

Background: HIV Nef disrupts chemotaxis of immune cells by targeting multiple steps.
Results: Nef recruits AIP4 E3 ligase, to ubiquitinate Gαi2 of heterotrimeric G-proteins for subsequent lysosomal degradation.
Conclusion: Nef impedes chemokine-signaling events via degradation of Gαi2.
Significance: Impaired G-protein signaling by HIV Nef-expressing lymphocytes probably contributes to immune dysfunction in AIDS.
Keywords: G-protein-coupled receptors (GPCR), G Proteins, Immunodeficiency, Lentivirus, Ubiquitin Ligase
Abstract
The HIV Nef protein is an important pathogenic factor that modulates cell surface receptor trafficking and impairs cell motility, presumably by interfering at multiple steps with chemotactic receptor signaling. Here, we report that a dominant effect of Nef is to trigger AIP4 E3 ligase-mediated Gαi2 ubiquitination, which leads to Gαi2 endolysosomal sequestration and destruction. The loss of the Gαi2 subunit was demonstrable in many cell types in the context of gene transfection, HIV infection, or Nef protein transduction. Nef directly interacts with Gαi2 and ternary complexes containing AIP4, Nef, and Gαi2 form. A substantial reversal of Gαi2 loss and a partial recovery of impaired chemotaxis occurred following siRNA knockdown of AIP4 or NEDD4 or by inhibiting dynamin. The N-terminal myristoyl group, 62EEEE65 motif, and 72PXXP75 motif of Nef are critical for this effect to occur. Nef expression does not affect a Gqi5 chimera where the five C-terminal residues of Gq are replaced with those of Gαi2. Lysine at position 296 of Gαi2 was identified as the critical determinant of Nef-induced degradation. By specifically degrading Gαi2, Nef directly subverts leukocyte migration and homing. Impaired trafficking and homing of HIV Nef-expressing lymphocytes probably contributes to early immune dysfunction following HIV infection.
Introduction
The recirculation of naive and memory lymphocytes, the trafficking of lymphocytes into lymphoid organs, and the appropriate localization of effector cells depend upon a regulated configuration of cell surface adhesion molecules and chemoattractants. Ligand-occupied chemoattractant receptors, trigger Gα subunits of the heterotrimeric G-protein Gi to exchange GTP for GDP. This leads to the release of Gαi-associated Gβγ subunits, activation of downstream effectors, and directed cell migration (1). T lymphocytes employ several different chemokine receptors. The chemokine receptors CCR7, CCR9, and CXCR4 (chemokine (CXC motif) receptor 4) along with their cognate ligands regulate T-cell ontogeny in the thymus (2–4), whereas CCR7 regulates the migration of peripheral T cells to lymphoid tissue during adaptive immune response (5, 6). Proinflammatory chemokine receptors like CXCR3 and CCR5 and their respective ligands regulate effector CD4 and CD8 T cell trafficking in peripheral tissues (7–10). During an adaptive immune response, activated paracortical T cells use CCR7 and CXCR5 to migrate to the follicle edge and eventually into the lymph node follicle, where they help coordinate B lymphocyte responses (11, 12). Impaired signaling through these different receptors can cause immune dysfunction and lymphoid tissue disorganization.
A hallmark of HIV infection is severe disruption of the cellular microenvironment of lymph nodes and the gastrointestinal associated lymphoid tissue. Although this occurs in part as a consequence of CD4+ T cell depletion, a major contributing factor is compromised chemoattractant receptor signaling (13, 14). Perinatal HIV infection results in profound T cell developmental abnormalities mimicking those observed in thymic dysplasia in DiGeorge syndrome (15). Implicating the HIV Nef protein as a major co-factor in the general pathology and immune dysfunction in HIV AIDS, transgenic mice expressing HIV-1 provirus or HIV Nef protein alone have a similar constellation of immune pathologies (16–18).
