

Background: Cethrax toxin from B. cereus inactivates mammalian cells through cytoplasmic ADP-ribosyltransferase activity.
Results: The crystal structure of Certhrax reveals that it has two domains, one that binds protective antigen and another that has ADP-ribosyltransferase activity.
Conclusion: Good inhibitors against the ADP-ribosyltransferase activity have been developed.
Significance: Certhrax may be an important virulence factor in B. cereus pathogenesis.
Keywords: ADP-ribosylation, Anthrax Toxin, Bacillus, Bacterial Toxins, Crystallography, Drug Development, Enzyme Inhibitors, Enzyme Kinetics, Bacterial Diseases, Enzyme Inhibitors
Abstract
We identified Certhrax, the first anthrax-like mART toxin from the pathogenic G9241 strain of Bacillus cereus. Certhrax shares 31% sequence identity with anthrax lethal factor from Bacillus anthracis; however, we have shown that the toxicity of Certhrax resides in the mART domain, whereas anthrax uses a metalloprotease mechanism. Like anthrax lethal factor, Certhrax was found to require protective antigen for host cell entry. This two-domain enzyme was shown to be 60-fold more toxic to mammalian cells than anthrax lethal factor. Certhrax localizes to distinct regions within mouse RAW264.7 cells by 10 min postinfection and is extranuclear in its cellular location. Substitution of catalytic residues shows that the mART function is responsible for the toxicity, and it binds NAD+ with high affinity (KD = 52.3 ± 12.2 μm). We report the 2.2 Å Certhrax structure, highlighting its structural similarities and differences with anthrax lethal factor. We also determined the crystal structures of two good inhibitors (P6 (KD = 1.7 ± 0.2 μm, Ki = 1.8 ± 0.4 μm) and PJ34 (KD = 5.8 ± 2.6 μm, Ki = 9.6 ± 0.3 μm)) in complex with Certhrax. As with other toxins in this family, the phosphate-nicotinamide loop moves toward the NAD+ binding site with bound inhibitor. These results indicate that Certhrax may be important in the pathogenesis of B. cereus.
Introduction
Pathogenic bacteria target host cells using an arsenal of toxic proteins that may alter cellular physiology through mechanisms such as protein modifications, pore formations, or proteolysis and may lead to cell death. Some bacterial toxins are responsible for the complete pathology of a disease, whereas others alter host immune response or aid in invasion of host cells (1). Renowned examples of bacterial toxins include diphtheria toxin, cholera toxin, botulinum toxin, and anthrax toxin.
Bacillus anthracis is famous as the causative agent of anthrax, which gained notoriety in 2001 after spores were sent through the United States Postal Service, infecting 22 people and killing five (2), and remains in the public eye as a potential bioterrorist threat. Inhalation anthrax is often fatal because diagnosis is not possible before antibiotic treatment becomes ineffective. Anthrax consists of three individually nontoxic proteins. Namely, the protective antigen (PA)2 is recruited to form heptamers on cell surface receptors, which can then bind anthrax lethal factor (LF) or edema factor (EF) to form a bipartite toxin. Endocytosis delivers the proteins to an endosome, where LF or EF can be translocated to the cytoplasm under acidic conditions. Once inside the cell, anthrax LF, which is a zinc metalloprotease, cleaves and inactivates MAPK kinases to interfere with cell signaling (3–5). Anthrax EF, which is a calcium- and calmodulin-dependent adenylyl cyclase, catalyzes the formation of cAMP and also affects cell signaling and ion fluxes (6, 7). Two plasmids, pXO1 and pXO2, are essential for anthrax toxicity. pOX1 contains genes for PA, LF, and EF, whereas pXO2 encodes a poly-γ-d-glutamic acid capsule that participates in virulence by evading the host immune system (8). These plasmids were thought to distinguish B. anthracis from other Bacillus species until recently, when related Bacillus cereus strains were found to contain similar plasmids.
B. cereus is a Gram-positive, spore-forming, rod-shaped bacterium, widely found in soil, dust, water, and the hospital environment. It has been widely accepted as a causative agent of emetic and diarrheal food poisoning. Because most B. cereus isolates are harmless, it is often dismissed as a nosocomial pest due to its ubiquitous nature. However, in immunocompromised patients, B. cereus may cause a number of illnesses, including septicemia, cutaneous infections, pneumonia, and periodontal disease (9). In recent years, more severe B. cereus infections have been reported in otherwise healthy individuals, involving overwhelming sepsis and sometimes fatal pulmonary infections resembling anthrax respiratory disease (10, 11). As a result, this bacterium is now gaining respect as a bona fide human pathogen. B. cereus G9241 is a strain 1 associated with severe pneumonia in previously healthy patients, has been well characterized, and has been shown to harbor a nearly complete pXO1 plasmid, similar to that of B. anthracis (11).
The mono-ADP-ribosyltransferase toxins (mARTs) are the principle causative agents of a wide range of diseases, including cholera, diphtheria, and whooping cough (12, 13). These toxic proteins bind NAD+, cleave it into ADP-ribose and nicotinamide components, and facilitate the covalent transfer of the ADP-ribose to a host cell protein target (usually), altering or inhibiting target activity. Due to this covalent addition, mART toxins may be solely responsible for or may contribute to the disease state caused by their native pathogenic acter. Toxins of this family have distinct and varied substrates, and even the protein residue modified is not consistent. mART enzymes lack global primary sequence homology but share a conserved fold pattern (SCOP code d.166.1.1.) in the catalytic domain and hallmark catalytic residues in the active site (13). Specifically, (i) a catalytic Arg preceded by an aromatic residue aids in NAD+ binding and scaffolding of the active site, (ii) a Ser-Thr-Ser motif on a β-strand stabilizes the NAD+ binding site, (iii) the ADP-ribosyl-turn-turn (ARTT) loop contains a catalytic Glu responsible for the ADP-ribosyltransferase activity and a Gln/Glu two residues upstream that may participate in substrate recognition, and (iv) a “phosphate-nicotinamide” (PN) loop contributes to NAD+ binding through hydrogen bonds with an Arg and aromatic residues. Historically, mART toxins have been divided into diphtheria-like (DT group) and cholera toxin-like (CT group), but they can be further divided based on their target substrate or domain organization (13, 14). Among others, the actin-targeting toxins include C2 toxin from Clostridium botulinum (15), ι-toxin from Clostridium perfringens (16), and Photox from Photorhabdus luminescens (17). These mART toxins share a secondary catalytic Glu residue on the ARTT loop and are usually AB binary toxins consisting of a mART domain and a cell-binding domain. By contrast, the C3-like toxins are small single domain enzymes targeting Rho GTPases. Examples include C3bot1 and C3bot2 from C. botulinum (18, 19); C3lim from C. limosum (20); and C3stau1, C3stau2, and C3stau3 from Staphylococcus aureus (21–23). Structures and biochemical data have allowed for reaction mechanisms to be proposed for several mARTs (24–28). Further identification and characterization of new mART toxins is increasing our collective understanding of how these enzymes work and, as a result, how to inhibit them.
In silico analysis has revealed a putative novel mART toxin, which we have named Certhrax, in the pathogenic B. cereus G4291 strain (29). We have cloned, purified, and characterized this enzyme to show that it is a mART toxin. Cytotoxicity studies in mouse macrophage cells indicate that, like anthrax LF, Certhrax gains entry into host cells via PA, and it is 60-fold more toxic than anthrax LF against this cell line. We have resolved the structure of Certhrax to 2.20 Å and show that it shares high structural similarity with two of the three domains in anthrax LF. Whereas anthrax LF includes an inactive mART domain and an active zinc metalloprotease domain, Certhrax differs by its active mART domain and lack of a metalloprotease domain. In the absence of the host protein target, Certhrax binds NAD+ and, like other mARTs, facilitates the scission of the NAD+ glycosidic bond between ADP-ribose and nicotinamide. Characterization of the glycohydrolase activity and NAD+-binding properties has allowed us to identify several inhibitory compounds against this toxin-catalyzed reaction. We present the structure of small molecule active site inhibitors, PJ34 and P6, in complex with Certhrax at 1.8 and 1.96 Å resolution, respectively. As the scientific and medical communities are becoming increasingly more aware of the serious infections caused by B. cereus, characterization of the first anthrax-like mART will help our understanding of the pathogenic behavior of these bacteria.
EXPERIMENTAL PROCEDURES
Unless otherwise noted, chemicals were purchased from Sigma-Aldrich.
Strains and Media
RAW264.7 murine macrophage cells (ATCC, TIB-71) were cultured in Dulbecco's modified Eagle's medium (DMEM) (Lonza, Basel, Switzerland) containing 10% fetal bovine serum (FBS) (Invitrogen) and penicillin (100 units/ml)-streptomycin (100 μg/ml) mixture (Lonza) in a water-jacketed CO2 incubator at 5% CO2, 37 °C in 25-cm2 culture flasks (Corning Glass). R3D cells, which were a kind gift of K. A. Bradley (UCLA), are an anthrax toxin-resistant cell line derived from RAW264.7 murine macrophage cells and were cultured in the same manner. Cells were routinely passaged once they reached 80–90% confluence. PA was a kind gift of Dr. Jeremy Mogridge (University of Toronto).
