

Background: RpPat acetylates many acyl-CoA synthetase enzymes.
Results: RpPat does not acetylate RpMatB but can acetylate chimeric versions of it that differ from the wild-type enzyme in a loop region >20 Å away from the acetylated lysine.
Conclusion: RpPat likely interacts with a large surface area of its substrates.
Significance: RpPat substrates cannot be predicted by a short acetylation motif alone.
Keywords: Acetyl-Coenzyme A, Enzyme Inactivation, Enzyme Structure, Post-translational Modification, Protein Acylation, Protein-Protein Interactions, Methylmalonyl-CoA Synthetase, Substrate Recognition
Abstract
Lysine acetylation is a post-translational modification that is important for the regulation of metabolism in both prokaryotes and eukaryotes. In bacteria, the best studied protein acetyltransferase is Pat. In the purple photosynthetic bacterium Rhodopseudomonas palustris, at least 10 AMP-forming acyl-CoA synthetase enzymes are acetylated by the Pat homologue RpPat. All bona fide RpPat substrates contain the conserved motif PX4GK. Here, we show that the presence of such a motif is necessary but not sufficient for recognition by RpPat. RpPat failed to acetylate the methylmalonyl-CoA synthetase of this bacterium (hereafter RpMatB) in vivo and in vitro, despite the homology of RpMatB to known RpPat substrates. We used RpMatB to identify structural determinants that are recognized by RpPat. To do this, we constructed a series of RpMatB chimeras that became substrates of RpPat. In such chimeras, a short region (11–25 residues) of RpMatB located >20 residues N-terminal to the acetylation site was replaced with the corresponding sequences from other AMP-forming acyl-CoA synthetases that were known RpPat substrates. Strikingly, the enzymatic activity of RpMatB chimeras was regulated by acetylation both in vitro and in vivo. Crystal structures of two of these chimeras showed that the major difference between them and wild-type RpMatB was within a loop region ∼23 Å from the acetylation site. On the basis of these results, we suggest that RpPat likely interacts with a relatively large surface of its substrates, in addition to the PX4GK motif, and that RpPat probably has relatively narrow substrate specificity.
Introduction
Lysine acetylation is a post-translational modification that occurs in all three domains of life (1, 2). In eukaryotes, histone tails are subject to many types of modifications, including lysine acetylation, and such modifications play an important role in gene expression and chromatin stability (3, 4). Because histone acetylation has been extensively studied, the modifying enzymes are often referred to as histone acetyltransferases, despite the fact that they can acetylate non-histone proteins as well. There are three groups of histone acetyltransferases as follows: the Gcn5-related N-acetyltransferases, the MYST family, and the p300 family (5, 6). Only Gcn5-related N-acetyltransferases are found in bacteria, and different family members acetylate a wide range of substrates containing primary amines, including small molecules, protein N-terminal amino groups, and lysine side chains (7).
The bacterial protein acetyltransferase Pat4 was first discovered as a regulator of the AMP-forming acetyl-CoA synthetase (Acs) enzyme in Salmonella enterica (8). In Escherichia coli, the homologue of SePat is known as Pka or YfiQ (9). Acs (EC 6.2.1.1; pfam 00501) activates acetate to acetyl-CoA (10). In S. enterica, Acs activity is abolished when residue Lys-609 is acetylated. Acs acetylation is reversed by the action of a sirtuin-type NAD+-dependent protein deacetylase called CobB (11, 12). In other bacteria, Pat homologues have been shown to acetylate different acyl-CoA synthetases and to propionylate the enzyme propionyl-CoA synthetase (13–15). A feature shared by all these protein substrates is the acetylation site, which is located within a conserved PX4GK motif located near the C terminus of these proteins. At present, it is not known which components of this motif are important for recognition or whether this motif is sufficient for acetylation by Pat.
There are reports of protein substrates of Pat that are not acyl-CoA synthetases and do not contain the PX4GK motif. For example, the E. coli Pat homologue Pka acetylates RNase R, and acetylation affects the stability of the protein rather than its enzymatic activity (9, 16). S. enterica Pat has been reported to acetylate a wide range of metabolic enzymes in addition to Acs, including glyceraldehyde-3-phosphate dehydrogenase (GapA), isocitrate dehydrogenase phosphatase/kinase (AceK), and isocitrate lyase (AceA) (17). We note that our group was unable to repeat these findings (14). Thus, whether Pat has broad or narrow substrate specificity remains an open question. Because it has been suggested that Pat may be a master regulator of bacterial metabolism (17), it is important to define the substrate range of Pat.
In this work, we investigated the substrate specificity of RpPat, the Pat homologue in the purple photosynthetic α-proteobacterium Rhodopseudomonas palustris. In this bacterium, RpPat acetylates 10 different acyl-CoA synthetases involved in the activation of short, medium, and long chain fatty acids and aromatic acids (13, 14). To date, all known substrates of RpPat are acyl-CoA synthetases, and all contain the PX4GK motif at the site of acetylation. R. palustris has two protein deacetylases, RpSrtN and RpLdaA, both of which can deacetylate and thus reactivate acyl-CoA synthetases (13, 14). We examined two acyl-CoA synthetases involved in the catabolism of dicarboxylic acids in R. palustris, namely pimeloyl-CoA synthetase (RpPimA) and methylmalonyl-CoA synthetase (RpMatB). For reasons explained below, we also studied benzoate:CoA ligase (BclM) from Burkholderia xenovorans LB400 (BxBclM).
RpPimA activates medium chain mono- and dicarboxylic acids (18), and RpMatB activates malonate (a C3 dicarboxylate) and methyl malonate (a related C4 dicarboxylate) (19). Here, we show that wild-type RpMatB is not a substrate of RpPat, despite the fact that RpMatB contains the catalytic lysine residue located within the PX4GK motif.
Using a combination of in vivo, in vitro, and structural approaches, we determined that in RpMatB the sequence of a structural loop in the C-terminal domain recognized by RpPat in its bona fide substrates is different. As a result of relatively minor changes in this sequence, RpPat does not modify RpMatB. In substrates of RpPat, the alluded region is surprisingly far away from the acetylation site, strongly suggesting that it may be difficult to identify RpPat substrates exclusively on the presence of the PX4GK motif. It is noteworthy that RpPat (and probably its homologues in other organisms) does not acetylate all AMP-forming acyl-CoA synthetases. These results and others reported elsewhere (14) continue to question the idea proposed by others that S. enterica Pat is not a very specific enzyme (17).
EXPERIMENTAL PROCEDURES
Bacterial Strains and Growth Conditions
All strains and plasmids used in this study are listed in supplemental Tables S1 and S2. E. coli strains were grown at 37 °C in lysogenic broth (LB, Difco) (18, 19). B. xenovorans LB400 was obtained from the Agricultural Research Service culture collection, United States Department of Agriculture, and was cultured at 30 °C on LB devoid of NaCl. All R. palustris strains were derivatives of R. palustris CGA009 (20) and were cultured at 30 °C in photosynthetic medium (PM) (21) supplemented with succinate (10 mm), benzoate (3 mm), and NaHCO3 (10 mm) or methyl malonate (10 mm) and vitamin B12 (150 nm). When used, ampicillin was at 100 μg ml−1; chloramphenicol was at 20 μg ml−1, and kanamycin was at 50 μg ml−1 (E. coli) or 75 μg ml−1 (R. palustris). Radiolabeled [1-14C]acetyl-CoA (54 mCi/mmol) was purchased from Moravek, and all other chemicals were from Sigma. For growth curves, R. palustris starter cultures were grown to full density photosynthetically on PM containing succinate and kanamycin in Balch tubes (22) flushed with N2. Cultures were diluted 1:16 into fresh medium supplemented with the appropriate carbon source and kanamycin. Cultures were incubated at 30 °C in light without shaking, and growth was monitored at 660 nm using a Spectronic Spec20D (Thermo). Three biological replicates of each strain were monitored, and each experiment was repeated twice.
Molecular Techniques
DNA manipulations were performed using standard techniques (23). Restriction endonucleases were purchased from Fermentas. DNA was amplified using PfuUltra II Fusion DNA polymerase (Stratagene), and site-directed mutagenesis was performed using the QuikChange kit (Stratagene). Plasmid DNA was purified using the Wizard Plus SV miniprep kit (Promega), and PCR products were purified using the Wizard SV gel and PCR clean-up kit (Promega). DNA sequencing reactions were resolved and analyzed at the University of Wisconsin-Madison Biotechnology Center. Oligonucleotide primer sequences are listed in supplemental Table S3. Plasmids were moved into R. palustris by electroporation.