Several mechanisms have been found by which Nef can interfere with chemoattractant receptor signaling. First, HIV and SIV Nef proteins have been shown to down-regulate chemokine receptor expression, presumably through enhanced receptor endocytosis (19, 20). This has been associated with a marked chemotactic arrest, presumably due to loss of the cognate receptor (19). Second, Nef can interfere with signal transduction pathways downstream of the receptors by affecting different intracellular signaling proteins involved in these pathways. These include the Ser/Thr kinase PAK; small GTPases, such as Rac and CDC42; and guanine nucleotide exchange factors, such as Vav and DOCK2-ELMO1 (21–24). In particular, Nef inhibited T lymphocyte chemotaxis toward CXCL12 (chemokine (CXC motif) ligand 12) by disrupting spatially organized cytoskeleton rearrangements through unregulated DOCK2-ELMO1-mediated Rac activation (24). Third, Nef has been linked to decreases in integrin expression, which impaired transmigration across endothelial cells (25). Fourth, Nef expression can alter the morphology of T lymphocytes and dendritic cells through changes in F-actin dynamics (26–28) by deregulating cofilin through PAK2 interaction (29) and RhoA interaction with diaphanous interacting protein and p190RhoAGAP (30). With the exception of the studies implicating reduced receptor expression, the majority of these studies indicate that the prominent effect of Nef on chemokine and chemoattractant receptor signaling is downstream of the major signal transducer, the heterotrimeric G-protein Gi.
Although Nef undoubtedly targets multiple sites in the chemoattractant signaling pathway, we have discovered a dominant effect of Nef because its expression results in a marked loss of steady-state levels of Gαi2 protein but not of other Gα or Gβγ subunits. Nef triggered AIP4 (atrophin-interacting protein 4) E3 ligase-mediated Gαi2 ubiquitination, leading to its endolysosomal sequestration and destruction. The consequence of the loss of Gαi2 is a marked impairment in chemoattractant receptor signaling and directed leukocyte migration.
EXPERIMENTAL PROCEDURES
Expression Plasmids
Expression plasmids for CD4, CD8, WT and mutant Nef alleles, Nef Cerulean, and bicistronic IRES2 plasmids encoding Nef and GFP have been described (31). Chemokine receptors, YFP-tagged Gαi subunits, and the Gqi5 chimera have been described (32–36). Mutations replacing each one of the three lysines at positions 296, 307, and 314 of Gαi2 (but not in Gαi3) with arginines were engineered by PCR mutagenesis of YFP-tagged Gαi2 (35). The GST-Nef construct was constructed by in-frame cloning of Nef ORF into the EcoRI-SalI sites of pGEX-4T1 (GE Healthcare). Plasmids expressing GST fused to full-length AIP4, AIP4ΔWW, and AIP4-WW-I-IV were generously supplied by Adriano Marchese (Stritch School of Medicine, Loyola University, Chicago) as were the expression plasmids for FLAG-ubiquitin, FLAG-tagged WT AIP4, or c-Myc-tagged WT or C830A AIP4 mutant. The GST fusion proteins were expressed in Escherichia coli strain XL-1 Blue (Stratagene) and purified according to the manufacturer's (GE Healthcare) instructions.
