

Background: The role of SFKs in G protein-coupled receptor-mediated platelet activation is not well understood.
Results: AYPGKF induced Gq/Ca2+-dependent SFK phosphorylation, and AYPGKF-elicited platelet activation was partially inhibited by PP2 but was completely abolished by PKC inhibitors plus SFK inhibitors.
Conclusion: Ca2+/SFKs/PI3K and PKC represent alternative pathways mediating AYPGKF-dependent platelet activation.
Significance: This work increases understanding of important SFK functions in platelet activation.
Keywords: Calcium, G Proteins, Platelets, Secretion, Thrombin, Src Family Kinase
Abstract
The Src family kinases (SFKs) play essential roles in collagen- and von Willebrand factor (VWF)-mediated platelet activation. However, the roles of SFKs in G protein-coupled receptor-mediated platelet activation and the molecular mechanisms whereby SFKs are activated by G protein-coupled receptor stimulation are not fully understood. Here we show that the thrombin receptor protease-activated receptor 4 agonist peptide AYPGKF elicited SFK phosphorylation in P2Y12 deficient platelets but stimulated minimal SFK phosphorylation in platelets lacking Gq. We have previously shown that thrombin-induced SFK phosphorylation was inhibited by the calcium chelator 5,5′-dimethyl-bis-(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (dimethyl-BAPTA). The calcium ionophore A23187 induced SFK phosphorylation in both wild-type and Gq deficient platelets. Together, these results indicate that SFK phosphorylation in response to thrombin receptor stimulation is downstream from Gq/Ca2+ signaling. Moreover, A23187-induced thromboxane A2 synthesis, platelet aggregation, and secretion were inhibited by preincubation of platelets with a selective SFK inhibitor, PP2. AYPGKF-induced thromboxane A2 production in wild-type and P2Y12 deficient platelets was abolished by PP2, and AYPGKF-mediated P-selectin expression, integrin αIIbβ3 activation, and aggregation of P2Y12 deficient platelets were partially inhibited by the PKC inhibitor Ro-31-8220, PP2, dimethyl-BAPTA, or LY294002, but were abolished by Ro-31-8220 plus PP2, dimethyl-BAPTA, or LY294002. These data indicate that Ca2+/SFKs/PI3K and PKC represent two alternative signaling pathways mediating Gq-dependent platelet activation.
Introduction
The Src family of nonreceptor tyrosine kinases consists of signaling enzymes that regulate cell growth, differentiation, cell adhesion, carcinogesesis, and immune cell function (1). At least six members of the Src family kinases (SFKs)3 are present in platelets, including c-Src, Lyn, Fgr, Fyn, Lck, and Yes (2–4). SFKs play distinct roles in various aspects of platelet activation. c-Src binds to the cytoplasmic domain of the integrin subunit β3 and plays an important role in integrin αIIbβ3-dependent outside-in signaling (5–7). SFKs also play important roles in the VWF/GPIb-IX-mediated platelet activation (8–11). The cytoplasmic domain of GPIbα interacts with several intracellular molecules including filamin, 14-3-3ζ, SFKs (c-Src and Lyn), and PI3K (12). SFKs appear to be upstream of PI3K and calcium elevation in response to VWF (8). Among different SFK isoforms, the importance of Lyn (and to a much less degree, c-Src) has been shown in GPIb-IX signaling (11, 13). Lyn and Fyn, which bind to the cytoplasmic domain of GPVI (14–16), have been implicated in the GPVI-immunotyrosine-activating motif-Syk signaling pathway. Therefore, activation of SFKs in platelets by stimulation of the collagen receptor GPVI is a very early signaling event. Upon cross-linking of GPVI, the immunotyrosine-activating motif of FcR γ-chain is tyrosine-phosphorylated by SFKs, leading to the activation of phospholipase C (PLC) γ2, the key effector enzyme in the GPVI signaling cascade (17, 18). Activation of PLC γ2 liberates the second messengers 1,2-diacylglycerol (DAG) and inositol 1,4,5-trisphosphate, which results in PKC activation and calcium release, respectively.
The role of SFKs in platelet activation induced by G protein-coupled receptors is highly controversial. It has been reported that selective inhibitors of SFKs have no effect, have a stimulatory effect, or have an inhibitory effect, in platelet activation in response to thrombin stimulation (19–22). Although an early study reported that SFKs are not required in thrombin-induced platelet aggregation (19), several other studies showed that SFKs (mainly Lyn kinase) stimulate platelet activation and secretion induced by thrombin (20, 21). However, a very recent study reported that the SFK Fyn activated downstream of G12/13 inhibits Gq-dependent intracellular calcium mobilization, PKC activation, and thrombin-induced platelet aggregation (22). SFKs also play a role in Gi-dependent platelet activation and TXA2 synthesis (23, 24).
Platelets express many G proteins, including Gq, G12/13, Gi/z, and Gs. G proteins are coupled to agonist receptors that stimulate platelet activation, with the exception of Gs, which is coupled to receptors for physiological platelet inhibitors (PGI2 and adenosine) that mediate inhibitory signals by stimulating adenylyl cyclase-dependent cAMP synthesis (25). The aim of this study was to investigate the molecular mechanisms by which SFKs were activated upon thrombin receptor stimulation and to establish roles of SFKs in Gq-dependent platelet activation. To accomplish this aim, we used a combination of various knock-out mice, as well as pharmacological approaches to dissect the signaling pathways mediating SFK phosphorylation and to examine the effects of selective SFK inhibitors on platelet activation in response to thrombin receptor stimulation. We demonstrate that activation of the Gq/Ca2+ pathway is sufficient to elicit SFK phosphorylation. Furthermore, we show that Ca2+/SFKs/PI3K synergizes with the PKC pathway to mediate Gq-dependent platelet secretion and aggregation. In addition, our data suggest that SFKs are direct effectors of Gi and play important roles in ADP-induced platelet activation.
EXPERIMENTAL PROCEDURES
Materials
α-Thrombin was purchased from Enzyme Research Laboratories (South Bend, IN). PAR4 peptide AYPGKF-NH2 and PAR1 peptide SFLLRN-NH2 were custom-synthesized at Biomatik USA, LLC (Wilmington, DE). A calpain inhibitor E64, ADP, Src inhibitor-1, and the P2Y1 antagonist MRS-2179 were from Sigma. Luciferase/luciferin reagent and collagen were from Chrono-log (Havertown, PA). Forskolin, Ro-31-8220, GÖ6983, PKCθ/δ inhibitor, PP2, and LY294002 were purchased from Calbiochem. Calcium chelator dimethyl-BAPTA, Fura-2/AM, and Pluronic F-127 were from Invitrogen. Rabbit monoclonal antibodies against Lyn, the phosphorylated Ser473 residue of Akt, and phosphorylated Src at residues Tyr416 were from Cell Signaling Technology (Beverly, MA). A mouse monoclonal antibody against Fyn, rabbit polyclonal antibodies against Gi, and protein A/G beads were from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). cAMP ELISA kit was from Amersham Biosciences. The TXB2 EIA kit was from Assay Designs (Ann Arbor, MI). Oregon Green 488-labeled fibrinogen was from Molecular Probes (Eugene, OR). FITC-conjugated rat anti-mouse CD62P (P-Selectin) monoclonal antibody was from BD Biosciences (San Diego, CA).
Animals
Mice deficient in Gq (26), P2Y12 (27), TP (28), and Lyn (21, 29) were generated as described previously. Littermate wild-type mice from heterozygous breeding were used as controls. The mice were bred and maintained in the University of Kentucky Animal Care Facility following institutional and National Institutes of Health guidelines after approval by the Animal Care Committee.
Preparation of Platelets
Blood was collected from the abdominal aorta of isofluorane-anesthetized mice (8–10 weeks) using volume of ACD (85 mm trisodium citrate, 83 mm dextrose, and 21 mm citric acid) as anticoagulant (30). For each experiment, blood was pooled from three to four mice of each genotype. The platelets were then washed twice with CGS (0.12 m sodium chloride, 0.0129 m trisodium citrate, 0.03 m d-glucose, pH 6.5). The platelets were resuspended in modified Tyrode's buffer (12 mm NaHCO3, 138 mm NaCl, 5.5 mm glucose, 2.9 mm KCl, 2 mm MgCl2, 0.42 mm NaH2PO4, 10 mm HEPES, pH 7.4) at 3 × 108/ml and incubated for 1 h at 22 °C before use.