Inhibitors
Inhibitors P1, P3, and P6 were a gift from Guilford Pharmaceuticals (Baltimore, MD), originally part of a small, directed poly(ADP-ribose) polymerase library. V23 and V30 were identified as potential inhibitors through a screen against a previously studied mART toxin (30) and purchased from Chembridge Corp. (San Diego, CA). The P6 derivatives, P6C, P6D, P6F, and P6G, were designed and synthesized by Sinova Inc. (Bethesda, MD).
Cloning, Expression, and Purification of Certhrax
Certhrax was overexpressed in Escherichia coli Rosetta(DE3)pLysS (Novagen Biosciences Inc., Madison, WI) cells and purified using immobilized metal affinity chromatography. In brief, the open reading frame encoding the 476-residue Certhrax (GI:122687420) or a Q429A/E431A mutant (QXE) was cloned into a modified pET-28+ vector with an N-terminal hexa-His tag and a tobacco etch virus protease site. E. coli Rosetta cells were transformed with plasmid and grown at 37 °C in 2-liter cultures of 2× YT containing kanamycin (30 μg/ml). After induction with 1 mm isopropyl 1-thio-β-d-galactopyranoside, expression occurred overnight at 15 °C. Harvested cells were resuspended in 10 mm HEPES, pH 7.5, 500 mm NaCl, and 10% glycerol (Lysis buffer) and lysed using an Emulsiflex-C3 high pressure homogenizer (Avestin Inc., Ottawa, Canada). Following centrifugation at 14,000 × g for 30 min, the supernatant was passed through an 8-ml DEAE column. Column flow-through was loaded to a Zn2+-charged HiTrapTM Chelating HP column (GE Healthcare) equilibrated with Lysis buffer containing 10 mm imidazole and was eluted with a 0–250 mm imidazole gradient. Certhrax was concentrated using an Amicon Ultra-15 10K filter (Millipore, Billerica, MA) to ∼5 mg/ml in Lysis buffer.
Purification of Certhrax for Crystallography
For crystallization, truncated full-length Certhrax (residues 18–471) and catalytic domain (residues 242–469) were cloned into pNIC-CH (GenBankTM accession number EF199843) and pET-MHL (GenBankTM accession number EF456735; an alanine residue of the C-terminal fusion tag was deleted through primer design) vectors, respectively, and the resulting plasmids were transformed into E. coli BL21 (DE3) for protein overexpression. Both E. coli transformants were cultured in TB medium at 37 °C until A600 of 2.5–3.0 and induced with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside at 18 °C overnight. These proteins were purified as above, and truncated Certhrax was further purified by a Q anion exchange column (elution with a linear gradient of 0–0.5 m NaCl) and size exclusion chromatography equilibrated with gel filtration buffer (10 mm HEPES, pH 7.5, 10% glycerol, 2.5 mm tris(2-carboxyethyl)phosphine). For the catalytic domain of Certhrax, the N-terminal His tag was removed by tobacco etch virus protease before dialysis, and the dialyzed sample was directly loaded onto a size exclusion column. Both full-length and catalytic domain samples were concentrated to 25–30 mg/ml and stored at −80 °C until use.
FITC Labeling of Certhrax
Certhrax was fluorescently labeled at primary amines with a DyLight 488 amine-reactive dye kit following the manufacturer's instructions (ThermoScientific, Rockford, IL). The ratio of protein labeled was calculated and found to be 1.1 mol of fluorophore/mol of Certhrax.
NAD+ Glycohydrolase Assay
NAD+ cleavage was monitored using a fluorescence assay on a Cary Eclipse fluorescence spectrophotometer (Varian). Certhrax (5 μm) was mixed well with various concentrations of etheno-NAD+ (0–100 μm) in 20 mm Tris, pH 7.9. Increasing fluorescence intensity was monitored at excitation and emission wavelengths of 305 and 405 nm, respectively, for 300 s.
KD, IC50, and Ki Determination
Intrinsic tryptophan fluorescence quenching of Certhrax was used to determine the binding dissociation constant, KD, for both NAD+ and the inhibitors as described previously (31), with slight modifications. Briefly, 1 μm Certhrax in 50 mm NaCl, 20 mm Tris-HCl, pH 7.9, was titrated with inhibitor while fluorescence was monitored using excitation and emission wavelengths of 295 and 340 nm, respectively, using a Cary Eclipse fluorescence spectrophotometer. For inhibition studies, NAD+ glycohydrolase activity (5 μm Certhrax, 100 μm etheno-NAD+) was measured with various inhibitor concentrations. IC50 was calculated by non-linear regression curve fitting using GraphPad Prism version 5 (La Jolla, CA). These values were converted to Ki values using the Cheng-Prusoff equation (32), Ki = IC50/(1 + [S]/Km), where [S] is the NAD+ concentration and Km is for the NAD+ substrate. Experimental Ki values were determined for inhibitors, PJ34 and P6F, from Dixon plots of the kinetic inhibition data.
LD50 for Certhrax against RAW264.7 and R3D Cells with and without PA
Both RAW264.7 and R3D cells were diluted with supplemented DMEM to a final density of 2 × 104 cells/ml, and 100-μl aliquots were used to seed a 96-well plate. PA was diluted in fresh medium and was added at a final concentration of 100 ng/ml. Recombinant purified Certhrax was serially diluted in medium and added at final concentrations of 0.25–100,000 pg/ml. A cycloheximide control (5 μg/ml) was included in each assay as a positive control as well as controls for 100% cell viability in the absence and presence of PA. The 96-well plates were incubated at 37 °C in 5% CO2 for 72 h. Cell viability was determined using a 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) dye-based colorimetric assay as described previously (33). LD50 values were determined by extrapolation from each corresponding line of best fit (sigmoidal function in GraphPad Prism version 5.04), and the average value was reported ± S.D.
EC50 is the measure of a drug's efficiency and represents the concentration of a compound where 50% of its maximal effect is observed. It was determined for inhibitor P6 using RAW264.7 cells cultured in the presence of 10 ng/ml Certhrax and 100 ng/ml PA in 96-well plates for 96 h. P6 was added to the growth medium to give final concentrations ranging from 0.5 to 100 μm. Cell viability was measured using the MTT assay above, data were fit to a sigmoidal function in Origin 6.1, and EC50 values were reported with errors representing the S.D.
Fluorescence Confocal Microscopy
RAW264.7 cells were grown to 90% confluence, diluted to 2 × 104 cells/ml, seeded on glass coverslips in 6-well plates, and incubated in the absence or presence of PA (100 ng/ml) with Certhrax (1 × 106 pg/ml) for varying time periods. (Note that cells infected for only 0–60 min with Certhrax were preincubated with PA for 15 min before the addition of Certhrax). Cells were washed with PBS and fixed with 4% paraformaldehyde and then immediately stained with 0.1 μg/ml DAPI before mounting with Dako fluorescent mounting medium (Roche Applied Science, S3023). Images were acquired using confocal microscopy (Leica Upright DM 6000B) at ×63 magnification, a pinhole of 1 Airy unit, the FITC excitation and emission range (511–572 nm), and a pixel resolution of 1024 × 1024 (scanning speed of 400 Hz). The acquisition software was Leica Applications Suite Advanced Fluorescence (LAS AF) version 1.6.0. Images were taken as z-stacks and ImageJ (version 1.44p/Java 1.6.0_20) was used to generate two-dimensional images using z-projection and average intensity.
Crystallization, Data Collection, and Refinement of Certhrax and Certhrax-Inhibitor Complexes
Crystallization was performed by sitting drop vapor diffusion at 18 °C. Full-length (truncated) Certhrax was mixed with suramin (5 μm; Enzo Life Sciences, Farmingdale, NY) and incubated overnight at room temperature. Elastase was added (1:100 mol/mol; Sigma) to the protein solution immediately before crystallization trays were dispensed. Crystals were obtained against reservoirs containing 2.4 m sodium malonate and 10 mm HEPES, pH 7.2. Crystals of the catalytic domain of Certhrax in complex with PJ34 were obtained after overnight incubation with 1 mm PJ34 before crystallization in 2.9 m ammonium sulfate and 0.1 m BisTris, pH 6.4, within 2 days. Both crystals were cryoprotected in a 50:50 mixture of Paratone-N and mineral oil and frozen in liquid nitrogen for their structure determination. Finally, for the complex with inhibitor P6, the inhibitor was soaked into crystals by first transferring the crystal to a 4-μl drop of reservoir solution for 5 min. Then 2 μl of 10 mm P6 was added to the drop and incubated for 30 min. Crystals were rinsed in paratone for cryoprotection and frozen with liquid nitrogen.
Data sets were collected for full-length Certhrax crystals (apo and P6-bound) at the Canadian Light Source (beamline 08ID (34)) and for the Certhrax catalytic domain with PJ34 on a Rigaku FR-E rotating anode (Table 1). Data were processed using DENZO/SCALEPACK (35) and XDS/SCALA (36). The Certhrax-PJ34 structure was solved using molecular replacement with C. limosum C3 exoenzyme (Protein Data Bank entry 3BW8 (37)) and MOLREP (38). The Certhrax-apo and Certhrax-P6 structures were solved using molecular replacement with the Certhrax-PJ34 structure, a structure of the Certhrax PA-binding domain,3 and PHASER (39). PHENIX (40), COOT (41), and/or O (42) were used for interactive rebuilding, REFMAC (43) was used for final refinement of the Certhrax-apo and Certhrax-PJ34 structures, and Phenix.refine was used for the Certhrax-P6 structure, and MolProbity (44) was used for validation of model geometry in all cases.