Antibodies and Reagents
The following reagents were obtained from commercial sources. Unconjugated or dye-conjugated mAbs against EEA1 (early endosomal antigen 1) actin were from BD Immunocytometry (San Diego, CA); unconjugated LAMP1 (H4A3) and LAMP2 (H4B4) mAbs were from the Developmental Studies Hybridoma Bank (University of Iowa, Iowa City, IA); unconjugated or Alexa 488-, 647-, allophycocyanin-, or phycoerythrin-conjugated CD4 and CD8 were from Invitrogen or R&D Systems; antibodies against AIP4 and NEDD4, murine anti-Gαi2 mAb, rabbit polyclonal antibodies against Gαi2 (sc-7276), Gαi3 (sc-262), Gαo (sc-387), Gαq/11 (sc-392), and Gα13 (sc-410), and goat polyclonal antibody against actin (sc-1615) were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA); anti-YFP mAb was from Clontech Corp.; and rabbit IgG and murine mAb against HA or FLAG epitopes were from Covance or Roche Applied Science and Sigma-Aldrich, respectively. A second rabbit polyclonal antibody against Gαo (catalog number 3975) was purchased from Cell Signaling Technology (Danvers, MA). Secondary antibodies to mouse, rabbit, goat, and human IgG conjugated to various Alexa dyes were from Invitrogen Corp., and HRP-conjugated goat anti-mouse, rabbit, or human IgG and donkey anti-goat IgG were from Pierce. Dyansore was purchased from Tocris Corp.; epoxomicin, forskolin, isoproterenol, and MG-132 were from EMD Biosciences. Dynasore and epoxomicin treatments were used at 80 and 25 μm concentrations, respectively, for 4 h at 37 °C, followed by staining for flow cytometry or immunoblotting.
Cells and Transfection
For this study, HeLa cells were used for microscopy, and T cell lines of Jurkat or CEM cells were used for phenotypic assays. HeLa cell transfection conditions have been described in detail before (69). The Department of Transfusion Medicine at the National Institutes of Health provided elutriated monocytes and leukocyte-enriched buffy coat from anonymous volunteers. Peripheral blood lymphocytes were purified as before (69), and cells were cultivated under standard conditions in RPMI medium or DMEM with 10% fetal calf serum (FCS) and 1% l-glutamine, as appropriate. Primary hematopoietic cells and cell lines in suspension (7–10 × 106 cells/100 μl) were nucleofected with 3–5 μg of DNA using an Amaxa Nucleofector as recommended by the manufacturer. In all cases, GFP or CD8 expression plasmid was included to check transfection efficiency as described (31). Cell viability was checked using a FACS-based live/killed assay kit using a Guava Easycyte flow cytometer as described by the manufacturer (Millipore Corp.). Experiments were rejected if cell viability was less than 75%. For biochemical experiments, cell numbers were adjusted to equivalent viability and transfection efficiency. However, for most non-FACS-based experiments, transfectants were purified by positive selection using CD8 immunoaffinity beads from Stem Cell Technologies as recommended by the manufacturer.
HIV Infection
For single-cycle HIV infection, recombinant viruses expressing mouse CD24 in place of vpR (CD24), CD24 with a vpU deletion (CD24U), or CD24 lacking both vpU and Nef (CD24UM1T) were generated by 293-T cell transfection (Trans-IT LT1 transfection reagent, Mirus Corp.) with the respective HIV proviruses and a VSV-G expression plasmid. Infection was allowed to proceed for 48–72 h to achieve >40% infection. Virus production, quantification, and T cell infection kinetics have been described before (33).
RNA-mediated Interference
Gαi1, Gαi2, Gαi3, and Gαo subunits were knocked down using the recommended sequence (37). AIP4 and NEDD4 were knocked down using UUUCAAUGCAGAAUUUCUGUGGUCC and UAGAGGAGAAGGUUCUUGUUGUUGC, respectively (Invitrogen). Protocols for siRNA transfection followed by plasmid expression have been described before (31).
