

Background: Mechanism of VEGFA transcription by complex interplay of β-catenin and PPAR-δ remains elusive.
Results: The VEGFA promoter was dynamically occupied by β-catenin, TCF-4, and PPAR-δ. β-Catenin also bound downstream region of the VEGFA gene.
Conclusion: β-Catenin is responsible for the formation of chromatin loops, which is relieved by PPAR-δ activation.
Significance: Proposed mechanism could provide clues for VEGFA-mediated colon cancer cell initiation.
Keywords: β-Catenin, Colon Cancer, Peroxisome Proliferator-activated Receptor (PPAR), Transcription Regulation, Vascular Endothelial Growth Factor (VEGF)
Abstract
Vascular endothelial growth factor A (VEGFA) mRNA is regulated by β-catenin and peroxisome proliferator activated receptor δ (PPAR-δ) activation in colon cancer cells, but the detailed mechanism remains to be elucidated. As chromatin loops are generally hubs for transcription factors, we tested here whether β-catenin could modulate chromatin looping near the VEGFA gene and play any important role for PPAR-δ activated VEGFA transcription. First, we identified the far upstream site as an important site for VEGFA transcription by luciferase assay and chromatin immunoprecipitation in colorectal carcinoma HCT116 cells. Chromatin conformation capture analysis also revealed the chromatin loops formed by the β-catenin bindings on these sites near the VEGFA gene. Dynamic association and dissociation of β-catenin/TCF-4/PPAR-δ on the far upstream site and β-catenin/NF-κB p65 on the downstream site were also detected depending on PPAR-δ activation. Interestingly, β-catenin-mediated chromatin loops were relieved by PPAR-δ activation, suggesting a regulatory role of β-catenin for VEGFA transcription. Based on these data, we propose a model for PPAR-δ-activated VEGFA transcription that relies on β-catenin-mediated chromatin looping as a prerequisite for the activation. Our findings could extend to other β-catenin regulated target genes and could provide a general mechanism and novel paradigm for β-catenin-mediated oncogenesis.
Introduction
Vascular endothelial growth factor A (VEGFA)2 is the key regulator that initiates angiogenesis. Many signaling pathways are interconnected to activate VEGFA expression during tumorigenesis (1). Various transcription factors, such as hypoxia-inducible factor 1-α (HIF1-α), are important for transcriptional activation on the VEGFA promoter (2, 3). Additionally, Wnt signaling activates VEGFA expression during early colonic neoplasia, so pathological β-catenin activation might enhance tumor angiogenesis (4–8). In the case of colorectal cancer cells, peroxisome proliferator-activated receptor-δ (PPAR-δ) is also reported to be involved in VEGFA expression in some cases (9–13). How PPAR-δ induces VEGFA gene expression in some, but not all, microenvironments should be understood.
β-Catenin is important in the development of colon cancer, so inactivation of adenomatous polyposis coli and/or a β-catenin mutation leads to inappropriate stabilization of β-catenin in sporadic colon cancer. Activated β-catenin translocates to the nucleus and complexes with T-cell factor (TCF) transcription factor, which is a vital step in carcinogenesis because it globally alters the expression of oncogenic target genes such as cyclin D1 and c-myc (14–16). PPAR is a member of the nuclear hormone receptor superfamily with diverse tissue distributions and distinct but overlapping biological functions (17, 18). Among three PPARs (PPAR-α, PPAR-δ, and PPAR-γ), PPAR-δ is mostly involved in lipid metabolism but also affects inflammatory response, angiogenesis, terminal differentiation, and proliferation (19). However, it still remains to be determined whether PPAR-δ plays any direct and critical role during cancer cell formation and progression (11, 20–26). Cross-talk between PPAR-δ and Wnt/β-catenin signaling has been proposed (27–29), which could be indicative of a complicated and intricate interplay between them. Therefore, deciphering the molecular interplay between PPAR-δ and β-catenin is important to clarify the nature of the pathway network for cancer formation and progression. More specifically, detailed transcriptional functions of β-catenin in conjunction with PPAR-δ activation could provide clues for the complex interaction between these transcriptional regulators in VEGFA transcription.
Transcription is mediated by the binding of specific transcription factors to regulatory sequences. Long range interactions between promoter and regulatory elements are important for high level expression of target genes and may cause looping of chromatin as well as protein-protein interactions (30, 31). Chromatin looping brings distant cis-acting elements into close proximity, allowing dynamic and flexible interactions between genomic loci as well as between regulatory elements near the gene. Therefore, chromatin looping could modulate the gene expression program in cells, which could operate depending on the cell condition or tissue microenvironment. Global screening of β-catenin target genes has revealed many β-catenin/TCF binding sites at upstream and downstream sites in some target genes (32–35). Therefore, β-catenin/TCF binding sites at upstream and downstream sites contribute to the formation of chromatin looping near the gene (36). In fact, the many faces of β-catenin suggest its role in the regulation of chromatin modification as well as looping (37, 38).
To solve the complicated mechanism behind β-catenin and PPAR-δ-mediated VEGFA transcription, here we hypothesized that β-catenin forms chromatin looping near the VEGFA genomic locus, and PPAR-δ regulates it to promote VEGFA transcription. As a first step, we identified repressive chromatin looping via β-catenin bindings in the far upstream region and downstream regions of the VEGFA gene. More significantly, such chromatin looping was opened and triggered for activation by treatment with a PPAR-δ agonist. These findings suggest that β-catenin and PPAR-δ modulate VEGFA transcription. Most importantly, these data provide evidence for a novel paradigm of β-catenin-mediated chromatin looping, which could explain the diverse roles of β-catenin on its various target genes.
EXPERIMENTAL PROCEDURES
Luciferase Reporters, Mutagenesis, and Expression Clones
The VEGFA promoter with wild-type and mutant TBE3 (denoted as −1.8 and −0.8 kb VEGFA reporter) were kind gifts from Dr. Daniel C. Chung (Harvard Medical School, Boston, MA (4)). The longer VEGFA promoter fragment (denoted as −2.3-kb VEGFA reporters) was amplified by genomic polymerase chain reaction (PCR) from HCT116 cells with primers and cloned into the pGL3 control vector. Site-directed mutagenesis was performed by mutagenic PCR to generate the mutant TBE1 reporter (TACAAAG → TAGGGAG). The downstream region of the VEGFA gene (fragment #12) was amplified by PCR primers and cloned into the downstream region of the luciferase gene driven by the −2.3-kb VEGFA promoter (denoted as 2.3-kb+DS). The mammalian expression clone for PPAR-δ was cloned into the pCMV-3xFLAG vector. All primers used for this experiment are listed in supplemental Table 1.