TABLE 1.
Crystallographic data and refinement statistics for B. cereus Certhrax structures
| Certhraxc(PJ34) | CerthraxFL(apo) | CerthraxFL(P6) | |
|---|---|---|---|
| Diffraction data | |||
| X-ray source | Rigaku FR-E | CLS beamline 08ID (34) | CLS beamline 08ID |
| Wavelength (Å) | 1.5418 | 0.9762 | 0.9762 |
| Unit cell parameters (Å) | a = 31.9, b = 70.2, c = 98.7 | a = 61.37, b = 95.49, c = 190.65 | a = 61.31, b = 100.29, c = 191.09 |
| Space group | P212121 | P212121 | P212121 |
| Resolution range (Å)a | 57.2–1.8 | 40–2.20 | 48.50–1.96 |
| (1.9–1.8) | (2.28–2.20) | (2.03–1.96) | |
| Data completeness (%) | 98.7 (97.0) | 98.2 (96.0) | 99.14 (98.0) |
| Rsym (%)b | 6.6 (42.9) | 10.4 (69.8) | 6.0 (89.6) |
| Redundancy | 3.6 | 6.8 | 5.6 |
| Average I/σ(I) | 41.1 (4.2) | 25.8 (2.2) | 19.7 (2.3) |
| Refinement statistics | |||
| Molecular replacement program | MOLREP (38) | PHASER (39) | PHASER |
| Molecular replacement model | Protein Data Bank entry 3BW8 | Certhraxc and Certhrax N-terminal domain structurec | CerthraxFL |
| Rwork (%)d/Rfree (%)e | 20.9/25.7 | 24.2/28.8 | 18.8/22.8 |
| No. of atoms | |||
| Protein | 1,628 | 6,735 | 7,301 |
| Ligand | 22 | 52 | |
| Water | 163 | 26 | 493 |
| r.m.s. deviation from ideal | |||
| Bond length (Å) | 0.010 | 0.012 | 0.015 |
| Bond angles (degrees) | 1.179 | 1.3 | 1.069 |
| B-Factors (Å2) | 14.10 | 59.90 | 46.0 |
| Protein | 26.6 | 52.9 | 46.3 |
| Ligand | 27.9 | 35.3 | |
| Water | 35.9 | 30.4 | 43.9 |
| Wilson | 11.8 | 46.5 | 30.3 |
| Ramachandran plot (%) | |||
| Favored | 99.0 | 98.4 | 98.0 |
| Outliers | 0.0 | 0.0 | 0.3 |
a Values in parenthesis are for the highest resolution shell.
b Rsym = Σ|(I − 〈I〉)|IΣ(I), where I is the observed intensity.
c X. Guan, unpublished results.
d Σ‖Fo| − |Fc‖Σ|Fo|, where |Fo| and |Fc| are the observed and calculated structure factor amplitudes, respectively.
e The Rfree value was calculated with a random 5% subset of all reflections excluded from refinement.
RESULTS
B. cereus Encodes an Anthrax-like mART Toxin
The 476-residue Certhrax protein is found on the pBC218 plasmid of B. cereus strain G9241. This two-domain protein consists of a PA-binding domain and a mART domain. Specifically, the C-terminal mART domain shares up to 34% sequence identity with C2- and C3-like mART toxins. Primary structure alignment with other members of the mART family indicates that Certhrax includes all of the crucial catalytic residues necessary for activity. Fig. 1A compares active site residues of the Certhrax mART domain with sequences of other mART toxins. It is evident that the classic mART catalytic Arg in region 1, known to aid in NAD+ binding; the Ser-Thr-Ser motif in Region 2 that forms the scaffold for the active site; and the catalytic Glu-X-Gln motif in Region 3 crucial for activity are all present in Certhrax.
FIGURE 1.

In silico identification of Certhrax as a mono-ADP-ribosyltransferase. A, sequence alignment of Certhrax with several other mART toxins. Characteristic active site residues are shown to be conserved in region 1 (catalytic arginine, orange), region 2 (STS motif, green), and region 3 (catalytic QXE/EXE motif, pink). B, domain organization of B. anthracis anthrax LF (top) as compared with B. cereus Certhrax (bottom). Each toxin contains a separate PA protein (white) and an enzyme component (gray) which includes a PA-binding domain (pink) and a mART domain (green). Anthrax LF includes an additional insert domain (blue) and a zinc metalloprotease domain. C, SDS-PAGE gel and anti-His tag Western blot showing purified Certhrax. Lane 1, molecular mass standards (Bio-Rad) in kDa; lane 2, purified Certhrax; lane 3, Western blot, purified Certhrax.
Certhrax also shares 31% identity and 51% similarity with anthrax LF from B. anthracis and has similar domain organization, which is highlighted in Fig. 1B. Each protein contains a PA-binding domain and a mART domain; however, the latter is inactive in the anthrax LF. In addition, the C terminus of anthrax LF harbors an insertion in the mART domain and a zinc metalloprotease domain that confers its toxic effects. Both of these are absent in Certhrax, suggesting that the two toxins, although sharing some common structure and cell entry mechanism, do not share the same mechanism of lethality (i.e. target protein).
Certhrax Is Lethal to Cells and Gains Entry to Host Cells via Protective Antigen
Certhrax was overexpressed in Rosetta cells and purified at a yield of ∼4 mg/liter culture using immobilized metal affinity chromatography. The purity level was visualized by SDS-PAGE, and the protein was positively identified by Western blot analysis using an anti-His tag (Fig. 1C).
Based on the similarity to anthrax LF and on the homologous PA-binding domain, we predicted that Certhrax would also require PA for cell entry. RAW264.7 cells were selected for Certhrax infection assays because they have also been used as the host target cells for anthrax LF toxin, and a PA-resistant cell line is available. An infection assay of RAW264.7 cells with varying concentrations of Certhrax (0–100,000 pg/ml) in the presence of 100 ng/ml PA gave an LD50 of 98.6 ± 4.6 pg/ml (Fig. 2A) as measured by an MTT colorimetric assay 72 h postinfection. In the absence of PA, cell growth was not impaired, and little cytotoxicity was observed, even at 10 ng/ml, or 100 times the Certhrax LD50 dose. Like Certhrax, anthrax LF is not toxic in the absence of PA (45). To further corroborate this result, R3D cells, which lack the PA receptor, were treated with Certhrax (Fig. 2B). The cells did not show significant lethality until treated with extremely high doses of Certhrax, even in the presence of PA, supporting the finding that Certhrax achieves cell entry similarly to anthrax via a PA-mediated mechanism. The toxicity of Certhrax to the R3D cells at high doses may be due to the presence of a low affinity receptor or to the ability of PA to form a pore-like structure in membranes that may facilitate translocation of Certhrax into the host cell cytoplasm (46). Similar studies with anthrax LF give an LD50 of 6.1 ng/ml (47) using the same RAW264.7 cell line and an identical concentration of PA. Comparison with the LD50 of Certhrax indicates that Certhrax is 60 times more toxic toward the RAW264.7 cell line than its infamous counterpart.
FIGURE 2.

Certhrax cytotoxicity. A, LD50 curve of Certhrax with RAW264.7 cells in the presence (squares) and absence (circles) of 100 ng/ml PA and increasing concentrations of Certhrax. B, LD50 experiment similar to that in A, using R3D cells lacking the protective antigen receptor in the presence (squares) and absence (circles) of 100 ng/ml PA and increasing concentrations of Certhrax. C, effect of Certhrax on mouse macrophage cell growth as indicated by percentage viability of cells 72 h following the addition of 100 ng/ml PA and increasing concentrations of wild-type Certhrax (dark blue) or QXE variant of Certhrax (light blue), where the catalytic Gln and Glu have each been substituted with Ala. In A and B, Certhrax concentrations ranged from 0 to 100,000 pg/ml, and in A–C, cell viability was measured using an MTT colorimetric assay (described under “Experimental Procedures”). Error bars, S.D.
Certhrax Is a Novel Bacterial mART Enzyme
The infection assay was then used to evaluate the mART activity of the C-terminal domain of Certhrax. A catalytically inactive Certhrax in which both the hallmark Gln-429 and Glu-431 residues were substituted with Ala (QXE variant) was prepared. Proper folding was monitored by measuring the maximum wavelength of intrinsic tryptophan emission, which was 339 nm for both wild-type and QXE Certhrax. The lethality of this QXE variant was compared with wild-type Certhrax activity (Fig. 2C). Although 2.5 ng/ml Certhrax was enough to kill 100% of RAW264.7 cells, the catalytically inactive Certhrax was much less toxic because the cells remained ∼100% viable even at a 100 ng/ml dose. This indicates that these active site residues are required for the mART activity and that this mART enzymatic activity is responsible for the observed growth-defective phenotype in mouse fibroblasts. Furthermore, this QXE variant enzyme was more than 5,000-fold less active in vitro as compared with the wild-type enzyme (data not shown).
