, 1340–1348 [DOI] [PubMed] [Google Scholar]
- 57. Taguchi A., Yanagisawa K., Tanaka M., Cao K., Matsuyama Y., Goto H., Takahashi T. (2008) Identification of hypoxia-inducible factor-1α as a novel target for miR-17–92 microRNA cluster. Cancer Res. 68, 5540–5545 [DOI] [PubMed] [Google Scholar]
- 58. Lei Z., Li B., Yang Z., Fang H., Zhang G. M., Feng Z. H., Huang B. (2009) Regulation of HIF-1α and VEGF by miR-20b tunes tumor cells to adapt to the alteration of oxygen concentration. PLoS ONE 4, e7629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cascio S., D'Andrea A., Ferla R., Surmacz E., Gulotta E., Amodeo V., Bazan V., Gebbia N., Russo A. (2010) miR-20b modulates VEGF expression by targeting HIF-1α and STAT3 in MCF-7 breast cancer cells. J. Cell. Physiol. 224, 242–249 [DOI] [PubMed] [Google Scholar]
- 60. Hua Z., Lv Q., Ye W., Wong C. K., Cai G., Gu D., Ji Y., Zhao C., Wang J., Yang B. B., Zhang Y. (2006) miRNA-directed regulation of VEGF and other angiogenic factors under hypoxia. PLoS ONE 1, e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dolt K. S., Mishra M. K., Karar J., Baig M. A., Ahmed Z., Pasha M. A. (2007) cDNA cloning, gene organization, and variant specific expression of HIF-1α in high altitude yak (Bos grunniens). Gene 386, 73–80 [DOI] [PubMed] [Google Scholar]
- 62. Ayala de la Peña F., Kanasaki K., Kanasaki M., Tangirala N., Maeda G., Kalluri R. (2011) Loss of p53 and acquisition of angiogenic microRNA profile are insufficient to facilitate progression of bladder urothelial carcinoma in situ to invasive carcinoma. J. Biol. Chem. 286, 20778–20787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hattori S., Hashimoto R., Miyakawa T., Yamanaka H., Maeno H., Wada K., Kunugi H. (2007) Enriched environments influence depression-related behavior in adult mice and the survival of newborn cells in their hippocampi. Behav. Brain Res. 180, 69–76 [DOI] [PubMed] [Google Scholar]
- 64. During M. J., Cao L. (2006) VEGF, a mediator of the effect of experience on hippocampal neurogenesis. Curr. Alzheimer Res. 3, 29–33 [DOI] [PubMed] [Google Scholar]
- 65. Kuzumaki N., Ikegami D., Tamura R., Hareyama N., Imai S., Narita M., Torigoe K., Niikura K., Takeshima H., Ando T., Igarashi K., Kanno J., Ushijima T., Suzuki T., Narita M. (2011) Hippocampal epigenetic modification at the brain-derived neurotrophic factor gene induced by an enriched environment. Hippocampus 21, 127–132 [DOI] [PubMed] [Google Scholar]
- 66. Nestler E. J., Barrot M., DiLeone R. J., Eisch A. J., Gold S. J., Monteggia L. M. (2002) Neurobiology of depression. Neuron 34, 13–25 [DOI] [PubMed] [Google Scholar]
- 67. Rampon C., Jiang C. H., Dong H., Tang Y. P., Lockhart D. J., Schultz P. G., Tsien J. Z., Hu Y. (2000) Effects of environmental enrichment on gene expression in the brain. Proc. Natl. Acad. Sci. U.S.A. 97, 12880–12884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Maxwell P. H., Wiesener M. S., Chang G. W., Clifford S. C., Vaux E. C., Cockman M. E., Wykoff C. C., Pugh C. W., Maher E. R., Ratcliffe P. J. (1999) The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399, 271–275 [DOI] [PubMed] [Google Scholar]
- 69. Jaakkola P., Mole D. R., Tian Y. M., Wilson M. I., Gielbert J., Gaskell S. J., Kriegsheim A. v., Hebestreit H. F., Mukherji M., Schofield C. J., Maxwell P. H., Pugh C. W., Ratcliffe P. J. (2001) Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292, 468–472 [DOI] [PubMed] [Google Scholar]