Intracellular [Ca2+] Measurements
Two different spectrofluorimetric assays were used. In the ratiometric assay, the relative ratio of fluorescence emitted at 510 nm after sequential excitation at 340 and 380 nm was measured as described (33), using Fura-2 AM (Invitrogen) as the calcium-binding dye in a fluorimeter (Photon Technology Inc., South Brunswick, NJ). Alternatively, intracellular Ca2+ levels were measured using a FLIPR 3 calcium assay kit (Molecular Devices) with a FlexStation III scanning fluorimeter as described (38), using Softmax Pro (Molecular Devices) for data acquisition and analysis. Single cell analysis of calcium flux was performed using the Leica SP5, using a ×40 oil immersion objective, numerical aperture 1.25. HeLa cells in 8-well chambers (LabTek chambers, Thermo Scientific) were cotransfected with CerFP or Nef-CerFP and YFP-tagged WT Gαi2 or Gαi3. At 18–24 h post-transfection, cells were loaded with Hanks' balanced saline solution containing Ca2+ and Mg2+ (Invitrogen) with 1% FBS and 4 μm Fluo-4 AM (Invitrogen). The cells were irradiated with a UV laser at 405 nm for Cerulean and with an argon laser at 488 nm for Fluo-4 and at 514 nm for YFP. Images were acquired in a 512 × 400-pixel format with the pinhole set at 2 airy units every 1–1.4 s for ∼200 s. The images were collected as line scan images, which provide the values of the calcium-dependent increase in fluorescence along a single spatial dimension as a function of time. For analysis, the regions of interest (ROIs) were drawn on the cells expressing Cerulean/Nef Cerulean. At least 10 ROIs were drawn for each field, and the calcium flux was measured in >5 fields for each experimental condition. The change in the intensities was analyzed using the Leica software, followed by graphing using EXCEL.
Affinity Selections
GST pull-downs of cellular extracts (107 cell equivalents in 1 ml) in 25 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EGTA, 0.5 mm MgCl2, and 0.5% (w/v) Triton X-100 (lysis buffer) with protease inhibitors (1 Complete tablet/ml; Roche Applied Science) was done using purified GST or the indicated GST fusion proteins immobilized on glutathione- agarose beads (GE Healthcare) and incubated for 2 h. The beads were washed three times with lysis buffer and once with lysis buffer without Triton X-100. Bound proteins were resolved by SDS-PAGE and detected by immunoblotting using antibodies against the respective targets. His6-tagged Nef protein was affinity-selected from extracts of corresponding transfectants using Ni2+-nitrilotriacetic acid-agarose beads (Qiagen Corp.), and FLAG-tagged proteins were selected using agarose bead immobilized M2 mAb (Sigma-Aldrich).
cAMP Assay
Intracellular cAMP levels were assayed using the Direct cAMP Enzyme Immunoassay kit (Assay Designs), following the manufacturer's instructions. The optical density was read at 405 nm in a FlexStation II microplate scanner, and the results were analyzed using Softmax Pro 5 (Molecular Devices, Sunnyvale, CA).
Flow Cytometry
The fluorescent dye-conjugated antibodies for CD4, CD8, and the chemokine receptors were obtained from BD Biosciences, R&D Systems, or eBioscience. One million cells were incubated with the appropriate antibodies in 100 μl of PBS containing 1% goat serum for 15 min at room temperature. The data acquisition was carried out using a FACSortTM (BD Biosciences) flow cytometer, and the analyses were done using FlowJo version 9.2. (Tree Star Inc., Ashland, OR). Flow cytometric detection of F-actin by phalloidin staining has been described (75).
Confocal Microscopy
Confocal microscopy was done as described (32) using a Leica TCS-NT/SP5 microscope (Leica, Exton, PA) equipped with a 100× oil immersion objective, numerical aperture 1.32. Images were collected using digital zooms up to ×8. Co-localization experiments were done with live (fluorescent proteins) or fixed cells for most experiments as described (31). The fluorochromes were excited using a UV laser at 405 nm for Cerulean, an argon laser at 488 nm for Alexa 488 and YFP, and a DPSS laser at 561 nm for Alexa 568 and 647. Images were processed using the Leica TCS-NT/SP5 software, Leica Lite, and Adobe Photoshop CS4.