Confocal Microscopy Imaging of Cell Entry
In order to visualize the host cell entry of Certhrax, RAW264.7 cells in the presence of 100 ng/ml PA were treated with Certhrax covalently labeled with a fluorophore, DyLight488. Cells were infected with Certhrax for various times before being fixed and imaged using confocal microscopy (Fig. 3, A–I). At 5 min postinfection (Fig. 3D), weak fluorescence signal was seen, indicating that Certhrax had entered the cells. By 10 min postinfection (Fig. 3E), Certhrax was starting to localize in specific regions and was no longer evenly dispersed throughout the cell. Concurrent DAPI staining showed that the Certhrax was extranuclear in its cellular location (Fig. 3, B and J). After 20 min (Fig. 3F), the toxin was aggregating to form specific punctate regions within the cell, increasing in concentration until 1 h had elapsed (Fig. 3G). This correlates with previous images of anthrax LF infection (48), where LF is not uniformly distributed throughout the cell but seems to be coalescing on certain cellular organelles. By 24 h postinfection (Fig. 3H), the signal remained similar to that after 1 h; Certhrax, like anthrax LF, requires PA for cell entry (48, 49), so we expect that the two toxins may follow a similar pathway of cell entry. However, Certhrax does not appear to behave identically to anthrax LF once inside the cell. By 90 min postinfection with anthrax LF, Zornetta et al. (48) observed a small amount of weak punctate staining, predominantly cytoplasmic. This is in stark contrast to Certhrax, which continues to show intense non-uniform staining even up to 24 h after treatment without an apparent loss in fluorescence intensity. Notably, although anthrax LF and EF also enter cells in an identical manner via the PA receptor, they are each localized in different parts of the cell by 90 min post-treatment. Considering that its mechanisms of killing and toxicity is unique, it is not surprising that Certhrax also shows cellular localization distinct from that of anthrax LF. Extensive microscopy studies with numerous antibodies for cellular markers at a variety of time points may give more insight into the cellular entry pathway of Certhrax.
FIGURE 3.

Certhrax host cell entry. A and I, cells under differential interference contrast at 0 min and 24 h, respectively; B and J, DAPI staining at 0 min and 24 h, respectively. C–H, fluorescence confocal microscopy of Certhrax infection of RAW264.7 cells. Cells were fixed and stained with DAPI at varying time points postinfection with DyLight488 fluorophore-labeled Certhrax in the presence of 100 ng/ml PA. C, 0 min; D, 5 min; E, 10 min; F, 20 min; G, 1 h; H, 24 h postinfection. Scale bar, 10 μm.
NAD+ Glycohydrolase Activity and NAD+ Binding of Certhrax
NAD+ glycohydrolase activity is present as a secondary enzymatic activity in most mART enzymes and represents the first step of the mART reaction, where OH− serves as the nucleophile in the absence of a target protein (amino acid nucleophile) (26). In the absence of a natural protein substrate for mART enzymatic activity, NAD+ glycohydrolase activity of Certhrax was characterized using an established fluorescence-based assay (50). Certhrax NAD+ glycohydrolase activity exhibited Michaelis-Menten behavior, where the kinetic parameters (Table 2) indicate a strong glycohydrolase activity for Certhrax (Km = 41.6 ± 10.9 μm, kcat = 8.7 ± 1.0 min−1). Although some mART enzymes do not exhibit glycohydrolase activity, those that do are generally not as active as that seen with Certhrax. For example, Certhrax glycohydrolase activity is more than 300-fold higher than that of exotoxin A from P. aeruginosa (kcat = 1.67 ± 0.5 h−1) (31). The addition of the natural protein substrate to the fluorescence assay would lead to a sharp increase in the activity observed because mART activity is much stronger than the residual glycohydrolase activity of these enzymes. However, the addition of various concentrations of classical C2- and C3-like mART substrates, including purified α-skeletal actin and RhoA, resulted in no change in the activity observed, indicating that these are not substrates for Certhrax as they are for C2-like and C3-like toxins. A tryptophan fluorescence quenching assay was used to quantify NAD+ binding by titrating NAD+ into Certhrax. The dissociation constant, KD, for Certhrax with NAD+, 52.3 ± 12.2 μm, indicates a binding affinity for the substrate comparable with other mARTs (Table 2). For example, the actin-targeting Photox from P. luminscens has a KD of 11.2 ± 0.3 μm (17), whereas C3 from C. botulinum has a KD of 60 ± 6 μm (51).
TABLE 2.
Kinetic parameters of Certhrax glycohydrolase activity
Kinetic parameters were determined as described under “Experimental Procedures.” All measurements represent the mean ± S.E. of at least six replicates, from at least three independent experiments.
| Parameter | Certhrax |
|---|---|
| Km (μm) | 42 ± 11 |
| kcat (min−1) | 8.7 ± 1.0 |
| kcat/Km (m−1 min−1) | 2.1 × 105 |
| KD(NAD+) (μm) | 52 ± 12 |
Certhrax Crystal Structure
The structure of full-length Certhrax was resolved to 2.20 Å in the apo- (substrate-free) form. As shown in Fig. 4A, the Certhrax structure includes a PA-binding domain (residues 1–242; red) at the N terminus and a mART domain (residues 243–476; green) (also see Fig. 4B) with α/β topology at the C terminus. Superposition with the structure of anthrax LF (Protein Data Bank entry 1J7N; Fig. 4C) illustrates tertiary structural similarity in the mART domain (r.m.s. deviation = 4.022 over 153 atoms). However, a closer look at important catalytic residues of Certhrax highlights their absence in anthrax LF (Fig. 4D), which renders its mART domain inactive. The PA-binding domain of each toxin also aligns closely (r.m.s. deviation = 1.692 over 164 atoms); however, the mART and PA domains are twisted with respect to one another when comparing Certhrax with anthrax LF. This N-terminal domain consists of a 12-helix bundle alongside a four-stranded β-sheet. Several regions of anthrax LF domain 1 have been shown to play a role in PA binding and are relatively conserved in Certhrax as well (52). Specifically, the region of maximal homology between anthrax EF and LF has previously been shown to play a role in PA binding (53). Structural alignment shows that this region is also highly conserved in Certhrax (Fig. 4E), where the sequence from Val-110 to Lys-116 in Certhrax differs by only a single amino acid from the homologous sequence from Val-147 to Lys-153 in anthrax LF and aligns closely in tertiary structure. This supports our in vivo data showing that Certhrax gains entry to host cells via PA, similar to anthrax LF and EF.
FIGURE 4.

Structure of Certhrax. A, crystal structure of Certhrax (no ligand bound). The PA-binding domain (pink) and mART domain (green) are shown in a schematic representation. B, catalytic domain of Certhrax. Hallmark catalytic residues are shown in a stick representation in orange (Arg-341; R341), magenta (Gln-429 (Q429) and Glu-341 (E431)), and yellow (STS motif 387–389). The PN loop is highlighted in purple, and the ARTT loop is highlighted in blue. C, superposition of Certhrax (pale colors) with anthrax LF (bold colors), aligned with respect to the mART domains. Domains are colored as in Fig. 1B with the additional anthrax LF insertion domain and zinc metalloprotease domain in blue and gray, respectively. D, structural alignment of hallmark active site residues in Certhrax mART domain (light green) and aligned residues of anthrax LF (dark green). The highlighted region is shown in stick format and in text below. The numbers in parentheses indicate the number of residues not shown in the amino acid sequence. E, structural alignment of residues important for PA binding in Certhrax (pink) and anthrax LF (red). Highlighted residues shown in a stick representation are shown in text sequence below.
In the catalytic domain, Certhrax is similar to other mART toxins (Fig. 4B). The ARTT loop (residues 424–430) connects β-strands β5 and β6, as in ι-toxin and C3bot (54, 55), and includes the QXE catalytic motif. The secondary Glu/Gln residue of mARTs, located two residues upstream from the primary catalytic Glu, may play a role in substrate recognition (37). In actin-targeting toxins that attach ADP-ribose to an arginine residue, this secondary position is a Glu (56). In the case of Certhrax, as in the Rho GTPase target enzymes that modify an asparagine residue, a conserved Gln is present at this position. Additionally, an aromatic (Tyr) residue lies in the middle of the loop in Certhrax and is conserved among actin- and Rho GTPase-targeting mARTs and the mammalian ecto-ADP-ribosyltransferases and is also thought to play a role in substrate binding (13). The Ser-Thr-Ser motif is found on a β-strand and forms a scaffold for the active site. These three residues (positions 387–389) participate in 10 hydrogen bonds with other sites within the protein, including catalytic residues Arg-341 and Gln-429, and adjacent β-strands to stiffen the active site. The PN loop (residues 390–404) connects β-strands β3 and β4 and begins immediately following the Ser-Thr-Ser motif. In the absence of NAD+, the PN loop is pulled away from the active site, which is not uncommon, and it contains conserved aromatic and arginine residues, as seen in actin-targeting and C3-like mARTs, which are known to contribute to NAD+ binding (27, 51). In alignment with other C3-like and actin-targeting mARTs, Certhrax contains an α-helix that interacts with NAD+ in the active site and includes most of the conserved sequence (YX6/7INX2L) that is not observed in toxins of other mART subgroups.