Plasmid Construction
R. palustris pat, pimA, and matB were amplified from R. palustris genomic DNA using the primers listed in supplemental Tables S2 and S3. The PCR products were cut with the specified restriction enzymes and ligated into the overexpression vector pTEV5 cut with the same enzymes (24). The resulting plasmids directed the synthesis of proteins with an N-terminal hexahistidine (His6) tag, which was removed using recombinant tobacco etch virus (rTEV) protease (25). B. xenovorans bclM (locus tag Bxe_A1419) was amplified from a single colony and cloned into pTEV5 using PIPE cloning (26). The chimeric genes were generated using overlap-extension PCR (27) or site-directed mutagenesis and were subsequently cloned into pTEV5. A detailed description of chimera construction can be found in the supplemental material.
Protein Overexpression
The plasmid encoding RpPat (pRpPAT13) was transformed into strain BL21/pLysS. The resulting strain was grown until early stationary phase and subcultured 1:100 in 3 liters of super broth supplemented with ampicillin (100 μg ml−1) and chloramphenicol (10 μg ml−1). Cultures were grown with shaking to an A600 ∼0.5, and protein expression was induced with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside. Cultures were grown overnight at 30 °C; cells were harvested by centrifugation at 8000 × g for 12 min in a Beckman Coulter Avanti J-20 XOI refrigerated centrifuge with a JLA-8.1000 rotor, and cell pellets were frozen at −80 °C for later use. All other overexpression plasmids were transformed into strain JE9314, a pka-deficient derivative of E. coli C41(λDE3) (28), to ensure that proteins were purified in their nonacetylated state. The resulting strains were grown until early stationary phase and subcultured 1:100 in 2 liters of lysogenic broth supplemented with ampicillin (100 μg ml−1). Protein expression was induced with isopropyl 1-thio-β-d-galactopyranoside as described above, and cell pellets were frozen at −80 °C for later use.
Protein Purification
Protein concentrations were determined using a NanoDrop 1000 spectrophotometer (ThermoFisher) and A280 molar extinction coefficients for each protein. Extinction coefficients were obtained from the Integrated Microbial Genomes website (29) or calculated using the ProtParam tool (30).
rTEV Protease
rTEV protease was purified as described previously (25).
R. palustris Pat
RpPat was overproduced as an rTEV-cleavable His6 N-terminal fusion protein. The cell pellet was resuspended in 30 ml of RpPat binding buffer (sodium phosphate buffer (50 mm, pH 7.5), NaCl (500 mm), imidazole (20 mm), tris(2-carboxyethyl)phosphine (TCEP, 0.5 mm)) and supplemented with lysozyme and deoxyribonuclease I (1 mg ml−1 each). Cells were lysed using a French pressure cell (two passes, 10 MPa). Debris was removed by centrifugation at 39,000 × g for 30 min at 4 °C, and the soluble fraction was passed through a 0.45-μm filter. His6-RpPat was purified by nickel-affinity purification using an FPLC (ÄKTA) equipped with a 5-ml HisTrap FF column. After binding to the column, the column was washed with 6% RpPat elution buffer (sodium phosphate buffer (50 mm, pH 7.5), NaCl (500 mm), imidazole (500 mm), TCEP (0.5 mm)) and eluted in a linear gradient up to 100% RpPat elution buffer. Fractions containing RpPat were pooled and incubated with rTEV (1:10 rTEV to protein ratio) for 3 h at room temperature to cleave the tag. RpPat was dialyzed three times against RpPat binding buffer lacking imidazole; the first dialysis buffer contained EDTA (1 mm). The His6 tag and His7-tagged rTEV protease were removed by passage over the HisTrap column again, using the same buffers as the first purification except that the binding buffer did not contain imidazole. Untagged RpPat eluted in the wash step containing 6% RpPat elution buffer. Fractions were pooled and dialyzed three times against RpPat storage buffer containing HEPES (50 mm, pH 7.5), glycerol (20%, v/v), and NaCl (500, 300, and 150 mm, successively), before drop freezing in liquid nitrogen and storage at −80 °C.
Other Proteins
All other proteins were purified in a similar manner, with the following modifications. Cell pellets were resuspended in MatB binding buffer (sodium phosphate buffer (50 mm, pH 8.0), NaCl (300 mm), imidazole (20 mm)) and lysed with a Sonic Dismembrator (Fisher) at power level 9, with 2-s pulses separated by 5-s breaks, for 1.5 min. His6-tagged proteins were purified from the soluble fraction by nickel-affinity purification using a 1.5-ml bed volume of nickel-nitrilotriacetic acid Superflow resin (Qiagen). After binding to the column, the column was washed with RpMatB washing buffer (sodium phosphate buffer (50 mm, pH 8.0), NaCl (300 mm), imidazole (30 mm)), and proteins were eluted with RpMatB elution buffer (sodium phosphate buffer (50 mm, pH 8.0), NaCl (300 mm), imidazole (250 mm)). rTEV was added (1:50 to 1:100 rTEV to protein ratio), and after a 3-h cleavage reaction at room temperature proteins were dialyzed against RpMatB dialysis buffer 1 (sodium phosphate buffer (50 mm, pH 8.0), NaCl (300 mm), EDTA (0.5 mm)), then RpMatB dialysis buffer 2 (sodium phosphate buffer (50 mm, pH 8.0), NaCl (300 mm)), and finally RpMatB binding buffer. The second purification step employed the same buffers as the first, and untagged proteins were eluted in the flow-through. Fractions were pooled and dialyzed against RpMatB storage buffer 1 (Tris (50 mm, pH 7.5), NaCl (100 mm), EDTA (0.5 mm), glycerol (20%, v/v)) and then RpMatB storage buffer 2 (Tris (50 mm, pH 7.5), NaCl (100 mm), glycerol (20%, v/v)) before drop-freezing in liquid N2.
Chimeras
The purification protocols for chimeras RpMatB-BxBclM B1K491A and B3K488A for crystallography were similar to the ones described above, except that the purified, untagged protein was dialyzed into HEPES buffer (50 mm, pH 7.5) before concentration to 10–12 mg ml−1 using Ultracel 10,000 molecular weight cutoff centrifugal filters (Amicon Ultra). Accessible lysine side chains were reductively methylated using formaldehyde and dimethylamine borane complex as described previously (31). The reaction was quenched by addition of Tris-HCl buffer (pH 7.5) to a final concentration of 500 mm, and the protein was dialyzed overnight against 2 liters of Tris-HCl buffer (50 mm, pH 7.5) containing dithiothreitol (DTT, 2 mm), before dialysis into Tris-HCl buffer (10 mm, pH 7.5). The protein concentration was adjusted to 10 mg ml−1 before drop-freezing in liquid nitrogen.
In Vitro Protein Acetylation Assay
Proteins were acetylated using radiolabeled acetyl-CoA as described previously (8). Reactions contained HEPES buffer (50 mm, pH 7.0), TCEP (1 mm), [1-14C]Ac-CoA (19 μm), protein substrate (3 μm), and RpPat (0.06 μm). Reactions (25 μl total volume) were incubated for 60 min at 30 °C. Aliquots (10 μl each) were resolved using SDS-PAGE and visualized by staining with Coomassie Blue R-250. Gels were dried and exposed overnight to a MultiPurpose Phosphor Screen (Packard Instrument Co.). Radioactivity associated with protein was quantified using a Typhoon FLA9000 Biomolecular Imager (GE Healthcare) and OptiQuant version 04.00 software (Packard Instrument Co.).
In Vitro Acyl-CoA Synthetase Activity Assay
Acyl-CoA synthetases (1.5 μm each) were individually incubated with RpPat (0.5 μm) plus or minus 50 μm acetyl-CoA for 1 h at 30 °C using the same buffer system described above for 14C acetylation assays. Acyl-CoA synthetase activity was quantified using an NADH consumption assay (32). Reactions contained HEPES (50 mm, pH 7.5), TCEP (1 mm), ATP (2.5 mm), CoASH (0.4 mm), MgCl2 (5 mm), phosphoenolpyruvate (3 mm), NADH (0.1 mm), pyruvate kinase (1 unit), myokinase (5 units), lactate dehydrogenase (1.5 units), and an organic acid substrate (0.2 mm). Reactions were started by the addition of acyl-CoA synthetase (30 nm), and changes in the absorbance at 340 nm were monitored for 8 min in a 96-well plate format using a Spectramax Plus UV-visible spectrophotometer (Molecular Devices).