Platelet Aggregation and Secretion
Platelet aggregation at 37 °C was measured by detecting changes in light transmission using a turbidometric platelet aggregometer (Chrono-Log) with stirring (1000 rpm). ATP release was measured by adding luciferin/luciferase reagent (3–12 μl) to 250 μl of a washed platelet suspension 1 min before stimulation.
Western Blot Analysis of Akt and Src Phosphorylation
Washed platelets were stimulated with agonists in a platelet aggregometer at 37 °C for 5 min and then solubilized in SDS-PAGE sample buffer. The platelets were preincubated with various inhibitors at the indicated concentrations or vehicle control at 37 °C for 5 min prior to the addition of the platelet agonists to examine the effects of inhibitors on SFK phosphorylation. Platelet or cell lysates were analyzed by SDS-PAGE on 4–15% gradient gels and immunoblotted using a polyclonal anti-Akt antibody, rabbit monoclonal antibodies specific for the phosphorylated Akt residue Ser473, or phosphorylated Src residue Tyr416.
Ca2+ Mobilization
Washed mouse platelets were incubated with 12.5 μm Fura-2/AM, 0.2% Pluronic F-127 for 45 min at 37 °C. After washing with CGS, the platelets were resuspended to 3 × 108/ml in Tyrode's solution. To determine the role of SFKs in agonist-induced calcium mobilization, the platelets were preincubated with Me2SO or PP2 (10 μm) for 5 min prior to addition of agonists. Continuous fluorescent measurements were analyzed by excitation at 340 and 380 nm, and emission was measured at 509 nm using a model LS55 luminescence spectrometer (PerkinElmer Cetus, Waltham, MA). The intracellular Ca2+ level was expressed as relative fluorescence calculated based on the ratio of emissions simultaneously using FL WINLAB 4.0 software (PerkinElmer Cetus).
Rap1b Activation
The washed platelets were resuspended in modified Tyrode's solution were preincubated with Me2SO or PP2 (10 μm) for 5 min and then stimulated with agonists for 5 min at 37 °C in the aggregometer and lysed with the addition of an equal volume of ice-cold lysis buffer (100 mm Tris, 400 mm NaCl, 5 mm MaCl2, 2% Nonidet P-40, 20% glycerol, 2 mm PMSF, 220 Kallikrein inhibitor unit/ml aprotinin, 1 mm Na3VO4, 1 μg/ml leupeptin, pH 7.4). A sample containing 50 μl of lysate was added with an equal volume of 2× SDS sample buffer for the determination of total Rap1b levels. Active Rap1b was precipitated by glutathione-linked agarose beads prebound to RalGDS RBD fused to GST. Precipitated GTP-Rap1b was detected by SDS-PAGE and immunoblotting with a rabbit polyclonal anti-Rap1b-specific antibody.
Measurement of TXA2
Washed platelets from wild-type or P2Y12 deficient mice were stimulated with agonists for 5 min at 37 °C in aggregometer and stopped by adding 10 mm EDTA (final concentration). The platelet-free supernatant fraction was diluted 1:50 with the assay buffer supplied in the TXB2 EIA kit. TXB2, a stable metabolite of TXA2, was measured using the manufacturer's protocol. To determine the role of SFKs in agonist-induced TXA2 production, platelets were preincubated with PP2 for 5 min prior to addition of agonists.
P-selectin Expression
Washed platelets were stimulated with AYPGKF for 5 min at 37 °C and fixed by adding paraformaldehyde. The platelets were then incubated with a FITC-conjugated anti-mouse P-selectin antibody at 22 °C for 30 min. P-selectin expression was analyzed by flow cytometer. The platelets were preincubated with various inhibitors for 5 min prior to the addition of AYPGKF to determine the role of PKC, SFKs, and Ca2+ in agonist-induced P-selectin expression.
Fibrinogen Binding Assay
Agonist-mediated integrin activation was examined by flow cytometry analysis of Oregon Green 488-labeled fibrinogen binding to integrin αIIbβ3. Briefly, washed platelets were incubated with Oregon Green-labeled fibrinogen and AYPGKF at 37 °C for 5 min in aggregometer with stirring and stopped by adding 1% (final concentration) paraformaldehyde. Fibrinogen binding was analyzed by flow cytometry. The platelets were preincubated with various inhibitors for 5 min prior to the addition of Oregon Green-labeled fibrinogen and AYPGKF to determine the role of PKC, SFKs, PI3K, and Ca2+ in agonist-induced integrin activation.
Immunoprecipitation and Western Blotting
Washed platelets were resuspended in modified Tyrode's buffer at 1 × 109/ml and solubilized by adding an equal volume of solubilization buffer (2% Triton X-100, 0.1 m Tris, 0.01 m EGTA, and 0.15 m NaCl, pH 7.4) containing 0.2 mm E64, 2 mm phenylmethylsulfonyl fluoride, and 220 Kallikrein inhibitor unit/ml aprotinin. After precleared with Sepharose 4B beads, 500 μl aliquots of each platelet lysate were immunoprecipitated with the indicated antibodies and protein A/G beads. Pulldown proteins were analyzed by SDS-PAGE and immunoblotted using antibodies against Gi, Lyn, or Fyn.
RESULTS
Activation of Gq-induced SFK Phosphorylation
Although SFKs have been implicated in thrombin-induced platelet activation, it is unknown whether or not Gq signaling contributes to SFK activation in response to thrombin receptor stimulation. Thrombin activates mouse platelets via its receptor PAR4, which does not couple directly to Gi. Thrombin-induced activation of Gi is secretion-dependent, mainly by secreted ADP through its receptor P2Y12. SFK activity is correlated directly with autophosphorylation and dephosphorylation of a tyrosine residue in the middle of the catalytic domain (Tyr416 for c-Src). Therefore, SFK activation can be detected by measuring the extent of SFK autophosphorylation by Western blotting with a rabbit monoclonal antibody that specifically recognizes the phosphorylated SFK residue Tyr416. The PAR4 activator, the peptide AYPGKF, elicited SFK phosphorylation in P2Y12 deficient platelets (Fig. 1A). PKC is essential for platelet secretion. Thus, platelets from P2Y12 deficient mice were preincubated with a selective PKC inhibitor Ro-31-8220 to further exclude the effect of secretion on AYPGKF-induced SFK phosphorylation. Treatment of P2Y12 deficient platelets with Ro-31-8220, which abolished secretion from dense granules, did not affect SFK phosphorylation (Fig. 1A). Thus, AYPGKF is able to induce Gi-independent SFK activation. PAR4 couples to Gq and G12/13. We therefore used Gq deficient mice to determine the role of Gq in AYPGKF-induced SFK activation. AYPGKF stimulated substantial SFK phosphorylation in wild-type platelets in the presence of Ro-31-8220 and/or the P2Y12 antagonist Cangrelor (also named AR-C69931MX) but induced minimal SFK phosphorylation in platelets lacking Gq (Fig. 1B). These results demonstrate that AYPGKF-induced SFK phosphorylation is mainly Gq-dependent. The serotonin receptor 5-HT2a only couples to Gq. Serotonin stimulated SFK phosphorylation in platelets from wild-type mice (Fig. 1C). Serotonin-elicited SFK phosphorylation was neither inhibited by Cangrelor or Ro-31-8220 (Fig. 1C) nor affected by knock-out of the TXA2 receptor TP that couples to G12/13 (Fig. 1D). Thus, serotonin-induced SFK phosphorylation is not dependent on Gi or G12/13 signaling.
FIGURE 1.

The Gq signaling and Ca2+ elicited SFK phosphorylation in platelets. A, washed platelets from wild-type (P2Y12+/+) and P2Y12 deficient (P2Y12−/−) mice were preincubated with Ro-31-8220 (RO) (5 μm) or dimethyl sulfoxide (DMSO) for 5 min and then stimulated with AYPGKF (500 μm) at 37 °C for 5 min with stirring. SFK phosphorylation was detected by Western blotting with a rabbit monoclonal antibody specifically recognizing the phosphorylated Src residue Tyr416. B, washed platelets from wild-type (Gq+/+) and Gq deficient (Gq−/−) mice were preincubated with Me2SO, Cangrelor (1 μm), Ro-31-8220, or Cangrelor plus Ro-31-8220 for 5 min, and then stimulated with AYPGKF (500 μm) at 37 °C for 5 min with stirring. C, washed platelets from C57B6 mice were preincubated with Me2SO, Cangrelor, or Ro-31-8220 for 5 min and then stimulated with serotonin (10 μm) at 37 °C for 5 min with stirring. D, washed platelets from TP deficient mice were stimulated with serotonin at 37 °C for 5 min with stirring. E, washed platelets from wild-type (Gq+/+) and Gq deficient (Gq−/−) mice were stimulated with A23187 (0.5 μm) at 37 °C for 5 min with stirring. F, washed platelets from TP deficient mice were preincubated with buffer, MRS2179 (MRS) (10 μm), Cangrelor, or MRS2179 plus Cangrelor for 5 min and then incubated with A23187 (0.5 μm) or buffer at 37 °C for 5 min with stirring.