Inhibition of Certhrax
A number of small molecule compounds were tested for inhibitory properties against Certhrax (Table 3). Several of these have previously been identified as poly-ADP-ribosyltransferase inhibitors (P1, P3, and P6), whereas others were discovered through a virtual screen for inhibitors against another mART toxin, cholix (V23 and V30) (30). Derivatives of P6 (P6C, P6D, P6F, and P6G) were synthesized in an attempt to improve and elucidate the properties of inhibitor binding and toxin inhibition. These inhibitors are competitive inhibitors against the NAD+ substrate for this mART toxin family, and inhibition plots two of the best inhibitors, P6F and PJ34, are shown in Fig. 5 (A and B). Experimental Ki values were determined from the data in Fig. 5, A and B, for PJ34 and P6F (14.7 and 2.7 μm, respectively). Those inhibitors showing the most promise were further characterized using fluorescence-based assays to determine the KD and IC50 for each compound (Table 4). Notably, inhibitor P6 shows the most promise as a lead compound for further development (KD = 1.7 ± 0.2 μm, Ki = 1.8 ± 0.4 μm). This compound differs by only a single oxygen atom from P6D, which is a weaker inhibitor (KD = 3.0 ± 0.3 μm, Ki = 22.5 ± 0.4 μm). Likewise, P6F and P6G differ by a hexameric ring in exchange for a chloride ion with a substantial difference in inhibition due to this change (for P6F, KD = 3.1 ± 0.3 μm, Ki = 3.6 ± 0.3 μm; for P6G, Ki = >295 μm).
TABLE 3.
Chemical structures and characteristics of small molecule inhibitors tested against Certhrax
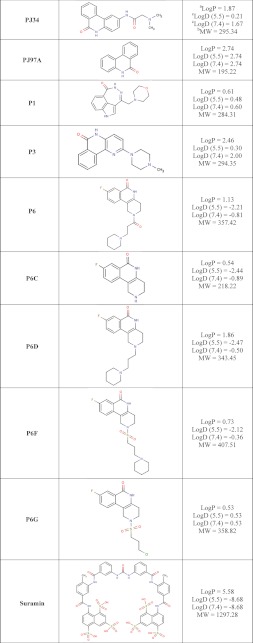
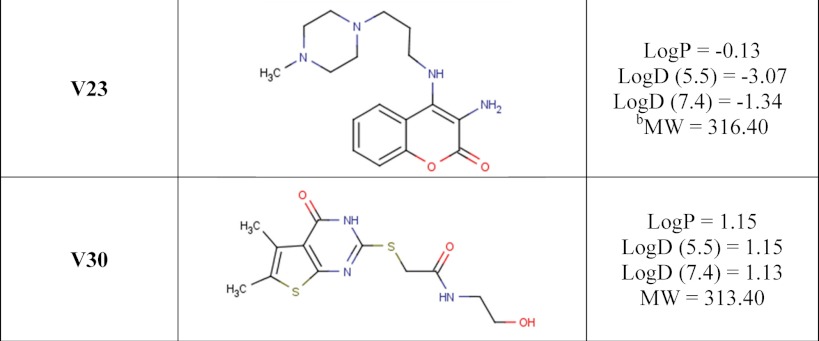
a The logP = log10 (partition coefficient), partition coefficient = [compound]octanol/[compound]water and was calculated (clogP) using an on-line clogP calculator by ChemAxon Ltd.
b,c The molecular weight (MW) (b) and logD pH (c) profiles were generated by the same on-line clogP calculator by ChemAxon Ltd. and represent the calculated log distribution coefficient, logD = log10 (distribution coefficient) where distribution coefficient, D = [microspecies]octanol/Σ[microspecies]water.
FIGURE 5.

Inhibition of Certhrax glycohydrolase activity. Shown are Lineweaver-Burk plots for Certhrax in the presence of various concentrations of inhibitors PJ34 (A) or P6F (B). The glycohydrolase activity of Certhrax was measured with 0 (circles), 6 (squares), 12 (diamonds), and 24 (triangles) μm inhibitor. vo indicates initial velocity. Error bars, S.E. C, inhibitor P6 protection from cytotoxicity of Certhrax-intoxicated cells as determined by half-maximal effective concentration (EC50). RAW264.7 cells were exposed to a lethal dose of Certhrax (10 ng/ml, 500 times LD50) at varying concentrations of compound P6. Error bars, S.D.
TABLE 4.
Binding and inhibition constants of 12 small molecule compounds with full-length Certhrax
| Inhibitor | KDa | IC50b | Kic |
|---|---|---|---|
| μm | μm | μm | |
| P6 | 1.7 ± 0.2 | 6.1 ± 1.2 | 1.8 ± 0.4 |
| P3 | 3.3 ± 0.4 | 7.2 ± 2.6 | 2.1 ± 0.8 |
| Suramin | 1.9 ± 0.7 | 10.6 ± 1.5 | 3.1 ± 0.4 |
| P6F | 3.1 ± 0.3 | 12.1 ± 1.1 | 3.6 ± 0.3 |
| P1 | 1.3 ± 0.2 | 13.0 ± 2.7 | 3.9 ± 0.8 |
| PJ97A | 1.6 ± 0.6 | 16.8 ± 1.6 | 5.0 ± 0.5 |
| PJ34 | 5.8 ± 2.6 | 32.3 ± 1.1 | 9.6 ± 0.3 |
| P6D | NDd | 76.1 ± 1.2 | 22.5 ± 0.4 |
| V30 | ND | 87.1 ± 1.3 | 25.8 ± 0.4 |
| P6C | ND | 121.3 ± 1.7 | 35.9 ± 0.5 |
| V23 | ND | 380.2 ± 3.3 | 112.4 ± 1.0 |
| P6G | ND | >1000 | >295 |
a KD values represent the mean ± S.E. from at least three independent experiments with at least nine replicates.
b IC50 values represent the mean ± S.E. from at least three independent experiments and at least six replicates.
c Ki values using the Cheng-Prusoff equation, Ki = IC50/(1 + [S]/Km), where [S] is the NAD+ concentration and Km is for the NAD+ substrate.
d ND, not determined.
To determine whether these inhibitors additionally have potential in vivo, the therapeutic effectiveness of the most potent inhibitor, P6, was determined in RAW264.7 cells by measuring the EC50 at a lethal concentration of Certhrax (Fig. 5C). The EC50 of P6 in the presence of 10 ng/ml toxin (100 times the LD50) was found to be 5.7 ± 1.2 μm (Table 3). The maximal effective concentration was ∼30 μm, which resulted in 90% cell viability. This indicates that the potency of P6 in vitro translates to in vivo characterization. Given previous studies that have also shown P6 as a potent inhibitor against exotoxin A of P. aeruginosa (30), we now have strong evidence that P6 is an ideal candidate for further development as a therapeutic agent and that it may have broad spectrum mART inhibition properties.
To further investigate the interactions of Certhrax with active site inhibitors, we determined the 1.96 Å structure of Certhrax in complex with inhibitor P6 (Fig. 6, A–C). This compound is buried in a cleft between the β1 and β3 strands. The P6 phenanthridinone ring is barely exposed to solvent due to enclosure by the ARTT and PN loops. The inhibitor is specifically stabilized in the active site by hydrogen bonds with Arg-342, located immediately beside the catalytic Arg-341, on the β1 sheet, Tyr-284 on the α3 helix, and an arene-arene interaction with Tyr-398 on the PN loop. The hexameric ring at the opposite end of the inhibitor is flanked on one side by the α3 helix but is otherwise solvent-exposed. Not surprisingly, this end of the molecule is less ordered, with higher crystallographic B-factors per residue. We have further determined the 1.8 Å structure of the mART domain of Certhrax (residues 242–468) in complex with inhibitor PJ34 (Fig. 6, D–F). In this case, the inhibitor is bound in the active site by hydrogen bonds with Arg-342 and a crystallographically ordered water molecule (Fig. 6D; note that the N,N-dimethyl acetamide tail of PJ34 is disordered and therefore not included in the model). Electron density allowed unambiguous placement of the PJ34 6-oxo-5,6-dyhydrophenanthridine moiety. However, we did not observe interpretable electron density for the glycyl substituent of PJ34, which we attribute to conformational disorder. Superposition of the mART domain of all three structures (Fig. 6G) shows that there are no large domain rearrangements due to inhibitor binding. The r.m.s. deviation of the apo catalytic domain with the P6-bound catalytic domain and with the PJ34-bound structure is 0.59 and 0.48 Å, respectively. Interestingly, comparison between the two inhibitor-bound structures gives a lower r.m.s. deviation of 0.33 Å when the mART domains are superimposed. The higher similarity of inhibitor-bound structures is in part due to a conserved loop movement upon inhibitor binding. The PN loop, spanning residues 390–398, includes Tyr-398, which forms an aromatic interaction with both inhibitor P6 and PJ34. At the furthest point, this loop moves 12 Å toward the active site to allow for the interaction. Comparison between the main chain atoms in the inhibitor-bound structures indicates that the PN loop position is conserved when an inhibitor is bound (r.m.s. deviation, 0.18), but superposition with the apo structure is significantly different (r.m.s. deviation for apo and PJ34 structures, 2.68 Å; for apo and P6 structures, 2.59 Å) (Fig. 6H). Other active site residues, including the Ser-Thr-Ser motif, the catalytic Arg-341, and the QXE motif, remain relatively unchanged despite inhibitor binding. This is in contrast to other mART enzymes, which show structural changes, specifically in the ARTT loop, upon substrate binding (51, 57, 58).
FIGURE 6.