Cell Culture, Transfection, and GW501516 Treatment
HCT116 human colorectal carcinoma cells (American Type Culture Collection, Manassas, VA) were cultured in denoted as 2.3 (Invitrogen) supplemented with 10% fetal bovine serum and 1× antimycotic (Invitrogen). Cells were transfected with expression clone and reporter constructs using Lipofectamine Plus according to the manufacturer's protocol (Invitrogen). PPAR-δ activation was achieved by GW501516 (10 μm) treatment after an 8-h serum starvation of HCT116 cells.
Luciferase Assay
Cells were transiently cotransfected with the firefly luciferase reporter (100 ng) and the Renilla luciferase reporter (10 ng) to normalize transfection efficiency. Luciferase activity was determined with a dual luciferase assay system (Promega, Madison, WI). Activity was determined using a Glomax 20/20 luminolmeter (Promega). Relative firefly luciferase activity was obtained by normalizing it to that of Renilla luciferase activity. Experiments were repeated at least three times, and values are expressed as the mean ± S.D.
Reverse Transcription Quantitative PCR (RT-qPCR) Analysis
Cells were harvested with TRIzol reagent (Molecular Research Center, Cincinnati, OH), and total mRNAs were isolated from cells. Total mRNA was reverse-transcribed with oligo(dT) (Fermentas, Burlington, ON, Canada) using RevertAid H Minus Reverse Transcriptase (Fermentas). The resulting cDNA was used in a Step ONE real-time PCR kit (Applied Biosystems, Foster City, CA). Quantitative PCR was performed with Dynamo Flash master mix (Finnzyme, Maahantuonti, Finland).
Quantitative Chromatin Immunoprecipitation (ChIP) and ChIP-qPCR Analyses
Cells were fixed in 1% formaldehyde, and glycine was added to stop the cross-linking reaction. Cells were lysed with immunoprecipitation assay buffer. Resuspended cells were sonicated and centrifuged, and the supernatant was collected. Protein G-Sepharose 4 Fast Flow beads (GE Healthcare) were blocked by tRNA. Immunoprecipitation was carried out for 12–24 h with each antibody. Normal IgG antibody was used as the control sample. After immunoprecipitation, precleared protein G-Sepharose beads were added and incubated for 2 h. The beads were collected and washed 3 times with Tris-EDTA buffer and then extracted with elution buffer consisting of 1% sodium dodecyl sulfate (SDS) and 0.1 m sodium bicarbonate. The eluents were heated to 65 °C for 6 h to reverse formaldehyde cross-linking. DNA fragments were purified by phenol extraction and ethanol precipitation. Purified DNA fragments were amplified with specific primers and visualized in agarose gel electrophoresis for ChIP analysis. Concomitantly, real-time qPCR was performed as described above for ChIP-qPCR analysis. The following antibodies were used for immunoprecipitation and Western blotting: anti-β-catenin (BD Transduction Laboratories), anti-TCF-4 (Santa Cruz Biotechnology, Santa Cruz, CA), anti-FLAG (Sigma), anti-nuclear factor κB (NF-κB) p65 (Santa Cruz Biotechnology), anti-β-actin (Abcam, Cambridge, MA), anti-α-tubulin (Abcam), and anti-Lamin B1 (Abcam).
Chromatin Conformation Capture (3C) Assay
The 3C protocol has been described previously (39). Briefly, cells were trypsinized, fixed with 1% formaldehyde for 10 min, and quenched with 125 mm glycine. The cells were washed with 1× phosphate-buffered saline and prelysed with prelysis buffer (10 mm Tris-HCl, pH 8.0, 10 mm NaCl, 0.2% Nonidet P-40, and 1× protease inhibitor mixture). Penetrated cells were washed with 1× restriction buffer, and SDS was added to each sample to a concentration of 0.1%. Each sample was incubated for 30 min at 37 °C and 900 rpm. The action of SDS was stopped by adding 1% Triton X-100; the resulting suspension was incubated for 1 h at 37 °C and 900 rpm. Next, the samples were reacted with 500 units of BamHI restriction enzyme (R0136T; New England BioLabs, Beverly, MA) overnight. The reaction was terminated by adding 2% SDS. Each sample was diluted in a reaction solution containing T4 DNA ligase (M0202L; New England BioLabs) overnight. Ligated DNA was gathered using standard phenol precipitation and ethanol precipitation methods. Prepared DNA was PCR amplified. Primers used for RT-PCR, ChIP-PCR, and 3C are listed in supplemental Table 1.
RESULTS
Far Upstream TCF Binding Element (TBE) Site Is Involved in VEGFA Transcription
We first analyzed the upstream region of VEGFA gene for putative β-catenin/TCF and PPAR-δ binding elements. Three TBE consensus sites (forward orientation, (T/A)(T/A)CAA(T/A)G; reverse orientation, C(T/A)TTG(T/A)(T/A)) were predicted (TBE1, −1863; TBE2, −1497; TBE3, −805 shown as solid boxes with numbers in Fig. 1A). To identify TBE sites that could be directly regulated by β-catenin, we analyzed these TBE sites by ChIP assay with anti-β-catenin and anti-TCF-4 antibodies in HCT116 colorectal carcinoma cell line. Concomitant and strong β-catenin and TCF-4 bindings were observed at TBE1 and TBE3 sites, but no significant occupancies of β-catenin and TCF-4 were found at the TBE2 site as shown by PCR analysis of precipitated genomic DNA (Fig. 1, B and C). ChIP-qPCR analysis also showed similar patterns of protein bindings on TBE sites (Fig. 1, D and E). To confirm the involvement of these TBE sites, we used luciferase reporters with a series of deletions in the upstream region of VEGFA gene. As shown in Fig. 1F, relative ratio of transcriptional activity was significantly increased as upstream TBE1 was included, suggesting an important role of TBE1 in VEGFA transcription. Evidently, TBE2 may not be a critical site for VEGFA transcription (Fig. 1F). In the case of TBE3, luciferase reporter assays with wild-type (CTTTGAT) or mutant (CTTTACT) TBE3 clearly demonstrated the critical role of the sequence (Fig. 1G), which was consistent with a previous report (4). In contrast, even though significant occupancies of β-catenin and TCF-4 were detected in the far upstream TBE1 site by ChIP analysis, luciferase reporters with wild-type (TACAAAG) or mutant (TAGGGAG) TBE1 site did not show the sequence dependence (Fig. 1H). These data suggest that far upstream TBE1 might be involved in VEGFA transcription in unusual way.
FIGURE 1.