Activation of SFKs by AYPGKF Was Downstream of Ca2+ Signaling but Independent of CalDAG-GEF Signaling
Thrombin-induced phosphorylation of SFKs in P2Y12 deficient platelets is inhibited by preincubation of platelets with the calcium chelator dimethyl-BAPTA (31), suggesting that Gq-dependent activation of SFKs is downstream of Ca2+ signaling. To extend these studies, we tested the ability of the calcium ionophore, A23187, to induce SFK phosphorylation. A23187 indeed stimulated SFK phosphorylation in platelets from wild-type and Gq deficient mice, respectively (Fig. 1E). Stimulation of platelets with A23187 causes ADP secretion and TXA2 synthesis. Therefore, SFK phosphorylation induced by A23187 was examined in platelets lacking TP in the presence of ADP receptor antagonists to exclude the possibility that A23187-mediated SFK phosphorylation is secondary to TXA2 and/or secreted ADP. A23187 induced SFK phosphorylation in TP deficient platelets in the presence of the ADP receptor antagonists Cangrelor plus MRS-2179 (blocking P2Y1) (Fig. 1F).
Lyn kinase plays important roles in thrombin-induced platelet secretion and aggregation (20, 21). In agreement with the role of Lyn kinase in thrombin-induced platelet activation, A23187 (Fig. 2, A and B) and AYPGKF (Fig. 2, C and D), respectively, induced SFK phosphorylation was diminished by Lyn deficiency.
FIGURE 2.

Lyn is one of the major SFK isoforms activated by Gq and Gq-mediated SFK activation is independent of CalDAG-GEFI/Rap1b signaling. A–D, washed platelets from wild-type (Lyn+/+) and Lyn deficient (Lyn−/−) mice were preincubated with Cangrelor at 37 °C for 5 min, and then stimulated with A23187 (A) or AYPGKF (C) for 5 min with stirring. Densitometry measurements from results in A and C were shown in B and D, respectively. The values were normalized with respect to Lyn+/+ platelets in the absence of agonists and Cangrelor and are expressed as relative phosphorylation (means ± S.D. from three separate experiments). Statistical significance was determined using Student's t test. *, p < 0.01 versus Lyn+/+. E, washed platelets from P2Y12 deficient (P2Y12−/−) mice or P2Y12/CalDAG-GEFI double knock-out mice (P2Y12−/−/CalDAG-GEFI−/−) were stimulated with AYPGKF at 37 °C for 5 min with stirring. SFK phosphorylation was detected by Western blotting with a rabbit monoclonal antibody specifically recognizing the phosphorylated Src residue Tyr416. F, washed platelets from P2Y12 deficient mice were preincubated with dimethyl sulfoxide (0.1%) or PP2 (10 μm) and then stimulated with AYPGKF (500 μm) at 37 °C for 5 min with stirring. GTP-bound Rap1 was precipitated with GST-RalGDS RBD bound to glutathione-agarose beads.
CalDAG-GEFI, which is downstream from Gq/Ca2+ signaling, plays an important role in PAR4-mediated Rap1 and αIIbβ3 activation (32). To determine whether PAR4-mediated SFK activation requires the CalDAG-GEFI signaling, AYPGKF-induced SFK phosphorylation was examined in platelets lacking CalDAG-GEFI. P2Y12 deficient mice were used to exclude involvement of P2Y12/Gi signaling. AYPGKF elicited similar levels of SFK phosphorylation in platelets from CalDAG-GEFI/P2Y12 double knock-out mice as that in P2Y12 deficient platelets (Fig. 2E). AYPGKF-induced Rap1b activation in P2Y12 deficient platelets was not affected by PP2 (Fig. 2F). Taken together, activation of SFKs by PAR4 stimulation is independent of CalDAG-GEFI signaling.
Activation of SFKs is an early signaling event for GPVI signaling (17). Accordingly, collagen-elicited elevation of Ca2+ was abolished by preincubation of platelets with PP2 (Fig. 3). To determine whether or not SFK signaling is involved in regulating Gq-mediated PLCβ activation, the effect of PP2 on elevation of Ca2+ in response to AYPGKF was examined. Elevation of Ca2+ elicited by AYPGKF was not affected by preincubation of platelets with PP2 (Fig. 3, A and C). Therefore, Gq-dependent SFK activation is downstream, not upstream, of Ca2+ signaling.
FIGURE 3.

Effects of PP2 on AYPGKF-elicited Ca2+ mobilization and A23187-induced aggregation and TXA2 generation. A--C, washed platelets from P2Y12 deficient platelets were labeled with 12.5 μm Fura-2/AM, 0.2% Pluronic F-127 and resuspended in Tyrode's solution. A and B, platelets were then preincubated with PP2 or dimethyl sulfoxide (DMSO) for 5 min and stimulated with AYPGKF (500 μm) (A) or collagen (5 μg/ml) (B). C, changes in the intracellular free calcium level were measured every 2 s and expressed as a ratio of fluorescence (FL) detected at 509-nm emission with excitation wavelengths of 340 and 380 nm. Summarized data from three experiments are shown. D, washed platelets from C57BL/6 mice were preincubated with PP2 or dimethyl sulfoxide for 5 min and stimulated with A23187 (100 nm) in a lumi-aggregometer at 37 °C. Real time ATP secretion and platelet aggregation were simultaneously recorded. E, washed platelets from TP+/+ or TP−/− mice were stimulated with A23187 in a lumi-aggregometer at 37 °C. Real time ATP secretion and platelet aggregation were simultaneously recorded. F, washed platelets from P2Y12 knock-out mice or wild-type controls were preincubated with PP2 or dimethyl sulfoxide for 5 min and stimulated with various concentrations of A23187 for 5 min at 37 °C with stirring. The reactions were stopped by adding 10 mm of EDTA (final concentration). Because TXA2 has a half-life of 37 s and is rapidly converted to the stable product TXB2, TXA2 was measured as TXB2 with TXB2 EIA kits. TXB2 production data were obtained from three tests. Statistical differences were examined by Student t test. The data are the means ± S.D. *, p < 0.001 versus dimethyl sulfoxide in the presence of same concentration of AYPGKF.
A23187-induced Platelet Activation and TXA2 Synthesis Required SFKs
Next, the effect of PP2 on A23187-induced platelet aggregation and secretion was examined to establish a role of SFKs in Gq/Ca2+-dependent platelet activation. Pretreatment of platelets with PP2 inhibited ATP release and aggregation induced by low dose (≤100 nm) (Fig. 3D) but not high dose (data not shown), A23187. Because low dose A23187-induced aggregation and secretion were also attenuated in TP deficient platelets (Fig. 3E), the effect of PP2 on A23187-induced TXA2 production was examined to address whether induction of TXA2 synthesis accounts for the stimulatory role of SFKs in A23187-mediated platelet activation. A23187 elicited less TXA2 production in P2Y12 deficient platelets than that in wild-type platelets (Fig. 3F), indicating that A23187-stimulated TXA2 synthesis through both P2Y12-dependent and -independent mechanisms. Pretreatment of P2Y12 deficient platelets with PP2 abolished A23187-induced TXA2 production (Fig. 3F). Similarly, AYPGKF-induced TXA2 production in P2Y12 deficient platelets was abolished by PP2 treatment (Fig. 4A). Thus, Gq/Ca2+-dependent TXA2 synthesis requires the SFK signaling.
FIGURE 4.

Effects of PP2 on AYPGKF-induced TXA2 synthesis and the effects of TP deficiency and PP2 on AYPGKF-induced platelet aggregation and ATP secretion. A, washed platelets from P2Y12 knock-out mice or wild-type controls were preincubated with PP2 or dimethyl sulfoxide (DMSO) for 5 min and stimulated with various concentrations of AYPGKF (AY) for 5 min at 37 °C with stirring. Statistical differences were examined by Student t test. The data are the means ± S.D. *, p < 0.001 versus dimethyl sulfoxide in the presence of same concentration of AYPGKF. B, washed platelets from TP+/+ or TP−/− mice were stimulated with AYPGKF (40 μm) in a lumi-aggregometer at 37 °C. C, washed platelets from P2Y12 knock-out mice were preincubated with PP2 or dimethyl sulfoxide for 5 min and stimulated with various concentrations of AYPGKF in a lumi-aggregometer at 37 °C. Real time ATP secretion and platelet aggregation were simultaneously recorded.