Structure of Certhrax with inhibitors bound in the active site. A, full-length Certhrax (pink, PA-binding domain; yellow, mART domain) in complex with inhibitor P6 (black). B, interactions with P6 in the Certhrax active site. Important active site residues are shown in a stick representation, and P6 is shown in a ball-and-stick representation (black). Arg-342 (orange) and Tyr-284 (blue) form hydrogen bonds with the inhibitor (dashed lines), whereas Tyr-398 (cyan) is involved in aromatic interactions. Other active site residues are shown in pink (QXE motif) and green (STS motif). C, electron density of P6 bound to Certhrax. A simulated annealing omit map around the inhibitor is shown in blue (2Fo − Fc contoured at 1σ). D, catalytic mART domain of Certhrax (light blue) in complex with inhibitor PJ34 (black). E, interactions with PJ34 in the Certhrax active site. Important active site residues are shown in a stick representation, and PJ34 is shown in a ball-and-stick representation (black). Arg-342 (orange) and a crystallographically ordered water (purple) form hydrogen bonds with the inhibitor (dashed lines), whereas Tyr-398 (cyan) is involved in aromatic interactions. Other active site residues are colored as in B. F, electron density of PJ34 bound to Certhrax. A simulated annealing omit map (2Fo − Fc based upon the 1σ rendering) around the inhibitor is shown in blue. G, superposition of the Certhrax mART domain from three separate crystal structures, including full-length apo Certhrax (green), full-length Certhrax in complex with P6 (yellow), and Certhrax catalytic domain in complex with PJ34 (blue). H, superposition of active site residues of three catalytic domain structures, colored as in G. Active site residues are shown in a stick representation, and loop 389–398, including Tyr-398, is also shown in color. Inhibitors P6 (black) and PJ34 (gray) are illustrated in a ball-and-stick representation.
DISCUSSION
In this study, we consider the activity of an enzyme from the pathogenic B. cereus strain G9241, which causes anthrax-like pneumonia in humans. Researchers are increasingly recognizing B. cereus as a dangerous human pathogen, and this strain is a prime example. In general, different species in the B. cereus group are defined by plasmid-encoded features. Here we identify Certhrax, which is encoded on the plasmid, pBC218, unique to strain G9241. Most clinical strains of B. cereus, including G9241, are often associated with pXO1-like plasmids, encoding homologs to the anthrax toxins (59). However, a pXO2-like plasmid, carrying genes for capsule synthesis and generally required for anthrax disease, is absent. Previous studies have shown that G9241 produces a capsule, but it does not react to poly-d-glutamic acid antibodies, as would be expected for a capsule produced by pXO2. Instead, pBC218 and pBCXO1 each encode separate capsular material (polysaccharide and hyaluronic acid, respectively) to aid in evasion of the host immune response. Both plasmids contribute to the anthrax-like disease in mice (60), and ΔpBC218 mutant was found to be 20–80-fold less virulent than the wild-type strain. We suggest that Certhrax may contribute to the virulence of this plasmid in this pathogenic strain.
It is difficult to say whether Certhrax or anthrax LF is the parental factor for the other toxin. Phylogenetic studies have shown that the B. cereus group of species is actually a single species, although the nomenclature does not reflect this (61). B. cereus G9241 occurs earlier on a phylogenetic tree of the B. cereus group than B. anthracis (62), yet the genes involved in anthrax disease are carried by extrachromosomal plasmids, which play an important role in Bacillus diversity and allow for horizontal gene transfer. In general, a pathogenic species will lose characteristics over time, as opposed to gaining them, but in this case, Certhrax is missing the metalloprotease domain as compared with anthrax LF, whereas anthrax LF lacks an active mART domain as compared with Certhrax, so this logic does not help to determine which toxin is the precursor to the other. In the case of an outbreak of anthrax-like infections, an understanding of the causative agents and the ability to identify a responsible strain will be crucial for timely disease control (63). Characterization of participating virulence factors is one important step in this understanding.
Herein, Certhrax is shown to gain entry to host cells via PA association, in a similar fashion to anthrax LF and EF. However, cytotoxicity studies with mammalian cells indicate that Certhrax is 60-fold more toxic than anthrax LF. Certhrax joins actin-targeting VIP2 and Rho GTPase-ribosylating C3cer as mART toxins originating from B. cereus. However, Certhrax is unique in its origin from the pathogenic G9241 strain. Certhrax shows some similarity in structure and function to both the actin- and Rho-targeting toxins but does not clearly align with a single one of these subgroups of mART toxins. Structurally, Certhrax shares the same core fold in the catalytic domain as other mARTs (CATH 3.90.176.10). The Certhrax ARTT loop contains a secondary catalytic Gln residue as in Rho-targeting toxins, and an aromatic residue mid-loop that is conserved in both actin- and Rho-targeting mARTs. However, an α3 helix that specifically interacts with NAD+ in other C2- and C3-like toxins is shorter in Certhrax. Additionally, a large movement of the ARTT loop upon NAD+ binding that is found in several other mART structures (51, 57, 58) does not occur upon binding of active site inhibitors in Certhrax. Furthermore, extensive attempts to identify the host protein target of Certhrax have so far been inconclusive. Typical mART substrates, such as eukaryotic elongation factor 2, actin, or RhoA, are not modified by Certhrax. We propose that Certhrax may belong to a new class of mARTs, bridging the well characterized C2- and C3-like mART families, but more intriguingly, also linking the mART toxins to anthrax LF. We are currently working with more exhaustive techniques to identify the natural substrate.
Structural alignment of Certhrax with other mART domains indicates that Certhrax aligns approximately as well with the inactive mART domain of anthrax LF (r.m.s. deviation, 3.9 Å) as with several other mART toxins (r.m.s. deviation, 1.9–2.5 Å). The primary difference remains the 80-residue insertion domain of anthrax LF, which interrupts the mART domain (64). This helical bundle domain is required for anthrax LF activity (47) but may not be required in Certhrax, where the metalloprotease domain is absent. The missing catalytic residues required for mART activity in anthrax LF begs the question whether Certhrax gained this function over time and lost the insertion and zinc metalloprotease domain or whether anthrax LF lost the mART activity and acquired additional domains. Perhaps the evolution was driven by the redundancy of two highly potent mechanisms of toxicity in a single protein. Alternatively, these two toxins may simply represent two different approaches to inactivate eukaryotic target proteins and disrupt host cell function.
Inhibition studies identified several compounds (competitive inhibitors of the NAD+ substrate) that will be useful as leads in the development of potent inhibitors against Certhrax. Specifically, inhibitor P6 binds and inhibits glycohydrolase activity with low micromolar affinity. Crystal structures of P6 and PJ34 in complex with Certhrax indicate that the inhibitors interact in a common fashion with residues within the binding/catalytic site, as would be expected for competitive inhibitors of the NAD+ substrate. Specifically, hydrogen bonding with Arg-342 and the movement of the PN loop allow an aromatic interaction with Tyr-398. The rearrangement of side chains for this interaction has been seen upon NAD+ binding with other mART toxins, such as C3bot and VIP2 (25, 51); however, in these cases, there was no large loop movement to position the aromatic side chains, as seen with Certhrax. Analysis of the binding mechanisms in these toxin-inhibitor complexes will be invaluable toward the development of potent therapeutics against Certhrax and related mART toxins. Moreover, because the active site of mART enzymes is reasonably conserved, the development of a powerful inhibitor for Certhrax may be applicable to several other enzymes in this family. Translation of the inhibitory effects to a cell-based system indicates that the P6 inhibitor is not toxic to the cells, can pass the cell membrane, and retains similar efficacy as demonstrated in vitro against the purified enzyme. Arising from these studies, P6 provides an excellent lead compound for the development of more potent inhibitors against Certhrax and other mART toxins.
During the initial stage of this work, the inhibitor suramin gained significant attention because crystals of full-length Certhrax were reproducibly obtained in the presence of this compound. Intriguingly, this compound shows relatively tight binding (KD = 1.9 ± 0.7), and inhibition of Certhrax (Ki = 3.1 ± 0.4). However, suramin is not seen in the crystal structures, and extensive co-crystallization trials with suramin produced only apo crystals. The role of suramin in crystallization and inhibition of full-length Certhrax remains an unsolved question.
Altogether, our observations contribute to the understanding of the toxicity of a pathogenic strain of B. cereus, G9241, and add a new member to the family of mART toxins. Continued work can lead to the development of more effective therapeutics that target these bacterial virulence factors and more efficient disease control in the event of an outbreak of anthrax-like disease.
Acknowledgments
We thank Dawn White for excellent technical assistance. We acknowledge Xinyu (Johnny) Guan for the crystallization of the N-domain of Certhrax4 and thank Limin Shen for providing multiple clones for Certhrax. We are grateful to Bum-Soo Hong for early work on the crystallography and to Kyung-Phil Kim for help with DNA sequencing. We thank Zachari Turgeon for initial work on Certhrax cytotoxicity and inhibitor development and testing. We also thank Dr. Robert Fieldhouse for bioinformatics analysis of Certhrax and for insightful discussions. Research described here was performed using beamline 08ID-1 at the Canadian Light Source, which is supported by the Natural Sciences and Engineering Research Council of Canada, the National Research Council Canada, the Canadian Institutes of Health Research, the Province of Saskatchewan, Western Economic Diversification Canada, and the University of Saskatchewan.