Mapping of upstream TBEs for β-catenin-mediated VEGFA transcription. A, shown is the predicted TBE forward, TBE reverse and PPAR-response elements (PPREs) in the upstream of VEGFA gene. Numbers in the boxes denote TBE sites from the upstream. 2 kb upstream to 1 kb downstream from the transcription start were analyzed for the predicted sites. Consensus sequences of the predicted TBEs in forward and reverse orientation are also shown by WebLogo3.0. B, ChIP analysis of TBE1, TBE2, and TBE3 in HCT116 colorectal carcinoma cells were grown in serum-free basal condition. Anti-β-catenin antibody (Ab) was employed in addition to IgG control antiserum. Input DNA was also shown to confirm the equal loading of the samples. IP, immunoprecipitate. C, ChIP analysis of TBE1, TBE2, and TBE3 with anti-TCF-4 antibody was as in B. D, ChIP-qPCR analysis for the TBEs using anti-β-catenin antibody is shown. Bound DNA was analyzed by quantitative real-time PCR and is presented as relative percent input of chromatin. Three independent analyses were performed; the results represent the average ± S.D. (***, p < 0.005). NC denotes negative control without antibody. E, ChIP-qPCR analysis for the TBEs using anti-TCF-4 antibody is shown. Bound DNA was analyzed by quantitative real-time PCR as in D. Three independent analyses were performed; the results represent the average ± S.D. (***, p < 0.005). F, a luciferase assay with various deletions in the upstream regions of VEGFA gene is shown. pGL2-VEGFA promoter reporters have −2.3 kb (from TBE1), −1.9 kb (from TBE2), and −0.8 kb (from TBE3) upstream fragment of VEGFA gene. HCT116 cells were transiently transfected with pGL2-VEGFA promoter reporter and pRL-TK and GW501516 treatment. Three independent experiments were performed, and the results represent the average ± S.D. (**, p < 0.01). G, shown is a luciferase assay with wild-type (Wt-TBE3) or mutant TBE3 sequences (Mt-TBE3) in the VEGFA promoter. The experiment was performed as in F and is shown in relative luciferase activities in comparison to that of wild type. Three independent experiments were performed; the results represent the average ± S.D. (***, p < 0.005). H, shown is a luciferase reporter assay with wild-type (Wt-TBE1) or mutant TBE1 site (Mt-TBE1) in the VEGFA promoter. The experiment was performed as in F and shown in relative luciferase activities in comparison to that of wild type. Three independent experiments were performed; the results represent the average ± S.D. (***, p < 0.005).
PPAR-δ Activation Leads to Dynamic Remodeling of β-Catenin, TCF-4, and PPAR-δ Protein Bindings on Upstream Sites
Even though TBE1 site has atypical TBE sequence, a strong positive VEGFA transcriptional activity was shown. So we questioned how TBE1 might be involved in VEGFA gene transcription. We performed ChIP analysis before and after PPAR-δ activation by GW501516 treatment in HCT116 cells. Intriguingly, two putative PPAR-response elements (PPREs) were found near TBEs (Fig. 1A), so we analyzed whether β-catenin and TCF-4 occupancy was regulated by PPAR-δ activation. Upon GW501516 treatment, the binding patterns of β-catenin and TCF-4 were changed on TBE1, with a drastic reduction in β-catenin occupancy but with a slight increase in TCF-4 binding (Fig. 2A). PPAR-δ binding was enhanced after GW501516 treatment if we compared the binding of FLAG-tagged PPAR-δ (Fig. 2A). Striking and dynamic remodeling of three proteins on TBE1 suggested that it was an important regulatory element for PPAR-δ-activated VEGFA transcription. In particular, opposite binding patterns of β-catenin (dissociation) and TCF-4 and PPAR-δ (association) were noticed after GW501516 treatment by ChIP-qPCR analysis (Fig. 2B). Such dynamic remodeling of transcription factors was in striking contrast to other TBEs in the VEGFA promoter. In the case of TBE3, PPAR-δ activation lead to enhanced β-catenin occupancy accompanied by TCF-4 DNA-binding protein (Fig. 2, A and C), implying the formation of an typically active form of β-catenin/TCF-4 transcription complex on TBE3. PPAR-δ also associated with TBE3 more than with TBE1 (Fig. 2, A and C). Because TBE1 demonstrated β-catenin dissociation upon PPAR-δ activation (Fig. 2, A and B), we then asked whether it was dependent on wild-type the TBE sequence. Significant GW501516-activated increase of luciferase activity was shown only when the TBE1 was wild type but not in mutant form (Fig. 2D). In contrast, TBE3 appeared not to be essential for GW501516-activated VEGFA transcription (Fig. 2E) even though it was critical for β-catenin-mediated basal VEGFA transcription (Fig. 1G). So far, TBE1 seemed to be important for PPAR-δ activated VEGFA transcription but with atypical binding behaviors of β-catenin, TCF-4, and PPAR-δ.
FIGURE 2.

Dynamic remodeling of β-catenin, TCF-4, and PPAR-δ bindings on upstream TBEs after GW501516 treatment. A, ChIP for TCF binding element 1 and 3 (TBE1 and TBE3) in the upstream of the VEGFA gene is shown. GW501516 treatment was applied to HCT116 cells as described above, and anti-β-catenin, anti-TCF-4, and anti-FLAG antibodies (for PPAR-δ) were employed. Western blot (W.B) analysis was also performed to confirm the expression of FLAG-PPAR-δ, equal use of the input and immunoprecipitation of each protein. Anti-β-actin antibody was used as a negative control. B, ChIP-qPCR for TBE1 of the VEGFA promoter is shown. GW501516 (GW) treatment was applied to HCT116 cells as described above, and anti-β-catenin, anti-TCF-4, and anti-FLAG antibodies were employed. Three independent analyses were performed; the results represent the average ± S.D. (***, p < 0.005). NC denotes negative control without antibody. C, ChIP-qPCR for TBE3 of the VEGFA promoter is shown. The experiment was performed as in B. Three independent analyses were performed; the results represent the average ± S.D. (***, p < 0.005). D, shown is a luciferase reporter assay with VEGFA natural promoter with wild-type (Wt-TBE1) or mutant TBE1 sites (Mt-TBE1). GW501516 was treated to HCT116 as described above. Three independent experiments were performed; the results represent the average ± S.D. (***, p < 0.005) of relative luciferase activities in comparison to that of DMSO-treated cells. E, shown is a luciferase reporter assay with VEGFA natural promoter containing wild-type (Wt-TBE3) or mutated TBE3 sites (Mt-TBEs). Three independent experiments were performed; the results represent the average ± S.D. (***, p < 0.005) of relative luciferase activities in comparison to that of DMSO-treated cells.