AYPGKF-induced Aggregation and Secretion Were Attenuated by TP Deficiency
Aggregation and secretion in response to low dose AYPGKF were reduced in TP deficient platelets (Fig. 4B), identifying a role of TXA2 in low dose thrombin-induced platelet activation. These results also suggest that induction of TXA2 synthesis is one mechanism accounting for the stimulatory role of SFKs in promoting platelet activation.
The Role of SFKs in PAR4-dependent Platelet Aggregation and Secretion
Although G12/13 signaling contributes to platelet shape change (33), the Gq signaling is responsible for thrombin-induced Ca2+ mobilization, secretion, and aggregation (26). Secreted ADP, primarily by activating the Gi signaling via P2Y12, contributes to PAR4-mediated platelet activation. Therefore, P2Y12 deficient mice were used to exclude the effect of the positive feedback mechanism and to establish a role of SFKs in Gq-dependent platelet activation. Preincubation of P2Y12 deficient platelets with PP2 inhibited aggregation and ATP release in response to low dose AYPGKF (≤100 μm) (Fig. 4C). However, aggregation and ATP release elicited by higher doses (≥250 μm) of AYPGKF were only slightly reduced by PP2 treatment (Fig. 4C). These results suggest the existence of an alternative pathway in Gq-dependent platelet activation under those conditions. In support of this view, AYPGKF-induced P-selectin expression (Fig. 5A) and integrin activation evident as fibrinogen binding (Fig. 5B) were reduced but not abolished by pretreatment of platelets with PP2 or dimethyl-BAPTA.
FIGURE 5.

Effects of PP2, Ro-31-8220, dimethyl-BAPTA, and LY294002 on AYPGKF-induced fibrinogen binding and P-selectin expression. A, washed platelets from P2Y12 knock-out mice were preincubated with dimethyl sulfoxide (DMSO), PP2, Ro-31-8220, dimethyl-BAPTA (10 μm) (BAPTA), Ro-31-8220 plus PP2, or Ro-31-8220 plus dimethyl-BAPTA for 5 min and stimulated with AYPGKF (500 μm) at 37 °C for 5 min with stirring and subsequently fixed with paraformaldehyde. Fixed platelets were incubated with a FITC-labeled monoclonal anti-mouse P-selectin antibody for 30 min at 22 °C. Surface expression of P-selectin was analyzed using flow cytometry. Data from a representative experiment and quantitative results from three experiments expressed as the mean fluorescence index (mean fluorescence intensity of platelets stimulated with an agonist/fluorescence intensity of unstimulated platelets) are shown. The statistical differences were examined by Student t test. The data are the means ± S.D. *, p < 0.005 versus dimethyl sulfoxide; #, p < 0.05 versus resting platelets (CON); +, p > 0.2 versus resting platelets. B, washed platelets from P2Y12 knock-out mice were preincubated with dimethyl sulfoxide, PP2, Ro-31-8220, dimethyl-BAPTA, LY294002 (20 μm) (LY), Ro-31-8220 plus PP2, Ro-31-8220 plus dimethyl-BAPTA, or Ro-31-8220 plus LY294002 for 5 min and stimulated with AYPGKF (500 μm) in the presence of FITC-labeled fibrinogen at 37 °C for 5 min with stirring and subsequently fixed with paraformaldehyde. Fibrinogen binding was analyzed using flow cytometry. Data from a representative experiment and quantitative results from three experiments are expressed as the mean fluorescence index (mean fluorescence intensity of platelets stimulated with an agonist/fluorescence intensity of unstimulated platelets) were shown. Statistical differences were examined by Student t test. The data are the means ± S.D. *, p < 0.05 versus dimethyl sulfoxide; #, p < 0.05 versus resting platelets (CON); +, p > 0.2 versus resting platelets.
PKC is known to be important for Gq-dependent platelet secretion and aggregation. Therefore, the effect of a non-isoform selective PKC inhibitor, Ro-31-8220, on AYPGKF-induced aggregation of P2Y12 deficient platelets was examined to confirm the role of PKC in Gq-mediated platelet activation. Although preincubation of platelets with Ro-31-8220 abolished AYPGKF-induced ATP release (Fig. 6A), Ro-31-8220 only partially inhibited platelet aggregation in response to high concentrations of AYPGKF (≥500 μm). Aggregation of washed platelets requires fibrinogen secreted from α granules and integrin activation. In agreement with the results that Ro-31-8220 only reduced aggregation, Ro-31-8220 inhibited but did not abolish AYPGKF-induced secretion of P-selectin expression (Fig. 5A) and fibrinogen binding (Fig. 5B). Based on these observations, we hypothesize that the Ca2+/SFK pathway and the PKC pathway represent two parallel pathways mediating Gq-dependent platelet activation. If this hypothesis is correct, AYPGKF-induced integrin activation, aggregation, and secretion in P2Y12 deficient platelets should be abolished by a PKC inhibitor plus a SFK inhibitor or dimethyl-BAPTA. Indeed, aggregation of P2Y12 deficient platelets, even in response to high concentrations of AYPGKF, was totally abolished by pretreatment of platelets with Ro-31-8220 (5 μm) plus dimethyl-BAPTA (10 μm) or with Ro-31-8220 plus PP2 (10 μm) (Fig. 6A). Ro-31-8220 at a lower concentration (1 μm) achieved similar effect as 5 μm (data not shown). AYPGKF-induced P-selectin expression (Fig. 5A) and fibrinogen binding (Fig. 5B) were abolished by pretreatment of platelets with Ro-31-8220 plus PP2. AYPGKF-elicited aggregation and secretion of P2Y12 deficient platelets were also abolished by Ro-31-8220 plus another Src inhibitor, the Src inhibitor 1 (Fig. 6B), or PP2 plus another pan-PKC inhibitor, GÖ6983 (Fig. 6C). The novel PKC isoforms PKC θ and δ have been implicated in PAR4/Gq-mediated platelet activation (34–37). A PKCθ/δ specific inhibitor slightly inhibited AYPGKF-induced platelet aggregation and secretion (Fig. 6D). Although the PKCθ/δ inhibitor plus PP2 markedly inhibited platelet aggregation and secretion, they did not abolish platelet aggregation and secretion elicited by AYPGKF. These results indicate a role of the novel PKC isoforms in PAR4-induced platelet activation but also suggest that other isoforms of PKC are involved in PAR4-mediated platelet activation. Dimethyl-BAPTA plus PP2 had no additive inhibitory effect on AYPGKF-induced aggregation as compared with dimethyl-BAPTA or PP2 alone (data not shown).
FIGURE 6.

Effects of Src and PKC inhibitors, dimethyl-BAPTA, and LY294002 on AYPGKF-elicited aggregation and ATP secretion of P2Y12 deficient platelets. A, washed platelets from P2Y12 knock-out mice were preincubated with dimethyl sulfoxide (DMSO), PP2, Ro-31-8220, dimethyl-BAPTA, LY294002, Ro-31-8220 plus PP2, Ro-31-8220 plus dimethyl-BAPTA, or Ro-31-8220 plus LY294002 for 5 min and stimulated with 500 μm of AYPGKF. B, washed platelets from P2Y12 knock-out mice were preincubated with dimethyl sulfoxide, Src inhibitor-1 (10 μm), Ro-31-8220, or Ro-31-8220 plus Src inhibitor-1 for 5 min and stimulated with 500 μm of AYPGKF. C, washed platelets from P2Y12 knock-out mice were preincubated with dimethyl sulfoxide, PP2, GÖ6983 (2 μm), or GÖ6983 plus PP2 for 5 min and stimulated with 500 μm of AYPGKF. D, washed platelets from P2Y12 knock-out mice were preincubated with dimethyl sulfoxide, PP2, PKCθ/δ inhibitor (2 μm), or the PKCθ/δ inhibitor plus PP2 for 5 min and stimulated with 500 μm of AYPGKF.
The Role of PI3K in PAR4-mediated Platelet Activation
We have recently shown that activation of the PI3K/Akt pathway elicited by Gq is downstream of Ca2+/SFK signaling (31). Thus, the effect of the PI3K inhibitor LY294002 on AYPGKF-induced aggregation and secretion of P2Y12 deficient platelets was examined to evaluate the role, if any, of SFKs in PI3K-mediated platelet activation. As with dimethyl-BAPTA and PP2, AYPGKF-elicited platelet aggregation and ATP release were only partially inhibited by pretreatment of platelets with LY294002 but were abolished by Ro-31-8220 plus LY294002 (Fig. 6A). Consistent with these results, fibrinogen binding to αIIbβ3 induced by AYPGKF was partially inhibited by preincubation of platelets with LY294002 but was totally abolished by Ro-31-8220 plus LY294002 (Fig. 5B).