Crystallization of RpMatB
Methylated RpMatB chimera B1K491A crystals were grown by mixing 2 μl of 10 mg/ml protein in Tris-HCl buffer ((10 mm, pH 7.5) containing TCEP (1 mm) and MgATP (10 mm)) with 2 μl of reservoir solution ((HEPES buffer (100 mm, pH 7.5), 200 mm potassium glutamate, polyethylene glycol 8000 (26% (w/v), propylene glycol (5%, w/v)) and hanging the resulting mixture over 500 μl of reservoir solution in Linbro plates at 25 °C. After 24 h, crystals were nucleated by streak seeding with previously grown crystals and allowed to grow for an additional week. For freezing, crystals were transferred to 100 μl of reservoir solution with the addition of MgATP (10 mm) and then transferred stepwise to a solution of polyethylene glycol 8000 (25%, w/v), propylene glycol (15%, w/v), ethylene glycol, HEPES buffer (100 mm, pH 7.5), potassium glutamate (200 mm), and MgATP (10 mm). Crystals were flash-frozen in liquid nitrogen.
Methylated RpMatB chimera B3K488A crystals were grown by mixing 2 μl of 10 mg/ml protein in Tris-HCl buffer (10 mm, pH 7.5) containing TCEP (1 mm), and MgATP (10 mm) with 2 μl of reservoir solution (triethanolamine (100 mm, pH 8.0), with magnesium sulfate (50 mm), monomethyl polyethylene glycol 5000 (21%, w/v), and glycerol (4%, w/v)) and hanging the resulting mixture over 500 μl of reservoir solution in Linbro plates at 25 °C. After 24 h, crystals were nucleated by streak seeding with previously grown crystals and allowed to grow for an additional week. For freezing, crystals were transferred to 100 μl of reservoir solution with the addition of MgATP (10 mm) and then transferred stepwise to a solution of monomethyl polyethylene glycol 5000 (25%, w/v), glycerol (18%, w/v), triethanolamine (100 mm, pH 8.0), magnesium sulfate (50 mm), and MgATP (10 mm). Crystals were flash-frozen in liquid nitrogen.
X-Ray Data Collection and Structural Refinement
X-ray diffraction data for all crystals were collected at the SBC 19-ID beam line (Advanced Photon Source, Argonne, IL). The data sets were integrated and scaled with the program HKL2000 (33). X-ray data collection statistics are given in Table 1.
TABLE 1.
Crystal structure data collection and refinement statistics
| Data collection | Chimera B1 | Chimera B3 |
|---|---|---|
| Space group | P61 | P61 |
| Cell dimensions | ||
| a, b, c | 299.3, 299.3, 47.9 Å | 173.2, 173.2, 47.9 Å |
| α, β, γ | 90, 90, 120° | 90, 90, 120° |
| Wavelength | 0.98 | 0.98 |
| Resolutiona | 25 to 2.0 Å (2.03 to 2.0 Å) | 25 to 2.0 Å (2.03 to 2.0 Å) |
| Rmergea | 6.3 (38.5) | 5.8 (30.1) |
| I/σIa | 11.9 (3.8) | 15.8 (6.1) |
| Completenessa | 99.8% (99.7%) | 99.9% (100%) |
| Redundancya | 5.1 (4.3) | 6.2 (6.1) |
| Refinement | ||
| Resolutiona | 25 to 2.0 Å (2.03 to 2.0 Å) | 25 to 2.0 Å (2.03 to 2.0 Å) |
| No. reflectionsb | 158,213 (8364) | 52,933 (2833) |
| Rwork / Rfreec | 0.219/0.254 | 0.148/0.178 |
| No. of atoms | ||
| Protein | 11,832 | 3958 |
| Water | 1586 | 535 |
| Ligand | 139 | 63 |
| Wilson B-factor | 23.0 | 25.3 |
| Average B-factor | ||
| Protein | 14.6 Å2 | 21.2 Å2 |
| Water | 40.8 Å2 | 41.4 Å2 |
| Ligand | 26.9 Å2 | 43.5 Å2 |
| Ramachandran | ||
| Most favored | 99.02% | 98.95% |
| Allowed | 1.95% | 1.87% |
| Disallowed | 0.14% | 0.0% |
| r.m.s.d. | ||
| Bond lengths | 0.008 Å | 0.005 Å |
| Bond angles | 1.325° | 1.169° |
| PDB accession no. | 4GXQ | 4GXR |
a Data in parentheses represent highest resolution shell.
b Data in parentheses represent the number of reflections used during refinement.
c Rfactor = Σ|Fobs − Fcalc|/Σ|Fobs|, where Rwork refers to the Rfactor for the data utilized in the refinement and Rfree refers to the Rfactor for 5% of the data that were excluded from the refinement.
Structural Determination
The structures of both methylated RpMatB chimeras were solved by molecular replacement using the program Phaser (34, 35) and the methylated RpMatBK488A structure (PDB accession number 4FUT) (36) as the search model. This was followed by iterative cycles of manual model building in Coot (37) and TLS and restrained refinement in Refmac 5.6 and Phenix.refine: 1.6_289 (38, 39). For both structures, TLS groups were defined such that each polypeptide chain within the asymmetric unit was divided into the N-terminal (residues 1–400) and C-terminal domains (residues 401–503 or 506). These two domains were naturally flexible during the catalytic cycle, and introduction of these TLS groups led to a 2% decrease in Rfree for both structures (21.88/25.44% R/Rfree with TLS compared with 23.10/27.61% R/Rfree for 4GXQ and 14.83/17.79% R/Rfree with TLS compared with 16.06/19.48% R/Rfree for 4GXR). Data processing and refinement statistics are presented in Table 1. All structural alignments were done with the program Superpose (40).
Structures have been deposited in the RCSB Protein Data Bank under accession codes 4GXQ and 4GXR for ATP-RpMatB chimera B1K491A and ATP-RpMatB chimera B3K488A, respectively.
RESULTS
In R. palustris, MatB Is Not Regulated by Acetylation
We tested whether RpPat could acetylate the acyl-CoA synthetases RpMatB, RpPimA, and BxBclM. RpMatB is 39% identical to RpPimA over 376 residues and 28% identical to BxBclM over 485 residues, suggesting that all three enzymes share many structural features, although they do exhibit different substrate specificities. RpPimA, RpMatB, and BxBclM contain the conserved lysine residue and surrounding motif PX4GK found in other AMP-forming acyl-CoA synthetases that are acetylated, such as the R. palustris benzoate:CoA ligase (RpBadA, Fig. 1A). We purified each enzyme, and each was tested as a substrate for RpPat using 14C-labeled acetyl-CoA as co-substrate (Fig. 1B). RpPat efficiently acetylated RpPimA and RpBclM, and when the conserved lysine residues (Lys-534 in RpPimA or Lys-520 in BxBclM) were changed to alanine, acetylation was no longer detectable, consistent with the expectation that this lysine was modified in both proteins. Surprisingly, RpPat did not acetylate RpMatB, despite its similarity to other acyl-CoA synthetases that are substrates of RpPat.
FIGURE 1.
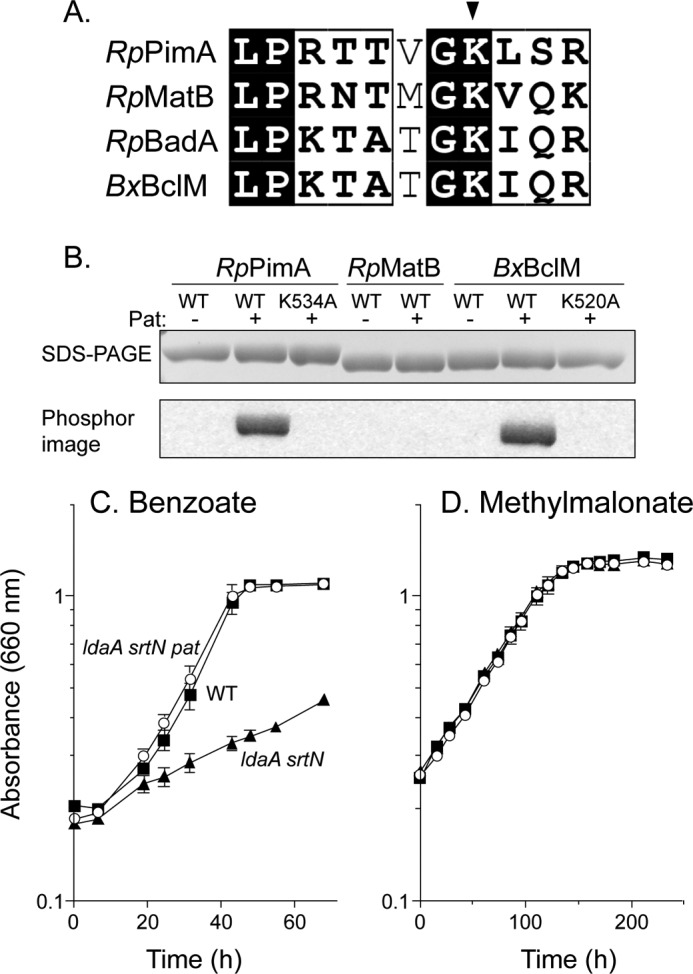
RpPat acetylates RpPimA and BxBclM but not RpMatB. A, alignment of the region around the lysine residue that is acetylated in RpBadA. Arrow indicates Lys-512 in RpBadA, corresponding to Lys-534 in RpPimA, Lys-488 in RpMatB, and Lys-520 in BxBclM. B, RpPimA, RpMatB, and BxBclM were incubated with [1-14C]acetyl-CoA and with or without RpPat. Changing Lys-534 of RpPimA or Lys-520 of BxBclM to alanine abolished acetylation. Top panel shows the acyl-CoA synthetases separated by SDS-PAGE, and bottom panel is the phosphorimage of the same gel. C, photosynthetic growth of R. palustris on benzoate is regulated by acetylation. A deacetylase-deficient strain (ΔldaA ΔsrtN, triangles) grew poorly because RpBadA was acetylated and rendered inactive, and deletion of pat (ΔldaA ΔsrtN Δpat, circles) restored growth. D, photosynthetic growth of R. palustris on methyl malonate was not regulated by acetylation. Wild-type (squares), deacetylase-deficient (ΔldaA ΔsrtN, triangles), and deacetylase- and pat-deficient (ΔldaA ΔsrtN Δpat, circles) strains all grew equally well.