- 70. Fiore R., Khudayberdiev S., Christensen M., Siegel G., Flavell S. W., Kim T. K., Greenberg M. E., Schratt G. (2009) Mef2-mediated transcription of the miR379-410 cluster regulates activity-dependent dendritogenesis by fine-tuning Pumilio2 protein levels. EMBO J. 28, 697–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bartel D. P. (2009) MicroRNAs. Target recognition and regulatory functions. Cell 136, 215–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yamakuchi M., Lotterman C. D., Bao C., Hruban R. H., Karim B., Mendell J. T., Huso D., Lowenstein C. J. (2010) p53-induced miRNA-107 inhibits HIF-1 and tumor angiogenesis. Proc. Natl. Acad. Sci. U.S.A. 107, 6334–6339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kai Z. S., Pasquinelli A. E. (2010) MicroRNA assassins. Factors that regulate the disappearance of miRNAs. Nat. Struct. Mol. Biol. 17, 5–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Marlatt M. W., Potter M. C., Lucassen P. J., van Praag H. (2012) Running throughout middle-age improves memory function, hippocampal neurogenesis, and BDNF levels in female C57Bl/6J mice. Dev. Neurobiol. 72, 943–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Koh S., Chung H., Xia H., Mahadevia A., Song Y. (2005) Environmental enrichment reverses the impaired exploratory behavior and altered gene expression induced by early-life seizures. J. Child Neurol. 20, 796–802 [DOI] [PubMed] [Google Scholar]
- 76. Gao J., Wang W. Y., Mao Y. W., Gräff J., Guan J. S., Pan L., Mak G., Kim D., Su S. C., Tsai L. H. (2010) A novel pathway regulates memory and plasticity via SIRT1 and miR-134. Nature 466, 1105–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Knierim J. J., Lee I., Hargreaves E. L. (2006) Hippocampal place cells. Parallel input streams, subregional processing, and implications for episodic memory. Hippocampus 16, 755–764 [DOI] [PubMed] [Google Scholar]
- 78. McEwen B. S. (2002) Sex, stress, and the hippocampus. Allostasis, allostatic load, and the aging process. Neurobiol. Aging 23, 921–939 [DOI] [PubMed] [Google Scholar]
- 79. Lisman J. E., Grace A. A. (2005) The hippocampal-VTA loop. Controlling the entry of information into long term memory. Neuron 46, 703–713 [DOI] [PubMed] [Google Scholar]
- 80. Cooper D. C., Klipec W. D., Fowler M. A., Ozkan E. D. (2006) A role for the subiculum in the brain motivation/reward circuitry. Behav. Brain Res. 174, 225–231 [DOI] [PubMed] [Google Scholar]
- 81. Winter C., von Rumohr A., Mundt A., Petrus D., Klein J., Lee T., Morgenstern R., Kupsch A., Juckel G. (2007) Lesions of dopaminergic neurons in the substantia nigra pars compacta and in the ventral tegmental area enhance depressive-like behavior in rats. Behav. Brain Res. 184, 133–141 [DOI] [PubMed] [Google Scholar]
- 82. Neeper S. A., Gómez-Pinilla F., Choi J., Cotman C. (1995) Exercise and brain neurotrophins. Nature 373, 109. [DOI] [PubMed] [Google Scholar]
- 83. Berchtold N. C., Kesslak J. P., Cotman C. W. (2002) Hippocampal brain-derived neurotrophic factor gene regulation by exercise and the medial septum. J. Neurosci. Res. 68, 511–521 [DOI] [PubMed] [Google Scholar]
- 84. Cotman C. W., Berchtold N. C., Christie L. A. (2007) Exercise builds brain health. Key roles of growth factor cascades and inflammation. Trends Neurosci. 30, 464–472 [DOI] [PubMed] [Google Scholar]
- 85. Martello G., Rosato A., Ferrari F., Manfrin A., Cordenonsi M., Dupont S. (2010) A microRNA targeting dicer for metastasis control. Cell 141, 1195–1207 [DOI] [PubMed] [Google Scholar]
- 86. Stranahan A. M., Khalil D., Gould E. (2007) Running induces widespread structural alterations in the hippocampus and entorhinal cortex. Hippocampus 17, 1017–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Duman C. H., Schlesinger L., Russell D. S., Duman R. S. (2008) Voluntary exercise produces antidepressant and anxiolytic behavioral effects in mice. Brain Res. 1199, 148–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Fournier N. M., Duman R. S. (2012) Role of vascular endothelial growth factor in adult hippocampal neurogenesis. Implications for the pathophysiology and treatment of depression. Behav. Brain Res. 227, 440–449 [DOI] [PMC free article] [PubMed] [Google Scholar]