β-Catenin Forms Chromatin Loops Near the VEGFA Gene, Which Are Relieved by PPAR-δ Activation
ChIP analyses demonstrated the distinct binding patterns of β-catenin/TCF/PPAR-δ on TBE1, and a luciferase assay identified upstream TBE1 as an important site for PPAR-δ-activated VEGFA transcription. However, it was unclear how TBE1 activated VEGFA transcription. Based on previous reports that β-catenin binds upstream and downstream TBE sites on some target genes (33, 35, 36), we reasoned that β-catenin could mediate the formation of chromatin loops near the VEGFA gene thru TBE1 binding. To test this, we designed a 3C experiment and looked for possible downstream BamHI fragments of the VEGFA gene mediating chromatin looping with TBE1. An upstream TBE1 site was fixed as an anchor, and other possible looping fragments were detected with downstream 3C primers (Fig. 3A). The BAC clone with the VEGFA gene was used to validate PCR primer efficiency and to normalize the 3C signal (Fig. 3B). Robust looping to fragments #8, #12, and #14 were observed when 3C experiments were performed with HCT116 cells grown in basal conditions. However, strikingly, when VEGFA transcription was activated by GW501516, chromatin looping at the TBE1 site was completely released (Fig. 3B). If looping frequency from the anchor fragment was shown as relative proximity from TBE1, fragment #12, which was located 20.2 kb downstream from TBE1, showed the strongest looping frequency (Fig. 3C). More importantly, chromatin loops were completely released in response to GW501516 treatment (Fig. 3, B and C). As chromatin loops are formed around the VEGFA gene, we then asked whether β-catenin is critical for looping. Chromatin looping was greatly reduced up to 60% after Β-CATENIN knockdown in comparison to control knockdown with siGFP (Fig. 3, D and E). These data clearly demonstrated that β-catenin is involved in the formation of chromatin loops around the VEGFA gene, which can be released by activating PPARδ.
FIGURE 3.

Chromatin looping near the VEGFA gene by β-catenin. A, shown is the design of 3C analysis near the VEGFA gene (−3 to +10 kb). BamHI restriction enzyme sites (black bars) and the resulting fragments are shown as numbers (#) and distances (kb) from the anchor primer (red arrow) at TBE1. The 3C reverse primers pairing the anchor primer are indicated as blue arrows, and the negative primer is shown as a gray arrow. B, 3C analysis with BAC template and with genomic DNA template is shown. Genomic DNAs were extracted from dimethyl sulfoxide or GW501516 (GW)-treated HCT116 cells, and 3C assays were performed. PCR products for detecting each 3C fragment are shown in the order of the genomic BamHI sites from the coding region (#8, #10, and #11) and the downstream regions (#12, #14, #15, and #17) of the VEGFA gene. The gene for glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a loading control (control), and the BAC template was used as a positive control. C, shown is a graphics presentation of the 3C data shown in B after normalizing for PCR efficiency with the BAC template and GAPDH. The x axis is arranged by the genomic distances from the anchor primer, and the y axis marks the relative proximity of the chromosome fragments. D, 3C analysis after β-catenin knockdown is shown. HCT116 cells were transfected with β-catenin small interfering RNA or green fluorescent protein (GFP) siRNA. The 3C assay was performed as described above, and GAPDH was used as loading control (control). β-Catenin protein knockdown was confirmed by Western blot analysis. β-Actin was used as the loading control. E, shown is a graphics representation of the 3C data from siRNA-transfected HCT116 cells. Relative proximity of each fragment #8 and #12 and GAPDH from β-catenin siRNA (siβ-cat)-transfected cells were compared with that of GFP siRNA.
β-Catenin Binds Downstream TBEs and Represses VEGFA Transcription
After identifying the role of β-catenin in chromatin loops around the VEGFA gene, we looked for putative TBEs located downstream of the gene and named them downstream TBEs (TBE-DS). As indicated in Fig. 4A, some prominent TBE sites (TBE-DS5 and TBE-DS6) were predicted in the most likely looping fragment, #12, which could be β-catenin/TCF-4 binding sites. To validate these candidate binding sites, we performed a ChIP analysis with β-catenin and TCF-4 antibodies (Fig. 4B). β-Catenin bound to TBE-DS without activation, which was somewhat enhanced by GW501516 treatment. Interestingly, TCF-4 binding to TBE-DS was not significantly higher in comparison to the negative control, suggesting that occupancy of β-catenin in TBE-DS is independent of TCF-4 binding (Fig. 4, B and C). Therefore, we concluded that β-catenin, but not TCF-4, bound downstream fragments via TBE-DS sites in fragment #12. However, it was unclear whether the VEGFA downstream region was important for gene transcription. Thus, we constructed −2.3 kb VEGFA promoter driven luciferase reporters with or without downstream fragment #12 containing TBE-DS (Fig. 4D). When downstream fragment #12 was included in the reporter (−2.3-kb + DS), a significant reduction in luciferase activity was noticed not only for GW501516-induced VEGFA transcription but also for basal level transcription. This suggested a repressive role for downstream fragment #12 on VEGFA transcription. These findings were consistent with the 3C and ChIP data for β-catenin-mediated chromatin looping, which further suggested a repressive role for downstream fragment #12. We also confirmed the repressive role of β-catenin for VEGFA transcription in HCT116 cells (data not shown). Taken together, these results demonstrate β-catenin-mediated chromatin looping and repression of transcription via TBE-DS sites in fragment #12.
FIGURE 4.

Repression of VEGFA transcription by the binding of β-catenin in downstream fragment #12. A, downstream TBE sites (TBE-DS) located in downstream fragments #12 are shown. BamHI Fragment #12 contained TBE-DS5 and TBE-DS6. B, ChIP analysis of TBE-DS within fragment #12 is shown. Anti-β-catenin and anti-TCF-4 antibodies were employed in addition to IgG control antiserum. Input DNA was also shown. HCT116 cells were grown in serum-free conditions and activated by GW501516. Western blot (W.B) analysis was also performed to confirm the equal use of the input and immunoprecipitation of each protein. C, ChIP-qPCR analysis of downstream TBEs (TBE-DS) is shown. TBE-DS was analyzed for binding with β-catenin (left) and TCF-4 (right). Three independent analyses were performed; the results represent the average ± S.D. (***p < 0.005). NC denotes negative control without antibody. D, shown is a luciferase assay with the VEGFA promoter reporters either with downstream fragment #12 (VEGFA promoter + DS) or without as above. Three independent analyses were performed; the results represent the average ± S.D. (***, p < 0.005).
Downstream Fragment Is Occupied by NF-κB p65 and Regulates VEGFA Transcription
Because β-catenin occupies the downstream fragments without accompanied TCF-4 (Fig. 4, B and C), we then asked how β-catenin mediates chromatin looping to downstream fragments. Because β-catenin does not contain a DNA binding domain, it is likely that a DNA-binding protein other than TCF-4 might be involved in the TBE-DS6 site binding. Thus, we searched for a β-catenin binding partner protein that could regulate VEGFA transcription. We tested whether NF-κB p65 played any role in VEGFA transcription, because it regulates β-catenin-mediated transcription (40) as well as PPAR-δ function (41). Appropriately, we analyzed the p65 binding sequence (GGGRNNYYC) near VEGFA gene and found one p65 consensus site in the downstream fragment #12 at the vicinity of TBE-DS6 (Fig. 5A). When we performed the ChIP assay using anti- NF-κB p65 antibody, strong occupancy of NF-κB p65 was observed near TBE-DS6 as shown, which could be enhanced by GW501516 treatment (Fig. 5, B and C). Additionally, NF-κB p65 overexpression significantly reduced the level of GW501516-induced VEGFA mRNA (Fig. 5D). These observations suggested that NF-κB p65 occupies fragment #12 downstream of the VEGFA gene, possibly near the TBE site (TBE-DS6), and represses VEGFA mRNA expression. Luciferase reporter analysis with downstream fragment #12 (−2.3-kb + DS) supported this notion, showing the repression of VEGFA transcription by the overexpression of NF-κB p65 not only for GW501516-treated cells but also in basal cells (Fig. 5E). Promiscuous NF-κB activation can be excluded because p65 nuclear translocation and/or IκB degradation was not elicited by β-catenin (data not shown). These collective observations were consistent with the view that NF-κB p65 negatively regulates VEGFA by binding to downstream TBE-DS6 along with β-catenin.