Growth of R. palustris on Methyl Malonate Is Not under Acetylation Control
We recently showed that matB is required for photoheterotrophic growth of R. palustris on methyl malonate as a carbon source (36). If RpMatB activity were regulated by acetylation in vivo, we would expect that a deacetylase-deficient strain (ΔsrtN ΔldaA) would have a growth defect, as is observed during growth on benzoate (Fig. 1C). This growth defect on benzoate occurs because RpBadA (benzoate:CoA ligase) is acetylated and inactivated by RpPat, and the deacetylases RpSrtN and RpLdaA are not present to reactivate RpBadA. As shown in Fig. 1D, an R. palustris ΔsrtN ΔldaA strain (JE11616, supplemental Table S1) grew as well as the wild-type strain on methyl malonate, a result that was consistent with our in vitro data, which suggested that RpMatB was not a substrate of RpPat.
We further confirmed that RpPat did not acetylate RpMatB. To do this, we quantified RpMatB activity after incubation with RpPat and acetyl-CoA (Table 2). Results of positive control experiments, where RpPimA or BxBclM substituted for RpMatB in the reaction mixture, showed a ≥99% loss of acyl-CoA synthetase activity upon acetylation. In contrast, RpMatB activity was unaffected by incubation with RpPat and acetyl-CoA.
TABLE 2.
Acyl-CoA synthetase enzyme activity
| Enzyme | Substrate | Unacetylated activitya | Acetylated activity | Decrease |
|---|---|---|---|---|
| % | ||||
| PimA | Octanoate | 6.9 ± 0.2 | 0.04 ± 0.02 | 99 |
| MatB | Methyl malonate | 8.6 ± 0.8 | 8.8 ± 1.01 | 0 |
| BclM | Benzoate | 6.2 ± 0.7 | 0.04 ± 0.03 | 99 |
| P9 chimera | Methyl malonate | 3.7 ± 0.2 | 0.05 ± 0.03 | 98 |
| B1 chimera | Methyl malonate | 6.1 ± 0.1 | 1.8 ± 0.32 | 71 |
| B2 chimera | Methyl malonate | 6.0 ± 0.2 | 2.1 ± 0.56 | 65 |
| B3 chimera | Methyl malonate | 6.8 ± 0.2 | 4.1 ± 0.39 | 40 |
a Activity was reported as micromoles of AMP min−1 mg−1.
Generation of Chimeric Variants of RpMatB That Can Be Acetylated by RpPat
All AMP-forming acyl-CoA synthetases consist of a large N-terminal domain and a smaller C-terminal domain connected by a flexible hinge region (41, 42). These enzymes activate fatty acids in two steps, an adenylation half-reaction and a thioester-forming half-reaction; to accomplish this, the C-terminal domain rotates ∼140° between two catalytic conformations (43, 44). The catalytic lysine residue that is acetylated in members of this family of enzymes is located within the C-terminal domain (Fig. 2A). When the enzyme is in the adenylation conformation, the catalytic lysine residue is buried in the active site in the cleft between the N- and C-terminal domains, although this lysine residue is surface-exposed when the enzyme rotates into the thioester-forming conformation. Thus, this lysine is likely only available for modification when the enzyme adopts the thioester-forming conformation.
FIGURE 2.

Construction of RpMatB chimeric proteins that can be acetylated. A, scheme of RpMatB N- and C-terminal domains drawn to scale. Arrowhead indicates location of conserved lysine residue Lys-488. B, acetylation of six RpMatB-RpPimA chimeras by RpPat using [1-14C]acetyl-CoA. Top panel shows SDS-polyacrylamide gel, and bottom panel shows the phosphorimage of the same gel. C, schematic representation of the six RpMatB-RpPimA chimeras P1–P6, in which a section of the C-terminal domain of RpMatB was replaced with the corresponding sequence from RpPimA (gray). C-terminal domains are drawn to match scale of A. Numbers in parentheses indicate the residue of RpMatB at which the fusion begins (i.e. first residue replaced by PimA). D, acetylation by RpPat of RpMatB chimeras containing an internal fragment of RpPimA. E, schematic representation of RpMatB-RpPimA chimeras P7–P11, containing an internal fragment of RpPimA (gray). Numbers in parentheses indicate residues of RpMatB that were replaced by the corresponding sequence of RpPimA. F, acetylation by RpPat of the RpMatB-BxBclM chimera B1. Changing the conserved lysine residue (Lys-488 in wild-type RpMatB numbering) in either chimera resulted in no acetylation by RpPat. G, schematic representation of the RpMatB-RpPimA chimera P9 and RpMatB-BxBclM chimera B1 that were the best identified substrates of RpPat. H, alignment of the C-terminal ends of RpMatB and the RpMatB-RpPimA P9 and RpMatB-BxBclM B1 chimeras, starting at Gly-431. The sequences derived from RpPimA and BxBclM are shaded, and RpMatB residue numbers are indicated with arrows.
To determine why RpPat could not acetylate RpMatB, we made a series of fusion proteins in which a portion of the C-terminal domain of RpMatB was replaced with the corresponding sequence of RpPimA (Fig. 2C). RpPat acetylated RpMatB-RpPimA chimeras that contained at least the last 62 residues of RpPimA (Fig. 2B, annotated as chimeras P1, P2, and P3). These chimeras included at least 43 RpPimA-derived residues upstream of the acetylated lysine, demonstrating that acetylation depended on more than just the motif immediately surrounding the acetylation site.
To determine whether this entire region upstream of the acetylated lysine was required to turn RpMatB into an RpPat substrate, we made a second set of chimeras in which an internal piece of RpMatB was replaced with the corresponding sequence from RpPimA (Fig. 2E). Strong acetylation was observed when residues 440–465 of RpMatB were replaced with the corresponding region of RpPimA (Fig. 2D, chimera P9). Acetylation still occurred at the conserved lysine residue (Lys-489 in the P9 chimera) because substitution of an alanine at this position abolished acetylation (Fig. 2F). These results showed that the primary sequence surrounding Lys-488 in RpMatB (Fig. 2H) would not block the modification of Lys-488 by RpPat, but a region spanning 49 to 23 residues N-terminal to this lysine was critical for recognition by RpPat.
RpPat also acetylated a similar RpMatB-BxBclM chimera (Fig. 2, F and G), although the segment of RpMatB replaced by BxBclM had to be adjusted slightly to yield a stable protein. In this chimera, referred to as RpMatB-BxBclM B1, residues 444–465 of RpMatB were replaced with the corresponding sequence of BxBclM (Fig. 2H). Substitution of the conserved active site lysine residue, Lys-491 in the RpMatB-BxBclM B1 chimera, abolished acetylation (Fig. 2F), demonstrating that acetylation occurred at the expected site.
RpMatB Chimeras Are Regulated by Acetylation in Vitro and in Vivo
Both the RpMatB-RpPimA P9 and RpMatB-BxBclM B1 chimeras retained enzymatic activity with methyl malonate, although the RpMatB-RpPimA P9 chimera was only ∼40% as active as wild-type RpMatB, and the RpMatB-BxBclM B1 chimera was ∼70% as active as wild-type RpMatB (Table 2). The RpMatB-RpPimA P9 chimera was efficiently acetylated by Pat and lost ∼99% of its activity upon acetylation, whereas the RpMatB-BxBclM B1 chimera lost ∼70% of its activity upon acetylation, under our assay conditions (Table 2).