The Role of SFKs in Gi-mediated Platelet Activation
It is well established that the P2Y12/Gi pathway plays a key role in agonist-induced activation of the PI3K/Akt pathway (31). Although it is believed that P2Y12/Gi-dependent activation of PI3K is mediated by the Gi βγ subunits (38), a previous study showed that Gi-dependent Akt phosphorylation in human platelets was inhibited by PP2 (39). Accordingly, ADP-induced Akt phosphorylation in Gq deficient platelets was abolished by preincubation of platelets with PP2 (Fig. 7A). These results suggest that P2Y12/Gi-dependent activation of PI3K/Akt is downstream from the SFK signaling. Presumably, SFKs are direct effectors of G proteins (40). Consistent with this view, Lyn (Fig. 7B) and Fyn (Fig. 7C) were pulled down from mouse platelet lysates by a rabbit polyclonal antibody against Gi. Gi was pulled down from mouse platelet lysates by antibodies against Lyn (Fig. 7D) or Fyn (Fig. 7E). Thus, Lyn and Fyn interact with Gi in platelets. ADP-induced SFK phosphorylation was significantly attenuated in Lyn deficient platelets (Fig. 7F), indicating that Lyn is one of the major isoforms of SFKs that is activated by the P2Y12/Gi pathway.
FIGURE 7.

ADP- or SFLLRN-induced SFK phosphorylation and the roles of SFKs in ADP- and SFLLRN-induced platelet activation. A, washed platelets from Gq deficient (Gq−/−) mice were preincubated with dimethyl sulfoxide (DMSO) or PP2 and then stimulated with ADP (10 μm) at 37 °C for 5 min with stirring and solubilized with SDS-PAGE sample buffer. Phosphorylation of Akt was detected by Western blotting with rabbit monoclonal antibodies specifically recognizing the phosphorylated Akt residues Ser473. B and C, Lyn and Fyn kinases were pulled down by an anti-Gi antibody. Washed platelets from C57BL/6J mice were lysed, and the cell extracts were incubated with rabbit IgG or a rabbit polyclonal antibody against Gi2 followed by incubation of protein A/G beads. The immunoprecipitates were analyzed by Western blotting with a rabbit polyclonal anti-Lyn antibody (B) or a mouse monoclonal anti-Fyn antibody (C). D and E, Gi was pulled down by anti-Lyn or -Fyn antibodies. Washed platelets from C57BL/6 mice were lysed, and the cell extracts were incubated with mouse IgG, a mouse monoclonal antibody against Lyn (D), or a mouse monoclonal antibody against Fyn (E) followed by incubation of protein A/G beads. Immunoprecipitates were analyzed by Western blotting with a rabbit polyclonal antibody against Gi. F, washed platelets from wild-type (Lyn+/+) and Lyn deficient (Lyn−/−) mice were preincubated with buffer or MRS-2179 for 5 min and then stimulated with ADP (10 μm) at 37 °C for 5 min with stirring. G, washed human platelets were preincubated with dimethyl sulfoxide or dimethyl-BAPTA for 5 min and then stimulated with SFLLRN (10 μm) at 37 °C for 5 min with stirring. Src phosphorylation was detected by Western blotting with a rabbit monoclonal antibody specifically recognizing the phosphorylated Src residue Tyr416. H, washed human platelets were labeled with 12.5 μm Fura-2/AM, 0.2% Pluronic F-127 and resuspended in Tyrode's solution. The platelets were then preincubated with PP2 or dimethyl sulfoxide for 5 min and stimulated with SFLLRN (10 μm). Changes in the intracellular free calcium level were measured every 2 s and expressed as a ratio of fluorescence (FL) detected at 509-nm emission with an excitation wavelength of 340 and 380 nm. I, washed human platelets were preincubated with dimethyl sulfoxide, PP2, Ro-31-8220, or Ro-31-8220 plus PP2 for 5 min and stimulated with 10 μm of SFLLRN in a lumi-aggregometer at 37 °C. IB, immunoblot; IP, immunoprecipitation.
The Role of SFKs in PAR1-dependent Platelet Activation
Thrombin activates human platelets mainly by the G protein-coupled receptors PAR1 and PAR4. Thus, the role of SFKs in PAR1-mediated platelet activation was investigated using the PAR1 agonist peptide SFLLRN. As we expected, SFLLRN stimulated SFK phosphorylation in washed human platelets (Fig. 7G). SFLLRN-induced SFK phosphorylation was inhibited by dimethyl-BAPTA. In contrast, SFLLRN-induced Ca2+ mobilization was not affected by PP2 (Fig. 7H). These results indicate that as with PAR4, PAR1-dependent SFK activation is downstream from Ca2+ signaling. We examined the effect of PP2 on SFLLRN-induced platelet aggregation and ATP release to identify the role of SFKs in PAR1-mediated platelet activation. Platelet aggregation in response to SFLLRN was partially inhibited by pretreatment of the platelets with PP2 or Ro-31-8220 but was abolished by PP2 plus Ro-31-8220 (Fig. 7I).
DISCUSSION
In this study, using a combination of knock-out mice and pharmacological approaches, we documented and characterized the function of two alternative pathways for eliciting Gq-dependent platelet secretion and aggregation. First, we discovered that SFKs could be activated by the Gq/Ca2+ pathway (Fig. 8). Our data indicate that Gq/Ca2+-dependent SFK activation plays an important role in AYPGKF-induced TXA2 synthesis. We further demonstrate that PKC and Ca2+/SFKs/PI3Ks represent two alternative pathways mediating Gq-dependent secretion and aggregation. Additionally, we show that Lyn and Fyn interact with Gi, and SFK activation is important for P2Y12/Gi-dependent Rap1b activation and platelet aggregation.
FIGURE 8.
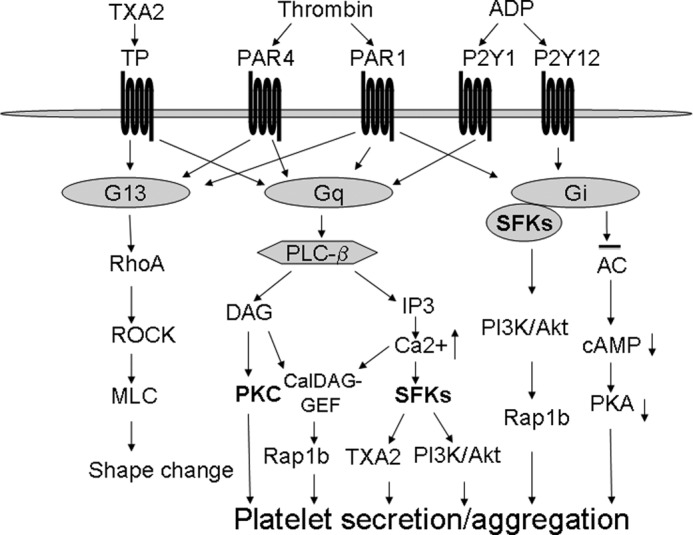
Important roles of SFKs in G protein-coupled receptor-induced platelet activation. Although the Gi pathway induces SFK activation by a direct interaction, Gq coupling receptors such as PAR1 and PAR4 induce SFK activation downstream from the PLCβ/Ca2+ signaling. SFKs play important roles in Gi- and Gq-dependent activation of the PI3K/Akt pathway, TXA2 synthesis, platelet secretion, and activation. The Ca2+/SFKs/PI3K pathway, together with the DAG/PKC pathway, mediates Gq-dependent platelet activation.
To our knowledge, this is the first documentation of the role of Gq in thrombin receptor-dependent SFK activation. PAR4 is a major thrombin receptor in mouse platelets, and PAR4 does not couple directly to Gi (41). AYPGKF-induced activation of the Gi pathway in mouse platelets depends mainly on ADP via P2Y12 signaling. Therefore, AYPGKF elicited SFK phosphorylation in P2Y12 deficient platelets, even in the presence of the PKC inhibitor Ro-31-8220, thereby abolishing platelet dense granule secretion, demonstrating that AYPGKF is able to stimulate Gi-independent SFK activation. Although thrombin failed to induce secretion and activation of Gq deficient platelets (26), it is able to induce G12/13 activation leading to shape change (33). Therefore, the data that AYPGKF elicited minimal SFK phosphorylation in Gq deficient platelets indicate that PAR4-mediated SFK phosphorylation is exclusively dependent on Gq signaling.