FIGURE 5.

Binding of NF-κB p65 in the downstream of VEGFA gene. A, shown is a diagram of VEGFA downstream fragment #12 containing NF-κB p65 consensus site next to TBE-DS6. B, shown is ChIP analysis of TBE-DS within fragment #12. Anti-NF-κB p65 antibody was employed in addition to IgG control antiserum. Input DNA was also shown. HCT116 cells were grown in serum-free condition and activated by GW501516 (GW). Western blot (W.B) analysis was also performed to confirm the equal use of the input and immunoprecipitation of NF-κB p65 protein. Anti-β-actin antibody was used as a negative control. C, shown is ChIP-qPCR analysis with anti-NF-κB p65 antibody for NF-κB site in downstream fragment #12 (TBE-DS6). Three independent analyses were performed; the results represent the average ± S.D. (***, p < 0.005). NC denotes negative control without antibody. D, shown is RT-qPCR analysis of VEGFA mRNA after p65 overexpression in conjunction with GW501516 treatment. Expression of NF-κB p65 protein was confirmed by Western blot analysis. E, luciferase (Luc.) assay with the VEGFA promoter reporters with downstream fragment #12 (−2.3-kb VEGFA promoter + DS) with or without p65 overexpression. Three independent analyses were performed; the results represent the average ± S.D. (***, p < 0.005).
DISCUSSION
The roles of the Wnt/β-catenin pathway and PPAR-δ have been proposed for VEGFA mRNA expression, but the mechanism for transcriptional regulation remains elusive and controversial (12, 13, 42–46). Here, we showed that VEGFA mRNA transcription can be regulated by PPAR-δ only when β-catenin-mediated chromatin loops are formed in HCT116 colorectal carcinoma cell line. Chromatin loops around VEGFA were released upon PPAR-δ activation so VEGFA transcription was activated. As summarized as a model in Fig. 6, chromatin loops were formed by β-catenin binding, probably by TCF-4 in upstream sites, as well as by NF-κB p65 in downstream sites. The model predicts that β-catenin mediates repressive looping and that PPAR-δ-specific ligands release the loops, creating an active transcription unit. In fact, β-catenin can sometimes repress transcription of target genes by binding to corepressors such as the histone deacetylase complex and affecting chromatin structure (37, 38, 47, 48).
FIGURE 6.
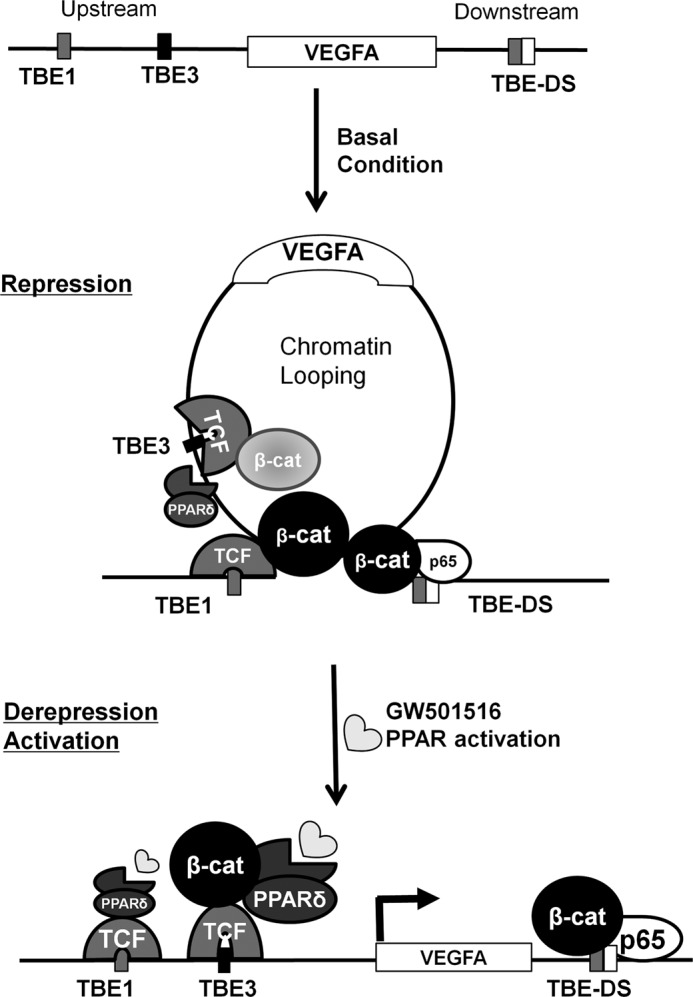
Schematic diagram of the proposed model. Shown is the model for β-catenin-mediated and PPAR-δ-activated vascular endothelial growth factor A (VEGFA) transcription. The chromatin loop is formed by β-catenin near the VEGFA gene in HCT116 cells grown in basal serum-free condition. β-Catenin occupies the far upstream TBE1 and TBE3 along with TCF-4 at upstream TBE sites and NF-κB p65 at downstream sites. At this basal state VEGFA transcription is repressed. Upon activation by GW501516, the chromatin loop is released by dissociation of β-catenin in TBE1 and association of PPAR-δ in upstream TBEs. VEGFA transcription is derepressed or activated by such remodeling of transcription factors near the gene that accompanied by the release of chromatin loop. Therefore, β-catenin-mediated chromatin loops near the VEGFA gene are prerequisite for PPAR-δ-activated VEGFA transcription in colorectal carcinoma, such as HCT116 cells.