The RpMatB-RpPimA P9 chimera, which retained 40% of the wild-type RpMatB activity, did not complement a ΔmatB strain growing photoheterotrophically on methyl malonate as a carbon source (Fig. 3). However, the same chimera did complement a ΔmatB Δpat strain (Fig. 3, circles), suggesting that in the ΔmatB pat+ strain the chimera was acetylated and rendered inactive. If this were true, we predicted that a ΔmatB strain co-expressing the genes encoding the chimera and the RpLdaA deacetylase would also grow, and this was the case (Fig. 3, open diamonds). These results indicated that the RpMatB-RpPimA P9 chimera could be regulated by acetylation in vivo.
FIGURE 3.
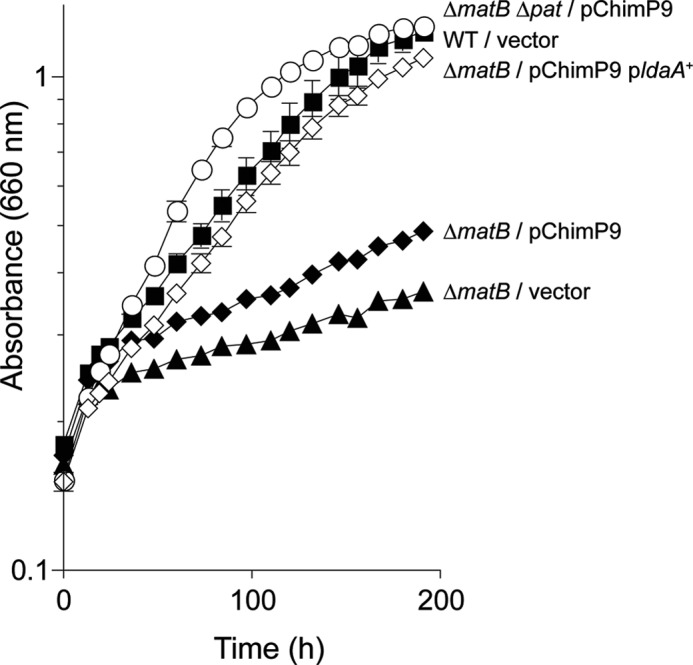
Activity of the RpMatB-RpPimA P9 chimera is controlled by acetylation in vivo. Absorbance was monitored during photosynthetic growth of R. palustris on methyl malonate (10 mm). Data points are averages of four replicates, and error bars represent standard deviations. Squares, wild-type (CGA009) harboring plasmid pBBR1MCS-2 as a vector control; filled triangles, strain JE13568 (ΔmatB/pBBR1MCS-2); filled diamonds, strain JE13912 (ΔmatB/pRpMATB25 (encodes the RpMatB-RpPimA P9 chimera)); circles, strain JE14288 (ΔmatB Δpat/pRpMATB25 (encodes the RpMatB-RpPimA P9 chimera)); empty diamonds, strain JE14957 (ΔmatB/pRpMATB45 (encodes the RpMatB-RpPimA P9 chimera and ldaA+).
Three-dimensional Crystal Structure of RpMatB-RpBclM Chimera B1
Because acyl-CoA synthetases show little sequence conservation in the region between residues 440 and 465 (RpMatB numbering, Fig. 2H), it was unclear why this region was important for recognition or acetylation by RpPat. We hypothesized that there was a structural difference between wild-type RpMatB and its chimeras, and we attempted to crystallize both the RpMatB-RpPimA P9 and RpMatB-BxBclM B1 chimeras so that we could compare them with RpMatB and BxBclM, whose structures have been previously published (36, 45). Attempts to crystallize either RpPimA or the RpMatB-RpPimA P9 chimera were not successful; therefore, only the RpMatB-BxBclM B1 chimera was pursued.
We obtained crystals of the RpMatB-BxBclM B1 chimera with ATP bound only after we chemically methylated the surface-exposed lysine residues. Methylation of lysine residues is known to affect packing within the crystal lattice without significantly changing the three-dimensional structure of the protein (46–48). This method has been used to obtain crystals of recalcitrant proteins, including RpMatB (36, 49–53). To avoid predictable problems stemming from chemical methylation of the active site lysine residue, this lysine (Lys-491) was changed to alanine.
The structure of the RpMatB-BxBclM B1 chimera was solved by molecular replacement, using RpMatBK488A with ATP bound as a search model. Three RpMatB-BxBclM B1 chimera monomers were found within the unit cell each having substantially the same structure, including that of the chimera region. Within the RpMatB-BxBclM B1 chimera structure, chain A compared with chain B and chain A compared with chain C have root mean square deviations (r.m.s.d.) of 0.46 Å over 3948 eq atoms and 0.50 Å over 3949 eq atoms, respectively. However, chain C does show some disorder in the C-terminal domain due to crystal packing. Fig. 4 shows an alignment of the C-terminal domains of the RpMatB-BxBclM B1 chimera, RpMatBK488A, and BxBclM. All three structures are very similar, having an r.m.s.d. of 0.46 Å over 501 eq α-carbons and 1.88 Å over 456 eq α-carbons when comparing RpMatB-BxBclM B1 chimera and BxBclM, respectively, with RpMatBK488A and differ significantly only within the chimeric region.
FIGURE 4.

Crystal structure of RpMatB-BxBclM chimera B1. The C-terminal domain of the RpMatB-BxBclM B1 chimera is shown aligned with the C-terminal domains of RpMatB (PDB accession number 4FUT (36)) and BxBclM (PDB accession number 2V7B (45)). The BxBclM-derived residues of the chimera are colored yellow, and the corresponding residues of RpMatB and BxBclM are shown in cyan and dark blue, respectively. The active site lysine residue of RpMatB was modeled from the RpMatB apo structure (PDB accession number 4FUQ (36)), and the PX4GK region surrounding this residue (active site loop) is shown in red. Arrows indicate the chimera loop and the extra turn in the α-helix of BxBclM and the B1 chimera compared with RpMatB. Figure was prepared with PyMOL (60).
The BxBclM-derived residues in the RpMatB-BxBclM B1 chimera encompass the end of a β-strand, a connecting loop (referred to here as the chimera loop), and an α-helix that is located on the opposite side of the domain from the acetylated lysine residue and active site loop (Fig. 4). Electron density maps for this region are well defined as shown in Fig. 5. Although this chimeric region is very similar to the BxBclM structure from which it is derived (having an r.m.s.d. of 1.56 Å over 207 eq atoms), it is distinct from the corresponding region of RpMatB. The most notable difference is that there is one extra turn in the α-helix of BxBclM and the chimera compared with RpMatB (four turns instead of three), and the chimera loop has a different spatial orientation. Based on this structural comparison, it seemed possible that RpPat made crucial contacts with either the extra turn in the α-helix and/or the chimera loop.
FIGURE 5.

Stereo view of the C-terminal domains of RpMatB-BxBclM B1 chimera and RpMatB-BxBclM B3 chimera. The chimeric regions are depicted as sticks superimposed on an electron density map calculated with coefficients of the form 2Fo − Fc contoured at 1σ. Density for the backbone residues is well defined, although density for side chains that do not make crystal contacts is weak (Val-465 and MLY 466 in the B1 chimera and Arg-449, Glu-450, and Phe-451 in the B3 chimera). Figure was prepared with PyMOL (60).
We generated a sequence alignment of RpMatB and the RpMatB-BxBclM B1 chimera based on the structural alignment shown in Fig. 4, which included a three-amino acid gap in the RpMatB sequence where the extra turn in the α-helix would be (Fig. 6A). To identify which parts of this chimera were important for acetylation by RpPat, we first focused on the last three turns in the α-helix, which were structurally very similar in RpMatB and the chimera. We constructed chimera RpMatB-BxBclM B2, in which the last 11 residues in the chimeric region were changed back to the corresponding sequence in RpMatB (Fig. 6A). Chimera RpMatB-BxBclM B2 was acetylated as well as the RpMatB-BxBclM B1 chimera (Fig. 6, B and C; Table 2), suggesting that the amino acid side chains on the last three turns of the α-helix of the chimera were not important for acetylation. Next, we eliminated the three residues (Glu-Ile-Leu) that comprised the extra turn of the α-helix to generate chimera RpMatB-BxBclM B3 (Fig. 6A). The latter was also acetylated by RpPat (Fig. 6, B and C), albeit not as efficiently as the RpMatB-BxBclM B1 chimera (40% decrease in activity upon acetylation compared with a 71% decrease in activity for the RpMatB-BxBclM B1 chimera, Table 2). These data suggested that RpPat recognized proteins that have either a three-turn or a four-turn α-helix in this position.
FIGURE 6.