The serotonin receptor 5-HTa2 only couples to Gq (42). We show that serotonin induced SFK phosphorylation in the presence of Ro-31-8220 and Cangrelor, respectively, and in TP deficient platelets. Thus, it is unlikely that serotonin-induced SFK phosphorylation is secondary to either the G12/13 pathway activated by TXA2 or to the Gi pathway activated by secreted ADP or other dense granule contents. Together, our data demonstrate unequivocally that activation of Gq is sufficient to activate SFKs. This conclusion does not exclude the possibility that the G12/13 signaling might also contribute to the agonist-induced SFK phosphorylation. We show that although AYPGKF (Fig. 1B) or a TXA2 analog U46619 (data not shown) elicited minimal SFK phosphorylation in Gq deficient platelets, they potentiated ADP-induced SFK phosphorylation in Gq deficient platelets (supplemental Fig. S1), suggesting a role of G12/13 in SFK phosphorylation. A previous study reported that thrombin could induce SFK phosphorylation in Gq deficient platelets (39). However, in that study, thrombin-induced SFK phosphorylation in Gq deficient platelets was not compared with wild-type mouse platelets; therefore the absence of the appropriate control makes it difficult to evaluate the contribution of Gq in thrombin-induced SFK phosphorylation.
SFKs are upstream of PLCγ activation upon GPVI stimulation (17). In contrast, SFKs are not required for Gq-elicited Ca2+ elevation (Fig. 3). We conclude that Gq-dependent SFK phosphorylation is downstream from Ca2+. This conclusion is supported by the results that AYPGKF-induced SFK phosphorylation in P2Y12 deficient platelets is inhibited by dimethyl-BAPTA (31). Our data are consistent with the findings that an increase of Ca2+ concentration can activate SFKs (43).
We next sought to establish a role of SFKs in Gq-mediated platelet activation. Our results suggest that although platelet activation in response to low dose AYPGKF requires SFK signaling, there is an alternative pathway mediating Gq-dependent platelet activation in response to high-dose AYPGKF. Gq-mediated PLCβ activation results in generation of inositol 1,4,5-trisphosphate and diacylglycerol, which promotes mobilization of intracellular calcium and activation of PKC, respectively (44). PKC is known to play a key role in Gq-dependent platelet secretion and aggregation (34, 36, 45, 46). PKC-dependent platelet activation requires its effector pleckstrin (47). AYPGKF at high concentrations induces aggregation of PKC inhibitor-treated platelets (Fig. 6), and thrombin can induce aggregation and integrin activation of pleckstrin deficient platelets (47). These data suggest a PKC-independent mechanism involved in thrombin-induced platelet activation.
In this study, we show that platelet aggregation, integrin αIIbβ3 ligand binding function, and P-selectin expression in response to high concentrations of AYPGKF peptide were partially inhibited by the PKC inhibitors, the calcium chelator dimethyl-BAPTA, or SFK inhibitors but were totally abolished by a PKC inhibitor plus dimethyl-BAPTA or a SFK inhibitor. Thus, we propose a new concept to explain Gq-mediated platelet activation that states PKC and Ca2+/Src represent two parallel signaling pathways mediating Gq-dependent aggregation and secretion from α granules (Fig. 8). Although both the PKC pathway and the Ca2+/SFK pathway are required for integrin activation, α granule secretion, and platelet aggregation in response to low dose of AYPGKF, each pathway is sufficient to elicit aggregation in response to high concentrations of the agonist.
Stimulation of platelets with AYPGKF induced TXA2 synthesis, which appears to be important for low dose AYPGKF-induced platelet aggregation and secretion. Our data suggest that there are P2Y12-dependent and -independent mechanisms responsible for PAR4-induced TXA2 synthesis. AYPGKF-induced TXA2 production in P2Y12 deficient platelets was abolished by PP2 treatment but not inhibited or even enhanced by Ro-31-8220 treatment (data not shown). These data demonstrate that the SFK pathway but not the PKC pathway is responsible for PAR4-mediated TXA2 synthesis.
Identifying the roles of SFKs and PKC in PAR4-mediated platelet activation does not exclude the possibility that thrombin-induced platelet activation may involve other signaling pathways. In this regard, CalDAG-GEFI has been shown to play a role in PAR4-mediated platelet activation (32). CalDAG-GEFI plays a key role in agonist-induced Rap1b activation (32, 48). We found that AYPGKF-induced Rap1b activation in P2Y12 deficient platelets is not affected by PP2 and that AYPGKF-induced phosphorylation of SFKs was not inhibited by CalDAG-GEFI deficiency (Fig. 2) (49). These results suggest that Gq-mediated Rap1b activation is independent of the SFK pathway (Fig. 8).
We reported recently that the Gq-dependent activation of the PI3K/Akt pathway requires the Ca2+/SFKs pathway, but not the PKC pathway (31). Similar to dimethyl-BAPTA and PP2, the PI3K inhibitor LY294002 partially inhibited AYPGKF-induced secretion and aggregation in P2Y12 deficient platelets. Aggregation of and secretion by P2Y12 deficient platelets were completely abolished by treatment with Ro-31-8220 plus LY294002. These findings are consistent with the previous report that the PI3K inhibitor LY294002 inhibited thrombin-induced aggregation of pleckstrin-null platelets (47). PAR4 forms homodimers in platelets, which is important for PAR4 signaling (50). PAR4 can also form heterodimers with P2Y12 in thrombin-stimulated platelets (51). Although the role of SFKs in the formation of PAR4 homodimer or heterodimer with P2Y12 is unclear, Lyn kinase, but not other SFK kinases, associates with arrestin-2 and PI3K α/β subunits in thrombin-stimulated platelets that may be important for thrombin-induced Akt phosphorylation (51). These data are consistent with the findings that Lyn is one of the major SFK isoforms that is activated by the Gq/Ca2+ signaling (Fig. 2) and that Lyn plays a role in thrombin-elicited Akt phosphorylation (20).
We conclude that SFKs can be activated by the Gi pathway in platelets. This conclusion is supported by the data demonstrating that 1) ADP-elicited SFK phosphorylation in wild-type platelets is abolished by a P2Y12 antagonist but not by a P2Y1 antagonist and 2) ADP stimulated SFK phosphorylation in Gq deficient but not in P2Y12 deficient platelets (supplemental Fig. S1). This conclusion is consistent with a previous report showing that either Gi or Gz signaling is sufficient to activate SFKs (24). We show that Lyn and Fyn interact with Gi in platelets, suggesting that SFKs are direct effectors of Gi in platelets.
Platelets express both type IA (PI3Kα and PI3Kβ) and IB (PI3Kγ) classes of PI3K (52). PI3Kγ is a major isoform of PI3Ks that is activated by Gi (38). PI3Kγ can be activated by Gβγ subunits of heterotrimeric G proteins in vitro; therefore it has been suggested that PI3Kγ relays signals from G protein-coupled receptors (53–55). Akt phosphorylation by Gi stimulation depends on the PI3K signaling (30). Our data show that phosphorylation of Akt in response to ADP stimulation of Gq deficient platelets or in wild-type platelets in the presence of a P2Y1 antagonist is inhibited by PP2, indicating that Gi-dependent PI3K/Akt activation is likely to be downstream from SFKs but not directly by Gβγ subunits. Our data show that ADP-induced fibrinogen binding, Rap1b activation, and aggregation were inhibited by PP2 (supplemental Fig. S2, A–C), suggesting important roles of SFKs in Gi-dependent platelet activation. However, preincubation of platelets with PP2 did not affect ADP-induced inhibition of cAMP production in forskolin-stimulated platelets (supplemental Fig. S2D) (56), suggesting that SFKs are not involved in ADP inhibition of adenylate cyclase.
In conclusion, Fig. 8 summarizes the findings that thrombin activates SFKs through the Ca2+ signaling that synergizes with the PKC pathway to mediate Gq-dependent platelet secretion and activation. Platelet activation includes a series of rapid positive feedback loops that greatly amplify activation signals and enable robust platelet recruitment and stabilization of thrombi at the site of vascular injury. Two important mechanisms for amplification are the secretion of granule contents (mainly ADP) and synthesis of TXA2 from cyclooxygenase 1 signaling. SFKs are required for PAR4-mediated TXA2 synthesis, whereas PKC is required for cargo release from dense granules including ADP. Our data also suggest that the SFK and PKC pathways are mutually compensatory for integrin activation and α granule release in response to PAR4 stimulation. Additionally, our data demonstrate that SFKs are direct effectors of Gi and play important roles in Gi-dependent activation of the PI3K/Akt pathway in platelets.