We have also shown that β-catenin binds to a downstream region and is important for forming chromatin looping near the C-reactive protein gene (36) via a protein-protein interaction with TCF-4 and/or NF-κB (40), as appears to have occurred for the VEGFA gene. This might be analogous to the estrogen-stimulated chromosome remodeling for estrogen receptor-β expression (49). Our model is also consistent with the proposal that β-catenin can act as a binary switch to simultaneously activate or repress lineage-specific genes, possibly by interacting with transcriptional regulators (50). Considering chromatin looping as a key mechanism for the β-catenin-mediated binary switch, condition-dependent regulation of the VEGFA gene can be envisioned. Such condition-dependent chromatin looping might be related to the developmental regulation of many genes in the genome (51). As PPAR-δ agonists switch the SUMOylation status of an associated transcription factor, it might be worthwhile to test whether such a posttranslational modification of β-catenin is also switched by a PPAR-δ agonist to release chromatin looping (52).
Contradictory findings have been reported for PPAR-δ expression levels in many cancer cell lines and human patient tissues (24–26, 53, 54), cell proliferation, and cancer progression (11, 20–23), which might reflect differences in the model systems including culture conditions. However, a number of studies have consistently suggested that PPAR-δ induces angiogenesis, possibly by activating VEGFA expression (7, 12, 55). Therefore, understanding the molecular details of PPAR-δ transactivation of VEGFA could shed light on possible roles of PPAR-δ in tumor formation. Complex networks with other signaling pathways could provide a way to reconcile contradictory results from different systems (25, 56, 57). Interplay with β-catenin signaling might be reasonable, because of cancer-specific activation of β-catenin (50, 58, 59), association with PPAR-δ overexpression (26, 60, 61), and cross-talk between TCF/β-catenin signaling and PPAR expression and activity (62, 63). Here, we provide evidence that PPAR-δ acts on the VEGFA gene along with β-catenin by regulating chromatin loops. This might be consistent with recent findings concerning the role of PPAR-δ as an activator of β-catenin transcription activity (29). Taken together, these data support the idea of required regulatory chromatin looping to activate VEGFA gene transcription, as suggested in the “chromatin hub model” (64), which might lead to β-catenin-mediated chromatin regulation as a general mechanism for other Wnt target genes (35, 65).
This work was supported by the National Research Foundation of Korea grants funded by Ministry of Education, Science, and Technology of Korea Grants 2012-0006254 and 2012-0000662.

This article contains supplemental Table 1.
- VEGFA
- vascular endothelial growth factor A
- PPAR
- peroxisome proliferator-activated receptor
- TCF
- T-cell factor
- qPCR
- quantitative PCR
- 3C
- chromatin conformation capture
- TBE
- TCF binding element
- DS
- downstream.
REFERENCES
- 1. Grothey A., Galanis E. (2009) Targeting angiogenesis. Progress with anti-VEGFA treatment with large molecules. Nat. Rev. Clin. Oncol. 6, 507–518 [DOI] [PubMed] [Google Scholar]
- 2. Semenza G. L. (2003) Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 3, 721–732 [DOI] [PubMed] [Google Scholar]
- 3. Wouters B. G., Koritzinsky M. (2008) Hypoxia signaling through mTOR and the unfolded protein response in cancer. Nat. Rev. Cancer 8, 851–864 [DOI] [PubMed] [Google Scholar]
- 4. Zhang X., Gaspard J. P., Chung D. C. (2001) Regulation of vascular endothelial growth factor by the Wnt and K-ras pathways in colonic neoplasia. Cancer Res. 61, 6050–6054 [PubMed] [Google Scholar]
- 5. Easwaran V., Lee S. H., Inge L., Guo L., Goldbeck C., Garrett E., Wiesmann M., Garcia P. D., Fuller J. H., Chan V., Randazzo F., Gundel R., Warren R. S., Escobedo J., Aukerman S. L., Taylor R. N., Fantl W. J. (2003) β-Catenin regulates vascular endothelial growth factor expression in colon cancer. Cancer Res. 63, 3145–3153 [PubMed] [Google Scholar]
- 6. Rao T. P., Kühl M. (2010) An updated overview on Wnt signaling pathways. A prelude for more. Circulation Res. 106, 1798–1806 [DOI] [PubMed] [Google Scholar]
- 7. Wagner N., Jehl-Piétri C., Lopez P., Murdaca J., Giordano C., Schwartz C., Gounon P., Hatem S. N., Grimaldi P., Wagner K. D. (2009) Peroxisome proliferator-activated receptor b stimulation induces rapid cardiac growth and angiogenesis via direct activation of calcineurin. Cardiovasc. Res. 83, 61–71 [DOI] [PubMed] [Google Scholar]
- 8. Goodwin A. M., D'Amore P. A. (2002) Wnt signaling in the vasculature. Angiogenesis 5, 1–9 [DOI] [PubMed] [Google Scholar]
- 9. Fauconnet S., Lascombe I., Chabannes E., Adessi G. L., Desvergne B., Wahli W. (2002) Differential regulation of vascular endothelial growth factor expression by peroxisome proliferator-activated receptors in bladder cancer cells. J. Biol. Chem. 277, 23534–23543 [DOI] [PubMed] [Google Scholar]
- 10. Stephen R. L., Gustafsson M. C., Jarvis M., Tatoud R., Marshall B. R., Knight D. (2004) Activation of peroxisome proliferator-activated receptor δ stimulates the proliferation of human breast and prostate cancer cell lines. Cancer Res. 64, 3162–3170 [DOI] [PubMed] [Google Scholar]
- 11. Zuo X., Peng Z., Moussalli M. J., Morris J. S., Broaddus R. R., Fischer S. M., Shureiqi I. (2009) Targeted genetic disruption of peroxisome proliferator-activated receptor δ and colonic tumorigenesis. J. Natl. Cancer Inst. 101, 762–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang D., Wang H., Guo Y., Ning W., Katkuri S., Wahli W., Desvergne B., Dey S. K., DuBois R. N. (2006) Cross-talk between peroxisome proliferator-activated receptor δ and VEGFA stimulates cancer progression. Proc. Natl. Acad. Sci. U.S.A. 103, 19069–19074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Röhrl C., Kaindl U., Koneczny I., Hudec X., Baron D. M., König J. S., Marian B. (2011) Peroxisome proliferator-activated receptor γ and β/δ mediate vascular endothelial growth factor production in colorectal tumor cells. J. Cancer Res. Clin. Oncol. 137, 29–39 [DOI] [PubMed] [Google Scholar]