Construction of a minimal RpMatB-BxBclM chimera. A, alignment of the C-terminal end of RpMatB, starting at residues 431, with the RpMatB-BxBclM chimeras (labeled B1–B7). Relevant secondary structure elements of RpMatB are shown above the alignment, and the red box indicates the active site lysine (Lys-488). Residues in the chimeras derived from BxBclM are highlighted in black. B, acetylation of each BxBclM chimera by RpPat using [1-14C]acetyl-CoA. C, amount of acetylation in B was quantified and normalized to the total acetylation of the RpMatB-BxBclM B1 chimera. Values represent averages and standard deviations of three experiments. D, alignment of the C-terminal domains of RpMatB and the RpMatB-BxBclM B1 and B3 chimeras. BxBclM-derived residues in the B1 chimera are indicated in yellow, BxBclM-derived residues in the B3 chimera are shown in orange, and the corresponding residues in wild-type RpMatB are shown in cyan. In all three structures, the catalytic loop and active site lysine are shown in red (lysine residue is modeled from the RpMatB apo structure). D was prepared with PyMOL (60).
Crystal Structure of RpMatB-RpBclM Chimera B3
We determined the crystal structure of the RpMatB-BxBclM B3 chimera to confirm that it did have a shortened α-helix. The RpMatB-BxBclM B3 chimera K488A variant was methylated prior to crystallization and crystallized with the same unit cell and space group as the previously reported ATP-RpMatBK488A (19). As expected, the RpMatB-BxBclM B3 chimera did have a shorter α-helix within the chimeric region, similar to that of wild-type RpMatB (Fig. 6D). Again electron density for this region was well defined as shown in Fig. 5. The chimera loop had a different conformation than both wild-type RpMatB and the RpMatB-BxBclM B1 chimera suggesting that it was not the backbone conformation of the loop but rather the disposition of amino acid side chains that is important for RpPat recognition.
The RpMatB-BxBclM B3 chimera has nine residues that differ from wild-type RpMatB. The first is a phenylalanine in place of valine at position 444, within the β-strand immediately preceding the chimera loop, which appears to be required for stability of the chimera. Two conserved valine residues and eight nonconserved residues follow this phenylalanine within the chimera loop that differs between RpMatB and the RpMatB-BxBclM B3 chimera (Fig. 6A). When the eight nonconserved residues were changed, two at a time, to the corresponding RpMatB sequence, the amount of acetylation decreased substantially (Fig. 6, B and C). The largest effect was seen when the first two pairs of amino acids were changed (chimeras RpMatB-BxBclM B4 and B5, changes in residues Leu-447 to Glu-450 of the chimera), which were each acetylated about 5-fold less than the RpMatB-BxBclM B1 chimera. These results showed that changes within the chimera loop strongly affected how well RpPat recognized the RpMatB-BxBclM chimeras.
Both the RpMatB-BxBclM B1 and B3 chimeras crystallized in the adenylation conformation, in which the acetylated lysine residue (Lys-488) is buried in the active site cleft between the N- and C-terminal domains. We have previously reported structures of RpMatB in the adenylation and apo conformations, and we generated a model of the thioester-forming conformation based on the structure of Streptomyces coelicolor MatB (36, 54). Fig. 7 shows a stereo view of the B3 chimera modeled in all three conformations, with the Lys-488 side chain modeled from the MatB apo structure. It is likely that RpPat interacts with acyl-CoA synthetases when they are in the thioester-forming conformation, as the active site lysine residue is most accessible in this state; however, modification may also be possible in the apo conformation.
FIGURE 7.

Stereo view of the accessibility of Lys-488 in the apo (A), adenylation (B), and thioesterification (C) conformations. The N-terminal domain is shown in white and the C-terminal domain in blue. The active site loop and Lys-488 are shown in red, and the chimera loop is shown in orange. The Lys-488 side chain is modeled from the MatB apo structure. The C-terminal domains are shown in the same orientation in all three conformations. The arrows indicate the direction and magnitude of rotation of the N-terminal domain relative to the C-terminal domain during the transition from one conformation to the next. In all cases, the C-terminal domain is from the RpMatB-BxBclM B3 chimera structure, and the N-terminal domains are either from wild-type MatB (apo and thioesterification, PDB accession code 4FUQ) or MatBK488A (adenylation, PDB accession code 4FUT). In each case the N- and C-terminal domains are aligned to RpMatB structures (apo, 4FUQ; adenylation, 4FUT) or to the S. coelicolor MatB structure (thioesterification, 3NYQ (54)). Figure was prepared with PyMOL (60).
From the model of the RpMatB-BxBclM B3 chimera in the thioester-forming conformation (Fig. 8A), it seems plausible that RpPat would interact with both Lys-488 in the active site loop and the tip of the chimera loop, because both loops protrude from the same face of the C-terminal domain. This information also suggested that RpPat recognized a large section of RpMatB as the two most extreme points of these loops (the hydroxyl of Ser-454 and methylene group of Met-486) were 35 Å apart within the crystal structure.
FIGURE 8.

RpPat recognition elements of the RpMatB-BxBclM B3 chimera. A, RpMatB-BxBclM B3 chimera modeled in the thioester-forming conformation. The N-terminal domain is shown in cyan, the C-terminal domain in white, the active site loop in red, the residues derived from BxBclM in orange, and additional residues changed in the RpMatB-BxBclM B1 chimera are shown in yellow. The acetylation site, Lys-488, was modeled from the RpMatB apo structure. B, surface electrostatic calculations for the RpMatB and RpMatB-BxBclM B3 chimera C-terminal domains were generated using PyMOL (60), with electropositive regions shown in blue and electronegative regions shown in red. Ribbon diagrams of the active site and chimera loop backbones are shown for reference, and residues within the chimera loop are labeled. The active site lysine residue (Lys-488) was mutated to an alanine in the RpMatB-BxBclM B3 chimera structure to aid crystallization. Figure was prepared with PyMOL (60).
Fig. 8B shows calculated surface electrostatic potentials for the C-terminal domains of MatB and the RpMatB-BxBclM B3 chimera. Both proteins have positively charged regions surrounding the active site lysine residue and then a negatively charged band in the upper part of the C-terminal domain. Interestingly, the RpMatB-BxBclM B3 chimera has another pronounced positively charged region consisting of Lys-448 and Arg-449 within the chimera loop, whereas the wild-type RpMatB structure is predominantly electronegative throughout the chimera loop region. These surface representations show substantial differences in both surface shape and charge in the chimera loop that may explain how RpPat can recognize the RpMatB-BxBclM B3 chimera but not wild-type RpMatB.
DISCUSSION
A Loop ∼20 Å Away from the Acetylation Site Is Critical for RpPat Recognition of Its Protein Substrate
Previous studies have shown that RpPat acetylates at least 10 different acyl-CoA synthetases in R. palustris, all of which contain a conserved PX4GK motif within the active site loop (13, 14). Notably, this motif is not sufficient for acetylation. For example, RpMatB is not a substrate of RpPat even though it shares extensive homology with other acyl-CoA synthetases and does contain the PX4GK motif preceding the catalytic lysine residue. The key difference in RpMatB appears to be the shape and electrostatic potential of a loop region located ∼20 Å away from the conserved active site lysine residue and >20 amino acids away in the protein sequence. Such a structure appears crucial for acetylation (Figs. 2 and 6). This loop region found in bona fide substrates of RpPat can replace the corresponding sequence of RpMatB to yield catalytically active RpMatB variants that are now subject to acetylation control (Figs. 2 and 3).
At present, the three-dimensional structure of RpPat or its homologues has not been reported. Hence, we can only speculate how RpPat may interact with its substrates. Results reported herein suggest that RpPat interacts with a large surface area of its substrates (Fig. 8). It seems possible that Pat simultaneously interacts with the active site lysine residue and the loop region identified by this work and that RpPat may require these two positively charged loop regions for substrate recognition. These results demonstrate that RpPat, and perhaps other Pat homologues, shows a great deal of specificity among acyl-CoA synthetases. These results further support recently reported data (14) that challenged suggestions by others that Pat homologues in E. coli and S. enterica acetylate a wide range of structurally diverse substrates (17, 55).
Insights into the Physiological Role of RpPat
RpMatB appears to function in methyl malonate metabolism in R. palustris, where the ultimate product of methyl malonate catabolism is likely the tricarboxylic acid cycle intermediate succinyl-CoA (36). Many, if not most, of the AMP-forming acyl-CoA synthetases that are regulated by RpPat either activate acetate or propionate directly to their corresponding acyl-CoA derivative or initiate the breakdown of longer chain fatty acids or aromatic acids to acetyl-CoA. It is plausible that RpPat is a sensor of acetyl-CoA or propionyl-CoA concentrations in the cell and uses these compounds to turn off the acyl-CoA synthetases at the beginning of these catabolic pathways. If this were the case, then we would hypothesize that RpPat only modifies acyl-CoA synthetases involved in acetyl-CoA or propionyl-CoA generation. One example of an acyl-CoA synthetase not involved in acetyl- or propionyl-CoA generation in R. palustris is RpMatB, which we have shown is not acetylated. Similarly, acyl-CoA synthetases involved in biosynthesis of natural products such as siderophores and antibiotics would not be regulated by RpPat. This hypothesis awaits further experimental work.