Acknowledgments
We thank Drs. Susan S. Smyth and Andrew J. Morris for insightful suggestions and critically reading the manuscript. We also thank Dr. Richard Charnigo for assistance with statistical analysis. Cangrelor was provided as a generous gift from the Medicines Company.
This work is supported by American Heart Association Midwest affiliate Grand-in-Aid 0855698G (to Z. L.) and in part by Centers of Biomedical Research Excellence (COBRE) in Obesity and Cardiovascular Disease Grant P20 RR021954 from the National Institutes of Health/National Center for Research Resources and the Lexington VA Medical Center.

This article contains supplemental Figs. S1 and S2.
- used are: SFK
- Src family kinase
- PAR
- protease-activated receptor
- dimethyl-BAPTA
- 5,5′-dimethyl-bis-(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid
- CalDAG-GEFI
- Ca2+ and diacylglycerol regulated guanine nucleotide exchange factor I
- PLC
- phospholipase C
- TXA2
- thromboxane A2
- TP
- TXA2 receptor
- GPVI
- glycoprotein VI.
REFERENCES
- 1. Martin G. S. (2001) The hunting of the Src. Nat. Rev. Mol. Cell Biol. 2, 467–475 [DOI] [PubMed] [Google Scholar]
- 2. Golden A., Nemeth S. P., Brugge J. S. (1986) Blood platelets express high levels of the pp60c-Src-specific tyrosine kinase activity. Proc. Natl. Acad. Sci. U.S.A. 83, 852–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Huang M. M., Bolen J. B., Barnwell J. W., Shattil S. J., Brugge J. S. (1991) Membrane glycoprotein IV (CD36) is physically associated with the Fyn, Lyn, and Yes protein-tyrosine kinases in human platelets. Proc. Natl. Acad. Sci. U.S.A. 88, 7844–7848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pestina T. I., Stenberg P. E., Druker B. J., Steward S. A., Hutson N. K., Barrie R. J., Jackson C. W. (1997) Identification of the Src family kinases, Lck and Fgr in platelets. Their tyrosine phosphorylation status and subcellular distribution compared with other Src family members. Arterioscler. Thromb. Vasc. Biol. 17, 3278–3285 [DOI] [PubMed] [Google Scholar]
- 5. de Virgilio M., Kiosses W. B., Shattil S. J. (2004) Proximal, selective, and dynamic interactions between integrin αIIbβ3 and protein tyrosine kinases in living cells. J. Cell Biol. 165, 305–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Flevaris P., Stojanovic A., Gong H., Chishti A., Welch E., Du X. (2007) A molecular switch that controls cell spreading and retraction. J. Cell Biol. 179, 553–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Arias-Salgado E. G., Lizano S., Sarkar S., Brugge J. S., Ginsberg M. H., Shattil S. J. (2003) Src kinase activation by direct interaction with the integrin beta cytoplasmic domain. Proc. Natl. Acad. Sci. U.S.A. 100, 13298–13302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kasirer-Friede A., Cozzi M. R., Mazzucato M., De Marco L., Ruggeri Z. M., Shattil S. J. (2004) Signaling through GP Ib-IX-V activates αIIbβ3 independently of other receptors. Blood 103, 3403–3411 [DOI] [PubMed] [Google Scholar]
- 9. Garcia A., Quinton T. M., Dorsam R. T., Kunapuli S. P. (2005) Src family kinase-mediated and Erk-mediated thromboxane A2 generation are essential for VWF/GPIb-induced fibrinogen receptor activation in human platelets. Blood 106, 3410–3414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Marshall S. J., Senis Y. A., Auger J. M., Feil R., Hofmann F., Salmon G., Peterson J. T., Burslem F., Watson S. P. (2004) GPIb-dependent platelet activation is dependent on Src kinases but not MAP kinase or cGMP-dependent kinase. Blood 103, 2601–2609 [DOI] [PubMed] [Google Scholar]
- 11. Yin H., Liu J., Li Z., Berndt M. C., Lowell C. A., Du X. (2008) Src family tyrosine kinase Lyn mediates VWF/GPIb-IX-induced platelet activation via the cGMP signaling pathway. Blood 112, 1139–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Du X. (2007) Signaling and regulation of the platelet glycoprotein Ib-IX-V complex. Curr. Opin. Hematol. 14, 262–269 [DOI] [PubMed] [Google Scholar]
- 13. Liu J., Pestina T. I., Berndt M. C., Jackson C. W., Gartner T. K. (2005) Botrocetin/VWF-induced signaling through GPIb-IX-V produces TxA2 in an αIIbβ3- and aggregation-independent manner. Blood 106, 2750–2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Suzuki-Inoue K., Tulasne D., Shen Y., Bori-Sanz T., Inoue O., Jung S. M., Moroi M., Andrews R. K., Berndt M. C., Watson S. P. (2002) Association of Fyn and Lyn with the proline-rich domain of glycoprotein VI regulates intracellular signaling. J. Biol. Chem. 277, 21561–21566 [DOI] [PubMed] [Google Scholar]
- 15. Ezumi Y., Shindoh K., Tsuji M., Takayama H. (1998) Physical and functional association of the Src family kinases Fyn and Lyn with the collagen receptor glycoprotein VI-Fc receptor γ chain complex on human platelets. J. Exp. Med. 188, 267–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schmaier A. A., Zou Z., Kazlauskas A., Emert-Sedlak L., Fong K. P., Neeves K. B., Maloney S. F., Diamond S. L., Kunapuli S. P., Ware J., Brass L. F., Smithgall T. E., Saksela K., Kahn M. L. (2009) Molecular priming of Lyn by GPVI enables an immune receptor to adopt a hemostatic role. Proc. Natl. Acad. Sci. U.S.A. 106, 21167–21172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Watson S. P., Auger J. M., McCarty O. J., Pearce A. C. (2005) GPVI and integrin alphaIIb beta3 signaling in platelets. J. Thromb. Haemost. 3, 1752–1762 [DOI] [PubMed] [Google Scholar]
- 18. Nieswandt B., Watson S. P. (2003) Platelet-collagen interaction. Is GPVI the central receptor? Blood 102, 449–461 [DOI] [PubMed] [Google Scholar]
- 19. Briddon S. J., Watson S. P. (1999) Evidence for the involvement of p59fyn and p53/56lyn in collagen receptor signalling in human platelets. Biochem. J. 338, 203–209 [PMC free article] [PubMed] [Google Scholar]
- 20. Cho M. J., Pestina T. I., Steward S. A., Lowell C. A., Jackson C. W., Gartner T. K. (2002) Role of the Src family kinase Lyn in TxA2 production, adenosine diphosphate secretion, Akt phosphorylation, and irreversible aggregation in platelets stimulated with γ-thrombin. Blood 99, 2442–2447 [DOI] [PubMed] [Google Scholar]
- 21. Li Z., Zhang G., Liu J., Stojanovic A., Ruan C., Lowell C. A., Du X. (2010) An important role of the SRC family kinase Lyn in stimulating platelet granule secretion. J. Biol. Chem. 285, 12559–12570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim S., Kunapuli S. P. (2011) Negative regulation of Gq-mediated pathways in platelets by G12/13 pathways through Fyn kinase. J. Biol. Chem. 286, 24170–24179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shankar H., Kahner B. N., Prabhakar J., Lakhani P., Kim S., Kunapuli S. P. (2006) G-protein-gated inwardly rectifying potassium channels regulate ADP-induced cPLA2 activity in platelets through Src family kinases. Blood 108, 3027–3034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dorsam R. T., Kim S., Murugappan S., Rachoor S., Shankar H., Jin J., Kunapuli S. P. (2005) Differential requirements for calcium and Src family kinases in platelet GPIIb/IIIa activation and thromboxane generation downstream of different G-protein pathways. Blood 105, 2749–2756 [DOI] [PubMed] [Google Scholar]
- 25. Li Z., Delaney M. K., O'Brien K. A., Du X. (2010) Signaling during platelet adhesion and activation. Arterioscler. Thromb. Vasc. Biol. 30, 2341–2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Offermanns S., Toombs C. F., Hu Y. H., Simon M. I. (1997) Defective platelet activation in Gαq-deficient mice. Nature 389, 183–186 [DOI] [PubMed] [Google Scholar]