- 14. Barker N., Clevers H. (2006) Mining the Wnt pathway for cancer therapeutics. Nat. Rev. Drug Discov. 5, 997–1014 [DOI] [PubMed] [Google Scholar]
- 15. Clevers H. (2006) Wnt/β-catenin signaling in development and disease. Cell 127, 469–480 [DOI] [PubMed] [Google Scholar]
- 16. Saif M. W., Chu E. (2010) Biology of colorectal cancer. Cancer J. 16, 196–201 [DOI] [PubMed] [Google Scholar]
- 17. Castrillo A., Tontonoz P. (2004) Nuclear receptors in macrophage biology. At the cross-roads of lipid metabolism and inflammation. Annu. Rev. Cell Dev. Biol. 20, 455–480 [DOI] [PubMed] [Google Scholar]
- 18. Berger J., Moller D. E. (2002) The mechanisms of action of PPARs. Annu. Rev. Med. 53, 409–435 [DOI] [PubMed] [Google Scholar]
- 19. Wang D., Dubois R. N. (2008) Peroxisome proliferator-activated receptors and progression of colorectal cancer. PPAR Res. 2008, 931074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Park B. H., Vogelstein B., Kinzler K. W. (2001) Genetic disruption of PPAR-δ decrease the tumorigenicity of human colon cancer cells. Proc. Natl. Acad. Sci. U.S.A. 98, 2598–2603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang D., Wang H., Shi Q., Katkuri S., Walhi W., Desvergne B., Das S. K., Dey S. K., DuBois R. N. (2004) Prostaglandin E2 promotes colorectal adenoma growth via transactivation of the nuclear peroxisome proliferator-activated receptor δ. Cancer Cell 6, 285–295 [DOI] [PubMed] [Google Scholar]
- 22. Reed K. R., Sansom O. J., Hayes A. J., Gescher A. J., Winton D. J., Peters J. M., Clarke A. R. (2004) PPAR-δ status and Apc-mediated tumourigenesis in the mouse intestine. Oncogene 23, 8992–8996 [DOI] [PubMed] [Google Scholar]
- 23. Harman F. S., Nicol C. J., Marin H. E., Ward J. M., Gonzalez F. J., Peters J. M. (2004) Peroxisome proliferator-activated receptor-δ attenuates colon carcinogenesis. Nat. Med. 10, 481–483 [DOI] [PubMed] [Google Scholar]
- 24. Kim D. J., Bility M. T., Billin A. N., Willson T. M., Gonzalez F. J., Peters J. M. (2006) PPARβ/δ selectively induces differentiation and inhibits cell proliferation. Cell Death Differ. 13, 53–60 [DOI] [PubMed] [Google Scholar]
- 25. Hollingshead H. E., Killins R. L., Borland M. G., Girroir E. E., Billin A. N., Willson T. M., Sharma A. K., Amin S., Gonzalez F. J., Peters J. M. (2007) Peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) ligands do not potentiate growth of human cancer cell lines. Carcinogenesis 28, 2641–2649 [DOI] [PubMed] [Google Scholar]
- 26. Foreman J. E., Chang W. C., Palkar P. S., Zhu B., Borland M. G., Williams J. L., Kramer L. R., Clapper M. L., Gonzalez F. J., Peters J. M. (2011) Functional characterization of peroxisome proliferator-activated receptor-β/δ expression in colon cancer. Mol. Carcinog. 50, 884–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Müller R. (2004) Cross-talk of oncogenic and prostanoid signaling pathways. J. Cancer Res. Clin. Oncol. 130, 429–444 [DOI] [PubMed] [Google Scholar]
- 28. Wang D., DuBois R. N. (2007) Inflammatory mediators and nuclear receptor signaling in colorectal cancer. Cell Cycle 6, 682–685 [DOI] [PubMed] [Google Scholar]
- 29. Han C., Lim K., Xu L., Li G., Wu T. (2008) Regulation of Wnt/β-catenin pathway by cPLA2α and PPAR-δ. J. Cell Biochem. 105, 534–545 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30. Li Q., Barkess G., Qian H. (2006) Chromatin looping and the probability of transcription. Trends Genet. 22, 197–202 [DOI] [PubMed] [Google Scholar]
- 31. Kleinjan D. A., van Heyningen V. (2005) Long range control of gene expression. Emerging mechanisms and disruption in disease. Am. J. Hum. Genet. 76, 8–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yochum G. S., McWeeney S., Rajaraman V., Cleland R., Peters S., Goodman R. H. (2007) Serial analysis of chromatin occupancy identifies β-catenin target genes in colorectal carcinoma cells. Proc. Natl. Acad. Sci. U.S.A. 104, 3324–3329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yochum G. S., Cleland R., Goodman R. H. (2008) A genome-wide screen for β-catenin binding sites identifies a downstream enhancer element that controls c-Myc gene expression. Mol. Cell. Biol. 28, 7368–7379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hatzis P., van der Flier L. G., van Driel M. A., Guryev V., Nielsen F., Denissov S., Nijman I. J., Koster J., Santo E. E., Welboren W., Versteeg R., Cuppen E., van de Wetering M., Clevers H., Stunnenberg H. G. (2008) Genome-wide pattern of TCF7L2/TCF-4 chromatin occupancy in colorectal cancer cells. Mol. Cell. Biol. 28, 2732–2744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yochum G. S., Sherrick C. M., Macpartlin M., Goodman R. H. (2010) A β-catenin/TCF-coordinated chromatin loop at MYC integrates 5′ and 3′ Wnt responsive enhancers. Proc. Natl. Acad. Sci. U.S.A. 107, 145–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Choi Y. S., Hur J., Jeong S. (2007) β-Catenin binds to the downstream region and regulates the expression C-reactive protein gene. Nucleic Acids Res. 35, 5511–5519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mosimann C., Hausmann G., Basler K. (2009) β-Catenin hits chromatin. Regulation of Wnt target gene activation. Nat. Rev. Mol. Cell Biol. 10, 276–286 [DOI] [PubMed] [Google Scholar]
- 38. Valenta T., Hausmann G., Basler K. (2012) The many faces and functions of β-catenin. EMBO J. 31, 2714–2736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dekker J. (2006) The three C's of chromosome conformation capture. Controls, controls, controls. Nat. Methods 3, 17–21 [DOI] [PubMed] [Google Scholar]
- 40. Hwang I., Choi Y. S., Jeon M. Y., Jeong S. (2010) NF-κB p65 represses β-catenin-activated transcription of cyclin D1. Biochem. Biophys. Res. Commun. 403, 79–84 [DOI] [PubMed] [Google Scholar]
- 41. Aarenstrup L., Flindt E. N., Otkjaer K., Kirkegaard M., Andersen J. S., Kristiansen K. (2008) HDAC activity is required for p65/RelA-dependent repression of PPAR δ-mediated transactivation in human keratinocytes. J. Invest. Dermatol. 128, 1095–1106 [DOI] [PubMed] [Google Scholar]