This study was funded by the Canadian Institutes of Health Research (to A. R. M. and H. W. P.), Cystic Fibrosis Canada (to A. R. M.), the Human Frontier Science Program (to A. R. M.), and a National Sciences and Engineering Research Council graduate fellowship (to D. V.). The Structural Genomics Consortium is a registered charity (Number 1097737) and receives funds from the Canadian Institutes for Health Research, the Canadian Foundation for Innovation, Genome Canada through the Ontario Genomics Institute, GlaxoSmithKline, Karolinska Institutet, the Knut and Alice Wallenberg Foundation, the Ontario Innovation Trust, the Ontario Ministry for Research and Innovation, Merck and Co., Inc., the Novartis Research Foundation, the Swedish Agency for Innovation Systems, the Swedish Foundation for Strategic Research, and the Wellcome Trust.
The atomic coordinates and structure factors (codes 4GF1, 4FK7, and 4FXQ) have been deposited in the Protein Data Bank (http://wwpdb.org/).
D. Visschedyk, A. Rochon, W. Tempel, S. Dimov, H.-W. Park, and A. R. Merrill, unpublished results.
X. Guan, unpublished results.
- PA
- protective antigen
- EF
- edema factor
- LF
- lethal factor
- mART
- mono-ADP-ribosyltransferase
- r.m.s.
- root mean square.
REFERENCES
- 1. Burns D. L., Barbieri J. T., Iglewski B. H., Rappuoli R. (2003) Bacterial Protein Toxins, American Society for Microbiology Press, Washington, D. C [Google Scholar]
- 2. Jernigan J. A., Stephens D. S., Ashford D. A., Omenaca C., Topiel M. S., Galbraith M., Tapper M., Fisk T. L., Zaki S., Popovic T., Meyer R. F., Quinn C. P., Harper S. A., Fridkin S. K., Sejvar J. J., Shepard C. W., McConnell M., Guarner J., Shieh W. J., Malecki J. M., Gerberding J. L., Hughes J. M., Perkins B. A. (2001) Bioterrorism-related inhalational anthrax. The first 10 cases reported in the United States. Emerg. Infect. Dis. 7, 933–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Duesbery N. S., Webb C. P., Leppla S. H., Gordon V. M., Klimpel K. R., Copeland T. D., Ahn N. G., Oskarsson M. K., Fukasawa K., Paull K. D., Vande Woude G. F. (1998) Proteolytic inactivation of MAP kinase kinase by anthrax lethal factor. Science 280, 734–737 [DOI] [PubMed] [Google Scholar]
- 4. Tonello F., Montecucco C. (2009) The anthrax lethal factor and its MAPK kinase-specific metalloprotease activity. Mol. Aspects Med. 30, 431–438 [DOI] [PubMed] [Google Scholar]
- 5. Vitale G., Pellizzari R., Recchi C., Napolitani G., Mock M., Montecucco C. (1998) Anthrax lethal factor cleaves the N terminus of MAPKKs and induces tyrosine/threonine phosphorylation of MAPKs in cultured macrophages. Biochem. Biophys. Res. Commun. 248, 706–711 [DOI] [PubMed] [Google Scholar]
- 6. Drum C. L., Yan S. Z., Bard J., Shen Y. Q., Lu D., Soelaiman S., Grabarek Z., Bohm A., Tang W. J. (2002) Structural basis for the activation of anthrax adenylyl cyclase exotoxin by calmodulin. Nature 415, 396–402 [DOI] [PubMed] [Google Scholar]
- 7. Shen Y., Zhukovskaya N. L., Guo Q., Florián J., Tang W. J. (2005) Calcium-independent calmodulin binding and two-metal ion catalytic mechanism of anthrax edema factor. EMBO J. 24, 929–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Candela T., Fouet A. (2006) Poly-γ-glutamate in bacteria. Mol. Microbiol. 60, 1091–1098 [DOI] [PubMed] [Google Scholar]
- 9. Bottone E. J. (2010) Bacillus cereus, a volatile human pathogen. Clin. Microbiol. Rev. 23, 382–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Avashia S. B., Riggins W. S., Lindley C., Hoffmaster A., Drumgoole R., Nekomoto T., Jackson P. J., Hill K. K., Williams K., Lehman L., Libal M. C., Wilkins P. P., Alexander J., Tvaryanas A., Betz T. (2007) Fatal pneumonia among metalworkers due to inhalation exposure to Bacillus cereus containing Bacillus anthracis toxin genes. Clin. Infect. Dis. 44, 414–416 [DOI] [PubMed] [Google Scholar]
- 11. Hoffmaster A. R., Ravel J., Rasko D. A., Chapman G. D., Chute M. D., Marston C. K., De B. K., Sacchi C. T., Fitzgerald C., Mayer L. W., Maiden M. C., Priest F. G., Barker M., Jiang L., Cer R. Z., Rilstone J., Peterson S. N., Weyant R. S., Galloway D. R., Read T. D., Popovic T., Fraser C. M. (2004) Identification of anthrax toxin genes in a Bacillus cereus associated with an illness resembling inhalation anthrax. Proc. Natl. Acad. Sci. U.S.A. 101, 8449–8454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Deng Q., Barbieri J. T. (2008) Molecular mechanisms of the cytotoxicity of ADP-ribosylating toxins. Annu. Rev. Microbiol. 62, 271–288 [DOI] [PubMed] [Google Scholar]
- 13. Holbourn K. P., Shone C. C., Acharya K. R. (2006) A family of killer toxins. Exploring the mechanism of ADP-ribosylating toxins. FEBS J. 273, 4579–4593 [DOI] [PubMed] [Google Scholar]
- 14. Fieldhouse R. J., Merrill A. R. (2008) Needle in the haystack. Structure-based toxin discovery. Trends Biochem. Sci. 33, 546–556 [DOI] [PubMed] [Google Scholar]
- 15. Aktories K., Bärmann M., Ohishi I., Tsuyama S., Jakobs K. H., Habermann E. (1986) Botulinum C2 toxin ADP-ribosylates actin. Nature 322, 390–392 [DOI] [PubMed] [Google Scholar]
- 16. Vandekerckhove J., Schering B., Bärmann M., Aktories K. (1987) Clostridium perfringens ι-toxin ADP-ribosylates skeletal muscle actin in Arg-177. FEBS Lett. 225, 48–52 [DOI] [PubMed] [Google Scholar]
- 17. Visschedyk D. D., Perieteanu A. A., Turgeon Z. J., Fieldhouse R. J., Dawson J. F., Merrill A. R. (2010) Photox, a novel actin-targeting mono-ADP-ribosyltransferase from Photorhabdus luminescens. J. Biol. Chem. 285, 13525–13534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Aktories K., Rösener S., Blaschke U., Chhatwal G. S. (1988) Botulinum ADP-ribosyltransferase C3. Purification of the enzyme and characterization of the ADP-ribosylation reaction in platelet membranes. Eur. J. Biochem. 172, 445–450 [DOI] [PubMed] [Google Scholar]
- 19. Nemoto Y., Namba T., Kozaki S., Narumiya S. (1991) Clostridium botulinum C3 ADP-ribosyltransferase gene. Cloning, sequencing, and expression of a functional protein in Escherichia coli. J. Biol. Chem. 266, 19312–19319 [PubMed] [Google Scholar]
- 20. Just I., Mohr C., Schallehn G., Menard L., Didsbury J. R., Vandekerckhove J., van Damme J., Aktories K. (1992) Purification and characterization of an ADP-ribosyltransferase produced by Clostridium limosum. J. Biol. Chem. 267, 10274–10280 [PubMed] [Google Scholar]
- 21. Wilde C., Just I., Aktories K. (2002) Structure-function analysis of the Rho-ADP-ribosylating exoenzyme C3stau2 from Staphylococcus aureus. Biochemistry 41, 1539–1544 [DOI] [PubMed] [Google Scholar]
- 22. Yamaguchi T., Hayashi T., Takami H., Ohnishi M., Murata T., Nakayama K., Asakawa K., Ohara M., Komatsuzawa H., Sugai M. (2001) Complete nucleotide sequence of a Staphylococcus aureus exfoliative toxin B plasmid and identification of a novel ADP-ribosyltransferase, EDIN-C. Infect. Immun. 69, 7760–7771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yamaguchi T., Nishifuji K., Sasaki M., Fudaba Y., Aepfelbacher M., Takata T., Ohara M., Komatsuzawa H., Amagai M., Sugai M. (2002) Identification of the Staphylococcus aureus etd pathogenicity island, which encodes a novel exfoliative toxin, ETD, and EDIN-B. Infect. Immun. 70, 5835–5845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Barth H., Preiss J. C., Hofmann F., Aktories K. (1998) Characterization of the catalytic site of the ADP-ribosyltransferase Clostridium botulinum C2 toxin by site-directed mutagenesis. J. Biol. Chem. 273, 29506–29511 [DOI] [PubMed] [Google Scholar]
- 25. Han S., Craig J. A., Putnam C. D., Carozzi N. B., Tainer J. A. (1999) Evolution and mechanism from structures of an ADP-ribosylating toxin and NAD complex. Nat. Struct. Biol. 6, 932–936 [DOI] [PubMed] [Google Scholar]
- 26. Jørgensen R., Wang Y., Visschedyk D., Merrill A. R. (2008) The nature and character of the transition state for the ADP-ribosyltransferase reaction. EMBO Rep. 9, 802–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sakurai J., Nagahama M., Hisatsune J., Katunuma N., Tsuge H. (2003) Clostridium perfringens ι-toxin, ADP-ribosyltransferase. Structure and mechanism of action. Adv. Enzyme Regul. 43, 361–377 [DOI] [PubMed] [Google Scholar]