In R. palustris MatB May Have Evolved to Evade Inactivation by RpPat
RpMatB is the first example to date of an acyl-CoA synthetase that is not regulated by RpPat in R. palustris. Changes in a relatively nonconserved loop region distant from the acetylation site greatly affect is susceptibility to acetylation, suggesting that RpMatB evolved to escape regulation by RpPat. A variant of S. enterica acetyl-CoA synthetase that is not susceptible to acetylation has also been generated by changing one residue near the C terminus of the enzyme from leucine to proline (56). Together, these findings suggest that there may be many ways for these enzymes to evolve such that they retain their enzymatic activity but cannot be modified by Pat. In addition, some microbes do not have Pat homologues, suggesting that they either have alternative acetyltransferases that have the same role (such as Bacillus subtilis (57), Mycobacterium tuberculosis (58), and Sulfolobus solfataricus (59)) or that these microbes do not regulate acyl-CoA synthetases by acetylation.
Closing Remarks
From a practical perspective, our results indicate that it is difficult to predict, on the basis of primary sequence alone, whether or not an AMP-forming acyl-CoA synthetase is regulated by acetylation. RpMatB is the first example of an acyl-CoA synthetase that contains the PX4GK motif within the active site loop that is not acetylated by RpPat, although there may be many others. To make the computational identification of RpPat substrates more difficult, there appears to be little sequence conservation within the “chimera loop” identified herein as needed for acetylation. Thus, the newly discovered region does not provide needed homology to facilitate the identification of acyl-CoA synthetases that are under acetylation control. As shown by this work, more research is needed to define the true extent of the bacterial lysine acetylome and to establish how many proteins are true substrates of RpPat and its homologues in other bacteria. Evidence is mounting that RpPat specializes in acetylating a subset of acyl-CoA synthetases in R. palustris, and it will be interesting to see if this holds true for Pat homologues in other organisms.
Acknowledgments
Use of the Advanced Photon Source, an Office of Science User Facility operated for the United States Department of Energy Office of Science by Argonne National Laboratory, was supported by the United States Department of Energy under Contract DE-AC02-06CH11357.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 GM62203 (to J. C. E.-S.) and R01 GM83987 (to I. R.).
This article was selected as a Paper of the Week.

This article contains supplemental Experimental Procedures, Tables S1–S3, and additional references.
The atomic coordinates and structure factors (codes 4GXQ and 4GXR) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- Pat
- protein acetyltransferase
- Acs
- acetyl-CoA synthetase
- MatB
- methylmalonyl-CoA synthetase
- rTEV
- recombinant tobacco etch virus
- TCEP
- tris(2-(17)carboxyethyl)phosphine
- r.m.s.d.
- root mean square deviation
- PDB
- Protein Data Bank.
REFERENCES
- 1. Kim G. W., Yang X. J. (2011) Comprehensive lysine acetylomes emerging from bacteria to humans. Trends. Biochem. Sci. 36, 211–220 [DOI] [PubMed] [Google Scholar]
- 2. Thao S., Escalante-Semerena J. C. (2011) Control of protein function by reversible Nϵ-lysine acetylation in bacteria. Curr. Opin. Microbiol. 14, 200–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Taverna S. D., Li H., Ruthenburg A. J., Allis C. D., Patel D. J. (2007) How chromatin-binding modules interpret histone modifications. Lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 14, 1025–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Oberdoerffer P., Michan S., McVay M., Mostoslavsky R., Vann J., Park S. K., Hartlerode A., Stegmuller J., Hafner A., Loerch P., Wright S. M., Mills K. D., Bonni A., Yankner B. A., Scully R., Prolla T. A., Alt F. W., Sinclair D. A. (2008) SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell 135, 907–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang L., Tang Y., Cole P. A., Marmorstein R. (2008) Structure and chemistry of the p300/CBP and Rtt109 histone acetyltransferases. Implications for histone acetyltransferase evolution and function. Curr. Opin. Struct. Biol. 18, 741–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Berndsen C. E., Denu J. M. (2008) Catalysis and substrate selection by histone/protein lysine acetyltransferases. Curr. Opin. Struct. Biol. 18, 682–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vetting M. W., S de Carvalho L. P., Yu M., Hegde S. S., Magnet S., Roderick S. L., Blanchard J. S. (2005) Structure and functions of the GNAT superfamily of acetyltransferases. Arch. Biochem. Biophys. 433, 212–226 [DOI] [PubMed] [Google Scholar]
- 8. Starai V. J., Escalante-Semerena J. C. (2004) Identification of the protein acetyltransferase (Pat) enzyme that acetylates acetyl-CoA synthetase in Salmonella enterica. J. Mol. Biol. 340, 1005–1012 [DOI] [PubMed] [Google Scholar]
- 9. Liang W., Deutscher M. P. (2012) Post-translational modification of RNase R is regulated by stress-dependent reduction in the acetylating enzyme Pka (YfiQ). RNA 18, 37–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Starai V. J., Escalante-Semerena J. C. (2004) Acetyl-coenzyme A synthetase (AMP forming). Cell. Mol. Life Sci. 61, 2020–2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Starai V. J., Celic I., Cole R. N., Boeke J. D., Escalante-Semerena J. C. (2002) Sir2-dependent activation of acetyl-CoA synthetase by deacetylation of active lysine. Science 298, 2390–2392 [DOI] [PubMed] [Google Scholar]
- 12. Starai V. J., Takahashi H., Boeke J. D., Escalante-Semerena J. C. (2003) Short-chain fatty acid activation by acyl-coenzyme A synthetases requires SIR2 protein function in Salmonella enterica and Saccharomyces cerevisiae. Genetics 163, 545–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Crosby H. A., Heiniger E. K., Harwood C. S., Escalante-Semerena J. C. (2010) Reversible Nϵ-lysine acetylation regulates the activity of acyl-CoA synthetases involved in anaerobic benzoate catabolism in Rhodopseudomonas palustris. Mol. Microbiol. 76, 874–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Crosby H. A., Pelletier D. A., Hurst G. B., Escalante-Semerena J. C. (2012) System-wide studies of N-lysine acetylation in Rhodopseudomonas palustris reveal substrate specificity of protein acetyltransferases. J. Biol. Chem. 287, 15590–15601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Garrity J., Gardner J. G., Hawse W., Wolberger C., Escalante-Semerena J. C. (2007) N-Lysine propionylation controls the activity of propionyl-CoA synthetase. J. Biol. Chem. 282, 30239–30245 [DOI] [PubMed] [Google Scholar]
- 16. Liang W., Malhotra A., Deutscher M. P. (2011) Acetylation regulates the stability of a bacterial protein. Growth stage-dependent modification of RNase R. Mol. Cell 44, 160–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang Q., Zhang Y., Yang C., Xiong H., Lin Y., Yao J., Li H., Xie L., Zhao W., Yao Y., Ning Z. B., Zeng R., Xiong Y., Guan K. L., Zhao S., Zhao G. P. (2010) Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 327, 1004–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bertani G. (1951) Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J. Bacteriol. 62, 293–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bertani G. (2004) Lysogeny at mid-twentieth century: P1, P2, and other experimental systems. J. Bacteriol. 186, 595–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Larimer F. W., Chain P., Hauser L., Lamerdin J., Malfatti S., Do L., Land M. L., Pelletier D. A., Beatty J. T., Lang A. S., Tabita F. R., Gibson J. L., Hanson T. E., Bobst C., Torres J. L., Peres C., Harrison F. H., Gibson J., Harwood C. S. (2004) Complete genome sequence of the metabolically versatile photosynthetic bacterium Rhodopseudomonas palustris. Nat. Biotechnol. 22, 55–61 [DOI] [PubMed] [Google Scholar]
- 21. Kim M.-K., Harwood C. S. (1991) Regulation of benzoate-CoA ligase in Rhodopseudomonas palustris. FEMS Microbiol. Lett. 83, 199–203 [Google Scholar]
- 22. Balch W. E., Wolfe R. S. (1976) New approach to the cultivation of methanogenic bacteria: 2-mercaptoethanesulfonic acid (HS-CoM)-dependent growth of Methanobacterium ruminantium in a pressurized atmosphere. Appl. Environ. Microbiol. 32, 781–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Moore D. D., Dowhan D. (2002) in Current Protocols in Molecular Biology (Ausubel F. M., Brent R., Kingston R. E., Moore D. D., Seidman J. G., Smith J. A., Struhl K., eds) pp. 2.0.1–2.12.17, John Wiley & Sons, Inc., Hoboken, NJ [Google Scholar]