- 42. Peeters L. L., Vigne J. L., Tee M. K., Zhao D., Waite L. L., Taylor R. N. (2005) PPARγ represses VEGFA expression in human endometrial cells: implication for uterine angiogenesis. Angiogenesis 8, 373–379 [DOI] [PubMed] [Google Scholar]
- 43. Chandra V., Huang P., Hamuro Y., Raghuram S., Wang Y., Burris T. P., Rastinejad F. (2008) Structure of the intact PPAR-γ-RXR-nuclear receptor complex on DNA. Nature 456, 350–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Allred C. D., Talbert D. R., Southard R. C., Wang X., Kilgore M. W. (2008) PPARγ as a molecular target of eicosapentaenoic acid in human colon cancer (HT-29) cells. J. Nutr. 138, 250–256 [DOI] [PubMed] [Google Scholar]
- 45. Ignatenko N. A., Babbar N., Mehta D., Casero R. A., Jr., Gerner E. W. (2004) Suppression of polyamine catabolism by activated Ki-ras in human colon cancer cells. Mol. Carcinog 39, 91–102 [DOI] [PubMed] [Google Scholar]
- 46. Babbar N., Ignatenko N. A., Casero R. A., Jr., Gerner E. W. (2003) Cyclooxygenase-independent induction of apoptosis by sulindac sulfone is mediated by polyamines in colon cancer. J. Biol. Chem. 278, 47762–47775 [DOI] [PubMed] [Google Scholar]
- 47. Billin A. N., Thirlwell H., Ayer D. E. (2000) β-Catenin-histone deacetylase interactions regulate the transition of LEF1 from a transcriptional repressor to an activator. Mol. Cell. Biol. 20, 6882–6890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Margariti A., Zampetaki A., Xiao Q., Zhou B., Karamariti E., Martin D., Yin X., Mayr M., Li H., Zhang Z., De Falco E., Hu Y., Cockerill G., Xu Q., Zeng L. (2010) Histone Deacetylase 7 controls endothelial cell growth through modulation of β-catenin. Circ. Res. 106, 1202–1211 [DOI] [PubMed] [Google Scholar]
- 49. Hsu P. Y., Hsu H. K., Singer G. A., Yan P. S., Rodriguez B. A., Liu J. C., Weng Y. I., Deatherage D. E., Chen Z., Pereira J. S., Lopez R., Russo J., Wang Q., Lamartiniere C. A., Nephew K. P., Huang T. H. (2010) Estrogen-mediated epigenetic repression of large chromatin regions through DNA looping. Genome Res. 20, 733–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Olson L. E., Tollkuhn J., Scafoglio C., Krones A., Zhang J., Ohgi K. A., Wu W., Taketo M. M., Kemler R., Grosschedl R., Rose D., Li X., Rosenfeld M. G. (2006) Homeodomain-mediated β-catenin-dependent switching events dictate cell-lineage determination. Cell 125, 593–605 [DOI] [PubMed] [Google Scholar]
- 51. Kim Y. J., Cecchini K. R., Kim T. H. (2011) Conserved, developmentally regulated mechanism couples chromatin looping and heterochromatin barrier activity at the homeobox gene A locus. Proc. Natl. Acad. Sci. U.S.A. 108, 7391–7396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Oishi Y., Manabe I., Tobe K., Ohsugi M., Kubota T., Fujiu K., Maemura K., Kubota N., Kadowaki T., Nagai R. (2008) SUMOylation of Krüppel-like transcription factor 5 acts as a molecular switch in transcriptional programs of lipid metabolism involving PPAR-δ. Nat. Med. 14, 656–666 [DOI] [PubMed] [Google Scholar]
- 53. Gupta R. A., Tan J., Krause W. F., Geraci M. W., Willson T. M., Dey S. K., DuBois R. N. (2000) Prostacyclin-mediated activation of peroxisome proliferator activated receptor δ in colorectal cancer. Proc. Natl. Acad. Sci. U.S.A. 97, 13275–13280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Takayama O., Yamamoto H., Damdinsuren B., Sugita Y., Ngan C. Y., Xu X., Tsujino T., Takemasa I., Ikeda M., Sekimoto M., Matsuura N., Monden M. (2006) Expression of PPAR-δ in multistage carcinogenesis of the colorectum. Implications of malignant cancer morphology. Br. J. Cancer 95, 889–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Moraes L. A., Piqueras L., Bishop-Bailey D. (2006) Peroxisome proliferator-activated receptors and inflammation. Pharmacol. Ther. 110, 371–385 [DOI] [PubMed] [Google Scholar]
- 56. Notterman D. A., Alon U., Sierk A. J., Levine A. J. (2001) Transcriptional gene expression profiles of colorectal adenoma, adenocarcinoma, and normal tissue examined by oligonucleotide arrays. Cancer Res. 61, 3124–3130 [PubMed] [Google Scholar]
- 57. Hao C. Y., Moore D. H., Wong P., Bennington J. L., Lee N. M., Chen L. C. (2005) Alteration of gene expression in macroscopically normal colonic mucosa from individuals with a family history of sporadic colon cancer. Clin. Cancer Res. 11, 1400–1407 [DOI] [PubMed] [Google Scholar]
- 58. Essers M. A., de Vries-Smits L. M., Barker N., Polderman P. E., Burgering B. M., Korswagen H. C. (2005) Functional interaction between β-catenin and FOXO in oxidative stress signaling. Science 308, 1181–1184 [DOI] [PubMed] [Google Scholar]
- 59. Kioussi C., Briata P., Baek S. H., Rose D. W., Hamblet N. S., Herman T., Ohgi K. A., Lin C., Gleiberman A., Wang J., Brault V., Ruiz-Lozano P., Nguyen H. D., Kemler R., Glass C. K., Wynshaw-Boris A., Rosenfeld M. G. (2002) Identification of a Wnt/Dvl/β-catenin → Pitx2 pathway mediating cell type-specific proliferation during development. Cell 111, 673–685 [DOI] [PubMed] [Google Scholar]
- 60. Kwak H., Hwang I., Kim J. H., Kim M. Y., Yang J. S., Jeong S. (2009) Modulation of transcription by the peroxisome proliferator-activated receptor-δ binding RNA aptamer in colon cancer cells. Mol. Cancer Ther. 8, 2664–2673 [DOI] [PubMed] [Google Scholar]
- 61. Wu C. W., Yu J., Sung J. J. (2009) Peroxisome proliferator-activated receptor δ and gastric cancer. Oncol. Rep. 22, 451–457 [PubMed] [Google Scholar]
- 62. Handeli S., Simon J. A. (2008) A small molecule inhibitor of Tcf/β-catenin signaling down-regulates PPARγ and PPAR-δ activities. Mol. Cancer Ther. 7, 521–529 [DOI] [PubMed] [Google Scholar]
- 63. He T. C., Chan T. A., Vogelstein B., Kinzler K. W. (1999) PPAR-δ is an APC-regulated target of nonsteroidal anti-inflammatory drugs. Cell 99, 335–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. de Laat W., Grosveld F. (2003) Spatial organization of gene expression. The active chromatin hub. Chromosome Res. 11, 447–459 [DOI] [PubMed] [Google Scholar]
- 65. Hampsey M., Singh B. N., Ansari A., Lainé J. P., Krishnamurthy S. (2011) Control of eukaryotic gene expression. Gene loops and transcriptional memory. Adv. Enzyme Regul. 51, 118–125 [DOI] [PMC free article] [PubMed] [Google Scholar]