- 28. Yates S. P., Jörgensen R., Andersen G. R., Merrill A. R. (2006) Stealth and mimicry by deadly bacterial toxins. Trends Biochem. Sci. 31, 123–133 [DOI] [PubMed] [Google Scholar]
- 29. Fieldhouse R. J., Turgeon Z., White D., Merrill A. R. (2010) Cholera- and anthrax-like toxins are among several new ADP-ribosyltransferases. PLoS Comput. Biol. 6, e1001029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Turgeon Z., Jørgensen R., Visschedyk D., Edwards P. R., Legree S., McGregor C., Fieldhouse R. J., Mangroo D., Schapira M., Merrill A. R. (2011) Newly discovered and characterized antivirulence compounds inhibit bacterial mono-ADP-ribosyltransferase toxins. Antimicrob. Agents Chemother. 55, 983–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yates S. P., Taylor P. L., Jørgensen R., Ferraris D., Zhang J., Andersen G. R., Merrill A. R. (2005) Structure-function analysis of water-soluble inhibitors of the catalytic domain of exotoxin A from Pseudomonas aeruginosa. Biochem. J 385, 667–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cheng Y., Prusoff W. H. (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor that causes 50 percent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22, 3099–3108 [DOI] [PubMed] [Google Scholar]
- 33. Turgeon Z., White D., Jørgensen R., Visschedyk D., Fieldhouse R. J., Mangroo D., Merrill A. R. (2009) Yeast as a tool for characterizing mono-ADP-ribosyltransferase toxins. FEMS Microbiol. Lett. 300, 97–106 [DOI] [PubMed] [Google Scholar]
- 34. Grochulski P., Fodje M. N., Gorin J., Labiuk S. L., Berg R. (2011) Beamline 08ID-1, the prime beamline of the Canadian Macromolecular Crystallography Facility. J. Synchrotron Radiat. 18, 681–684 [DOI] [PubMed] [Google Scholar]
- 35. Otwinowski Z., Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 36. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vogelsgesang M., Aktories K. (2006) Exchange of glutamine-217 to glutamate of Clostridium limosum exoenzyme C3 turns the asparagine-specific ADP-ribosyltransferase into an arginine-modifying enzyme. Biochemistry 45, 1017–1025 [DOI] [PubMed] [Google Scholar]
- 38. Vagin A., Teplyakov A. (1997) MOLREP. An automated program for molecular replacement. J. Appl. Crystallogr. 30, 1022–1025 [Google Scholar]
- 39. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Adams P. D., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., Terwilliger T. C. (2002) PHENIX. Building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 [DOI] [PubMed] [Google Scholar]
- 41. Emsley P., Cowtan K. (2004) Coot. Model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 42. Jones T. A., Zou J. Y., Cowan S. W., Kjeldgaard M. (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47, 110–119 [DOI] [PubMed] [Google Scholar]
- 43. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 44. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity. All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Singh Y., Leppla S. H., Bhatnagar R., Friedlander A. M. (1989) Internalization and processing of Bacillus anthracis lethal toxin by toxin-sensitive and -resistant cells. J. Biol. Chem. 264, 11099–11102 [PubMed] [Google Scholar]
- 46. Petosa C., Collier R. J., Klimpel K. R., Leppla S. H., Liddington R. C. (1997) Crystal structure of the anthrax toxin protective antigen. Nature 385, 833–838 [DOI] [PubMed] [Google Scholar]
- 47. Quinn C. P., Singh Y., Klimpel K. R., Leppla S. H. (1991) Functional mapping of anthrax toxin lethal factor by in-frame insertion mutagenesis. J. Biol. Chem. 266, 20124–20130 [PubMed] [Google Scholar]
- 48. Zornetta I., Brandi L., Janowiak B., Dal Molin F., Tonello F., Collier R. J., Montecucco C. (2010) Imaging the cell entry of the anthrax edema and lethal toxins with fluorescent protein chimeras. Cell Microbiol. 12, 1435–1445 [DOI] [PubMed] [Google Scholar]
- 49. Abrami L., Kunz B., Deuquet J., Bafico A., Davidson G., van der Goot F. G. (2008) Functional interactions between anthrax toxin receptors and the WNT signaling protein LRP6. Cell Microbiol. 10, 2509–2519 [DOI] [PubMed] [Google Scholar]
- 50. Armstrong S., Merrill A. R. (2001) Application of a fluorometric assay for characterization of the catalytic competency of a domain III fragment of Pseudomonas aeruginosa exotoxin A. Anal. Biochem. 292, 26–33 [DOI] [PubMed] [Google Scholar]
- 51. Ménétrey J., Flatau G., Stura E. A., Charbonnier J. B., Gas F., Teulon J. M., Le Du M. H., Boquet P., Menez A. (2002) NAD binding induces conformational changes in Rho ADP-ribosylating Clostridium botulinum C3 exoenzyme. J. Biol. Chem. 277, 30950–30957 [DOI] [PubMed] [Google Scholar]
- 52. Melnyk R. A., Hewitt K. M., Lacy D. B., Lin H. C., Gessner C. R., Li S., Woods V. L., Jr., Collier R. J. (2006) Structural determinants for the binding of anthrax lethal factor to oligomeric protective antigen. J. Biol. Chem. 281, 1630–1635 [DOI] [PubMed] [Google Scholar]
- 53. Gupta P., Singh A., Chauhan V., Bhatnagar R. (2001) Involvement of residues 147VYYEIGK153 in binding of lethal factor to protective antigen of Bacillus anthracis. Biochem. Biophys. Res. Commun. 280, 158–163 [DOI] [PubMed] [Google Scholar]
- 54. Han S., Arvai A. S., Clancy S. B., Tainer J. A. (2001) Crystal structure and novel recognition motif of rho ADP-ribosylating C3 exoenzyme from Clostridium botulinum. structural insights for recognition specificity and catalysis. J. Mol. Biol. 305, 95–107 [DOI] [PubMed] [Google Scholar]
- 55. Tsuge H., Nagahama M., Nishimura H., Hisatsune J., Sakaguchi Y., Itogawa Y., Katunuma N., Sakurai J. (2003) Crystal structure and site-directed mutagenesis of enzymatic components from Clostridium perfringens ι-toxin. J. Mol. Biol. 325, 471–483 [DOI] [PubMed] [Google Scholar]
- 56. Laing S., Unger M., Koch-Nolte F., Haag F. (2011) ADP-ribosylation of arginine. Amino Acids 41, 257–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bell C. E., Eisenberg D. (1996) Crystal structure of diphtheria toxin bound to nicotinamide adenine dinucleotide. Biochemistry 35, 1137–1149 [DOI] [PubMed] [Google Scholar]
- 58. Li M., Dyda F., Benhar I., Pastan I., Davies D. R. (1996) Crystal structure of the catalytic domain of Pseudomonas exotoxin A complexed with a nicotinamide adenine dinucleotide analog. Implications for the activation process and for ADP ribosylation. Proc. Natl. Acad. Sci. U.S.A. 93, 6902–6906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rasko D. A., Rosovitz M. J., Økstad O. A., Fouts D. E., Jiang L., Cer R. Z., Kolstø A. B., Gill S. R., Ravel J. (2007) Complete sequence analysis of novel plasmids from emetic and periodontal Bacillus cereus isolates reveals a common evolutionary history among the B. cereus group plasmids, including Bacillus anthracis pXO1. J. Bacteriol. 189, 52–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Oh S. Y., Budzik J. M., Garufi G., Schneewind O. (2011) Two capsular polysaccharides enable Bacillus cereus G9241 to cause anthrax-like disease. Mol. Microbiol. 80, 455–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Papazisi L., Rasko D. A., Ratnayake S., Bock G. R., Remortel B. G., Appalla L., Liu J., Dracheva T., Braisted J. C., Shallom S., Jarrahi B., Snesrud E., Ahn S., Sun Q., Rilstone J., Okstad O. A., Kolstø A. B., Fleischmann R. D., Peterson S. N. (2011) Investigating the genome diversity of B. cereus and evolutionary aspects of B. anthracis emergence. Genomics 98, 26–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Stenfors Arnesen L. P., Fagerlund A., Granum P. E. (2008) From soil to gut. Bacillus cereus and its food poisoning toxins. FEMS Microbiol. Rev. 32, 579–606 [DOI] [PubMed] [Google Scholar]
- 63. Salter S. J. (2011) You cannot B. cereus. Nat. Rev. Microbiol. 9, 83. [DOI] [PubMed] [Google Scholar]
- 64. Pannifer A. D., Wong T. Y., Schwarzenbacher R., Renatus M., Petosa C., Bienkowska J., Lacy D. B., Collier R. J., Park S., Leppla S. H., Hanna P., Liddington R. C. (2001) Crystal structure of the anthrax lethal factor. Nature 414, 229–233 [DOI] [PubMed] [Google Scholar]