- 24. Rocco C. J., Dennison K. L., Klenchin V. A., Rayment I., Escalante-Semerena J. C. (2008) Construction and use of new cloning vectors for the rapid isolation of recombinant proteins from Escherichia coli. Plasmid 59, 231–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Blommel P. G., Fox B. G. (2007) A combined approach to improving large-scale production of tobacco etch virus protease. Protein Expr. Purif. 55, 53–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Klock H. E., Koesema E. J., Knuth M. W., Lesley S. A. (2008) Combining the polymerase incomplete primer extension method for cloning and mutagenesis with microscreening to accelerate structural genomics efforts. Proteins 71, 982–994 [DOI] [PubMed] [Google Scholar]
- 27. Horton R. M., Ho S. N., Pullen J. K., Hunt H. D., Cai Z., Pease L. R. (1993) Gene splicing by overlap extension. Methods Enzymol. 217, 270–279 [DOI] [PubMed] [Google Scholar]
- 28. Miroux B., Walker J. E. (1996) Overproduction of proteins in Escherichia coli. Mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 260, 289–298 [DOI] [PubMed] [Google Scholar]
- 29. Markowitz V. M., Korzeniewski F., Palaniappan K., Szeto E., Werner G., Padki A., Zhao X., Dubchak I., Hugenholtz P., Anderson I., Lykidis A., Mavromatis K., Ivanova N., Kyrpides N. C. (2006) The integrated microbial genomes (IMG) system. Nucleic Acids Res. 34, D344–D348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gasteiger E., Gattiker A., Hoogland C., Ivanyi I., Appel R. D., Bairoch A. (2003) ExPASy. The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 31, 3784–3788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rayment I. (1997) Reductive alkylation of lysine residues to alter crystallization properties of proteins. Methods Enzymol. 276, 171–179 [PubMed] [Google Scholar]
- 32. Reger A. S., Carney J. M., Gulick A. M. (2007) Biochemical and crystallographic analysis of substrate binding and conformational changes in acetyl-CoA synthetase. Biochemistry 46, 6536–6546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 34. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Collaborative Computational Project No. 4 (1994) The CCP4 suite. Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 36. Crosby H. A., Rank K. C., Rayment I., Escalante-Semerena J. C. (2012) Structure-guided expansion of the substrate range of methylmalonyl-CoA synthetase (MatB) of Rhodopseudomonas palustris. Appl. Environ. Microbiol. 78, 6619–6629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Emsley P., Cowtan K. (2004) Coot. Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 38. Skubák P., Murshudov G. N., Pannu N. S. (2004) Direct incorporation of experimental phase information in model refinement. Acta Crystallogr. D Biol. Crystallogr. 60, 2196–2201 [DOI] [PubMed] [Google Scholar]
- 39. Adams P. D., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., Terwilliger T. C. (2002) PHENIX. Building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 [DOI] [PubMed] [Google Scholar]
- 40. Krissinel E., Henrick K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 [DOI] [PubMed] [Google Scholar]
- 41. Gulick A. M., Starai V. J., Horswill A. R., Homick K. M., Escalante-Semerena J. C. (2003) The 1.75 Å crystal structure of acetyl-CoA synthetase bound to adenosine-5′-propylphosphate and coenzyme A. Biochemistry 42, 2866–2873 [DOI] [PubMed] [Google Scholar]
- 42. Gulick A. M. (2009) Conformational dynamics in the acyl-CoA synthetases, adenylation domains of nonribosomal peptide synthetases, and firefly luciferase. ACS Chem. Biol. 4, 811–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Reger A. S., Wu R., Dunaway-Mariano D., Gulick A. M. (2008) Structural characterization of a 140° domain movement in the two-step reaction catalyzed by 4-chlorobenzoate:CoA ligase. Biochemistry 47, 8016–8025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kochan G., Pilka E. S., von Delft F., Oppermann U., Yue W. W. (2009) Structural snapshots for the conformation-dependent catalysis by human medium-chain acyl-coenzyme A synthetase ACSM2A. J. Mol. Biol. 388, 997–1008 [DOI] [PubMed] [Google Scholar]
- 45. Bains J., Boulanger M. J. (2007) Biochemical and structural characterization of the paralogous benzoate CoA ligases from Burkholderia xenovorans LB400. Defining the entry point into the novel benzoate oxidation (box) pathway. J. Mol. Biol. 373, 965–977 [DOI] [PubMed] [Google Scholar]
- 46. D'Arcy A., Stihle M., Kostrewa D., Dale G. (1999) Crystal engineering. A case study using the 24-kDa fragment of the DNA gyrase B subunit from Escherichia coli. Acta Crystallogr. D Biol. Crystallogr. 55, 1623–1625 [DOI] [PubMed] [Google Scholar]
- 47. McElroy H. E., Sisson G. W., Schoettlin W. E., Aust R. M., Villafranca J. E. (1992) Studies on engineering crystallizability by mutation of surface residues of human thymidylate synthase. J. Cryst. Growth 122, 265–272 [Google Scholar]
- 48. Rypniewski W. R., Holden H. M., Rayment I. (1993) Structural consequences of reductive methylation of lysine residues in hen egg white lysozyme. An X-ray analysis at 1.8-Å resolution. Biochemistry 32, 9851–9858 [DOI] [PubMed] [Google Scholar]
- 49. Kim Y., Quartey P., Li H., Volkart L., Hatzos C., Chang C., Nocek B., Cuff M., Osipiuk J., Tan K., Fan Y., Bigelow L., Maltseva N., Wu R., Borovilos M., Duggan E., Zhou M., Binkowski T. A., Zhang R. G., Joachimiak A. (2008) Large-scale evaluation of protein reductive methylation for improving protein crystallization. Nat. Methods 5, 853–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kobayashi M., Kubota M., Matsuura Y. (1999) Crystallization and improvement of crystal quality for x-ray diffraction of maltooligosyl trehalose synthase by reductive methylation of lysine residues. Acta Crystallogr. D Biol. Crystallogr. 55, 931–933 [DOI] [PubMed] [Google Scholar]
- 51. Kurinov I. V., Uckun F. M. (2003) High resolution x-ray structure of potent anti-HIV pokeweed antiviral protein-III. Biochem. Pharmacol. 65, 1709–1717 [DOI] [PubMed] [Google Scholar]
- 52. Rayment I., Rypniewski W. R., Schmidt-Bäse K., Smith R., Tomchick D. R., Benning M. M., Winkelmann D. A., Wesenberg G., Holden H. M. (1993) Three-dimensional structure of myosin subfragment-1: a molecular motor. Science 261, 50–58 [DOI] [PubMed] [Google Scholar]
- 53. Schubot F. D., Waugh D. S. (2004) A pivotal role for reductive methylation in the de novo crystallization of a ternary complex composed of Yersinia pestis virulence factors YopN, SycN, and YscB. Acta Crystallogr. D Biol. Crystallogr. 60, 1981–1986 [DOI] [PubMed] [Google Scholar]
- 54. Hughes A. J., Keatinge-Clay A. (2011) Enzymatic extender unit generation for in vitro polyketide synthase reactions. Structural and functional showcasing of Streptomyces coelicolor MatB. Chem. Biol. 18, 165–176 [DOI] [PubMed] [Google Scholar]
- 55. Lima B. P., Antelmann H., Gronau K., Chi B. K., Becher D., Brinsmade S. R., Wolfe A. J. (2011) Involvement of protein acetylation in glucose-induced transcription of a stress-responsive promoter. Mol. Microbiol. 81, 1190–1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Starai V. J., Gardner J. G., Escalante-Semerena J. C. (2005) Residue Leu-641 of acetyl-CoA synthetase is critical for the acetylation of residue Lys-609 by the protein acetyltransferase enzyme of Salmonella enterica. J. Biol. Chem. 280, 26200–26205 [DOI] [PubMed] [Google Scholar]
- 57. Gardner J. G., Grundy F. J., Henkin T. M., Escalante-Semerena J. C. (2006) Control of acetyl-coenzyme A synthetase (AcsA) activity by acetylation/deacetylation without NAD+ involvement in Bacillus subtilis. J. Bacteriol. 188, 5460–5468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nambi S., Basu N., Visweswariah S. S. (2010) Cyclic AMP-regulated protein lysine acetylases in mycobacteria. J. Biol. Chem. 285, 24313–24323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Brent M. M., Iwata A., Carten J., Zhao K., Marmorstein R. (2009) Structure and biochemical characterization of protein acetyltransferase from Sulfolobus solfataricus. J. Biol. Chem. 284, 19412–19419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Delano W. L. (2002) The PyMOL Molecular Graphics System, DeLano Scientific LLC, San Carlos, CA [Google Scholar]