- 27. Foster C. J., Prosser D. M., Agans J. M., Zhai Y., Smith M. D., Lachowicz J. E., Zhang F. L., Gustafson E., Monsma F. J., Jr., Wiekowski M. T., Abbondanzo S. J., Cook D. N., Bayne M. L., Lira S. A., Chintala M. S. (2001) Molecular identification and characterization of the platelet ADP receptor targeted by thienopyridine antithrombotic drugs. J. Clin. Invest. 107, 1591–1598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Thomas D. W., Mannon R. B., Mannon P. J., Latour A., Oliver J. A., Hoffman M., Smithies O., Koller B. H., Coffman T. M. (1998) Coagulation defects and altered hemodynamic responses in mice lacking receptors for thromboxane A2. J. Clin. Invest. 102, 1994–2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chan V. W., Meng F., Soriano P., DeFranco A. L., Lowell C. A. (1997) Characterization of the B lymphocyte populations in Lyn-deficient mice and the role of Lyn in signal initiation and down-regulation. Immunity 7, 69–81 [DOI] [PubMed] [Google Scholar]
- 30. Li Z., Zhang G., Le Breton G. C., Gao X., Malik A. B., Du X. (2003) Two waves of platelet secretion induced by thromboxane A2 receptor and a critical role for phosphoinositide 3-kinases. J. Biol. Chem. 278, 30725–30731 [DOI] [PubMed] [Google Scholar]
- 31. Xiang B., Zhang G., Liu J., Morris A. J., Smyth S. S., Gartner T. K., Li Z. (2010) A Gi-independent mechanism mediating Akt phosphorylation in platelets. J. Thromb. Haemost. 8, 2032–2041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cifuni S. M., Wagner D. D., Bergmeier W. (2008) CalDAG-GEFI and protein kinase C represent alternative pathways leading to activation of integrin αIIbβ3 in platelets. Blood 112, 1696–1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Klages B., Brandt U., Simon M. I., Schultz G., Offermanns S. (1999) Activation of G12/G13 results in shape change and Rho/Rho-kinase-mediated myosin light chain phosphorylation in mouse platelets. J. Cell Biol. 144, 745–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nagy B., Jr., Bhavaraju K., Getz T., Bynagari Y. S., Kim S., Kunapuli S. P. (2009) Impaired activation of platelets lacking protein kinase C-θ isoform. Blood 113, 2557–2567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cohen S., Braiman A., Shubinsky G., Ohayon A., Altman A., Isakov N. (2009) PKCθ is required for hemostasis and positive regulation of thrombin-induced platelet aggregation and α-granule secretion. Biochem. Biophys. Res. Commun. 385, 22–27 [DOI] [PubMed] [Google Scholar]
- 36. Chari R., Getz T., Nagy B., Jr., Bhavaraju K., Mao Y., Bynagari Y. S., Murugappan S., Nakayama K., Kunapuli S. P. (2009) Protein kinase Cδ differentially regulates platelet functional responses. Arterioscler. Thromb. Vasc. Biol. 29, 699–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cohen S., Braiman A., Shubinsky G., Isakov N. (2011) Protein kinase C-θ in platelet activation. FEBS Lett. 585, 3208–3215 [DOI] [PubMed] [Google Scholar]
- 38. Hirsch E., Bosco O., Tropel P., Laffargue M., Calvez R., Altruda F., Wymann M., Montrucchio G. (2001) Resistance to thromboembolism in PI3Kγ-deficient mice. FASEB J. 15, 2019–2021 [DOI] [PubMed] [Google Scholar]
- 39. Kim S., Jin J., Kunapuli S. P. (2006) Relative contribution of G-protein-coupled pathways to protease-activated receptor-mediated Akt phosphorylation in platelets. Blood 107, 947–954 [DOI] [PubMed] [Google Scholar]
- 40. Ma Y. C., Huang J., Ali S., Lowry W., Huang X. Y. (2000) Src tyrosine kinase is a novel direct effector of G proteins. Cell 102, 635–646 [DOI] [PubMed] [Google Scholar]
- 41. Faruqi T. R., Weiss E. J., Shapiro M. J., Huang W., Coughlin S. R. (2000) Structure-function analysis of protease-activated receptor 4 tethered ligand peptides. Determinants of specificity and utility in assays of receptor function. J. Biol. Chem. 275, 19728–19734 [DOI] [PubMed] [Google Scholar]
- 42. Offermanns S. (2006) Activation of platelet function through G protein-coupled receptors. Circ. Res. 99, 1293–1304 [DOI] [PubMed] [Google Scholar]
- 43. Zhao Y., Sudol M., Hanafusa H., Krueger J. (1992) Increased tyrosine kinase activity of c-Src during calcium-induced keratinocyte differentiation. Proc. Natl. Acad. Sci. U.S.A. 89, 8298–8302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Brass L. F., Manning D. R., Cichowski K., Abrams C. S. (1997) Signaling through G proteins in platelets. To the integrins and beyond. Thromb. Haemost. 78, 581–589 [PubMed] [Google Scholar]
- 45. Konopatskaya O., Gilio K., Harper M. T., Zhao Y., Cosemans J. M., Karim Z. A., Whiteheart S. W., Molkentin J. D., Verkade P., Watson S. P., Heemskerk J. W., Poole A. W. (2009) PKCα regulates platelet granule secretion and thrombus formation in mice. J. Clin. Invest. 119, 399–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Han J., Lim C. J., Watanabe N., Soriani A., Ratnikov B., Calderwood D. A., Puzon-McLaughlin W., Lafuente E. M., Boussiotis V. A., Shattil S. J., Ginsberg M. H. (2006) Reconstructing and deconstructing agonist-induced activation of integrin αIIbβ3. Curr. Biol. 16, 1796–1806 [DOI] [PubMed] [Google Scholar]
- 47. Lian L., Wang Y., Flick M., Choi J., Scott E. W., Degen J., Lemmon M. A., Abrams C. S. (2009) Loss of pleckstrin defines a novel pathway for PKC-mediated exocytosis. Blood 113, 3577–3584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Crittenden J. R., Bergmeier W., Zhang Y., Piffath C. L., Liang Y., Wagner D. D., Housman D. E., Graybiel A. M. (2004) CalDAG-GEFI integrates signaling for platelet aggregation and thrombus formation. Nat. Med. 10, 982–986 [DOI] [PubMed] [Google Scholar]
- 49. Zhang G., Xiang B., Ye S., Chrzanowska-Wodnicka M., Morris A. J., Gartner T. K., Whiteheart S. W., White G. C., 2nd, Smyth S. S., Li Z. (2011) Distinct roles for Rap1b protein in platelet secretion and integrin αIIbβ3 outside-in signaling. J. Biol. Chem. 286, 39466–39477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. de la Fuente M., Noble D. N., Verma S., Nieman M. T. (2012) Mapping human protease-activated receptor 4 (PAR4) homodimer interface to transmembrane helix 4. J. Biol. Chem. 287, 10414–10423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li D., D'Angelo L., Chavez M., Woulfe D. S. (2011) Arrestin-2 differentially regulates PAR4 and ADP receptor signaling in platelets. J. Biol. Chem. 286, 3805–3814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rittenhouse S. E. (1996) Phosphoinositide 3-kinase activation and platelet function. Blood 88, 4401–4414 [PubMed] [Google Scholar]
- 53. Stoyanov B., Volinia S., Hanck T., Rubio I., Loubtchenkov M., Malek D., Stoyanova S., Vanhaesebroeck B., Dhand R., Nürnberg B. (1995) Cloning and characterization of a G protein-activated human phosphoinositide-3 kinase. Science 269, 690–693 [DOI] [PubMed] [Google Scholar]
- 54. Stephens L. R., Eguinoa A., Erdjument-Bromage H., Lui M., Cooke F., Coadwell J., Smrcka A. S., Thelen M., Cadwallader K., Tempst P., Hawkins P. T. (1997) The Gβγ sensitivity of a PI3K is dependent upon a tightly associated adaptor, p101. Cell 89, 105–114 [DOI] [PubMed] [Google Scholar]
- 55. Zhang J., Benovic J. L., Sugai M., Wetzker R., Gout I., Rittenhouse S. E. (1995) Sequestration of a G-protein βγ subunit or ADP-ribosylation of Rho can inhibit thrombin-induced activation of platelet phosphoinositide 3-kinases. J. Biol. Chem. 270, 6589–6594 [DOI] [PubMed] [Google Scholar]
- 56. Hardy A. R., Jones M. L., Mundell S. J., Poole A. W. (2004) Reciprocal cross-talk between P2Y1 and P2Y12 receptors at the level of calcium signaling in human platelets. Blood 104, 1745–1752 [DOI] [PubMed] [Google Scholar]