FRET Assay
FRET interactions between Nef-CerFP and YFP-tagged Gαi2 or Gαi3 were evaluated using the Leica acceptor photobleaching software to examine in live or fixed cells in the Leica SP5 confocal microscope with a ×60, numerical aperture 1.4 oil immersion objective in a 512 × 512-pixel format using a DPSS laser at 442 nm for Cerulean and an argon laser at 514 nm for YFP. The exposure times were kept equal within each series of images and chosen such that all pixel intensities were within the linear range. A ×63 oil lens was used unless mentioned otherwise. ROIs were selected from individual fields visualized at zoom factors between 1 and 8 (Z1–Z8), and 50–80% YFP photobleaching was achieved in the selected ROIs using the 514-nm laser at 100% power for 45 s at ×8 optical zoom. Each pixel of the image contains data corresponding to an emission spectrum resulting from both CFP and YFP. The fluorescence intensities of donor and acceptor were measured before and after photobleaching in the ROIs drawn at different cellular organelles and membranes. The diameter of the ROIs was kept uniform throughout the analysis at 8 μm. 10 ROIs were drawn for each part of a single cell, and at least 10 different cells were taken for each FRET efficiency calculation. Only regions photobleached at >50% were considered for analysis. FRET efficiency was calculated using the equation EF = (Ipostbleach − Iprebleach)/Ipostbleach, where I is the average CFP fluorescence intensity after the subtraction of the background.
Chemotaxis Assays
End-point chemotaxis was determined using the transwell system with membranes of 6.5-mm diameter and 5.0-μm pore size in RPMI containing 10 mm HEPES and 1% FBS as described (32). The ratios of migrated cells were determined from the number of cells in the lower and upper chambers counted in a cell sorter after the addition of a known number of fluorescent reference particles (Spherotech, Inc., Libertyville, IL) (38). Flow cytometric detection of F-actin by phalloidin staining has been described (38).
Statistical Analyses
The statistical analyses were performed using GraphPad Prism version 5.0. A paired t test or one-way analysis of variance test was performed as appropriate, to determine the significance of the observed differences between the paired or unpaired samples. A value of p < 0.05 was considered to be significant in all of the analyses. The graphs were generated using either Microsoft Excel or GraphPad Prism, and the bars represent mean ± S.E.
RESULTS
Nef-induced Chemotaxis Defect in T Cells and Monocytes
Nef has been shown to impair chemotaxis toward CXCL12 in lymphoid cell lines, presumably through mechanisms other than depletion of cell surface CXCR4 (24, 25, 29, 39, 40). To investigate how Nef subverts chemotactic responses downstream of chemokine receptors, we selected cells for study in which Nef only modestly (<50%) reduced receptor expression. These included the T cell lines, CEM and Jurkat, human peripheral blood mononuclear cells (PBMCs), and human monocytes. Despite the relatively modest reduction in receptor levels (data not shown), Nef expression still markedly inhibited CXCL12-, CCL19-, CCL2-, or formylmethionylleucylphenylalanine-induced chemotaxis (Fig. 1, A–C).
FIGURE 1.

Nef inhibits migration of Jurkat T cells (A1–A4), fresh PBMCs (B), and monocytes (C) toward CXCL12 in a transwell assay and F-actin accumulation in response to CXCL12 or CCL2 (D). Relative (%) chemotaxis of Jurkat cells expressing different Nef alleles (and gated for GFP co-expression) at the optimal peak levels of CXCL12 for cells is illustrated by the histogram (mean with S.E., n = 3; *, p < 0.01) in A1, and a representative CXCL12 dose-response profile of chemotaxis of Nef(−) or Nef(+) cells (n = 3) is illustrated in A2. Relative migration potential of Jurkat cells expressing GFP alone (Vec) or with WT NA7, M20A, P72A, E62A, or L164A mutant from Nef-GFP IRES vector toward CXCR4 is shown as a function of GFP expression in A3 (n = 3). *, p < 0.05; **, p < 0.01, when compared with plasmid-transfected cells. A4, relative (%) chemotaxis of Jurkat cells transfected with CD8 alone or with WT NA7 or NL4–3 Nef allele or M20A, P72A, E62A, or L164A NL4–3 Nef mutant. Chemotaxis data are shown for CD8(+) cells (n = 3). *, p < 0.01 compared with respect to null controls. B, the histogram on the left shows chemotaxis potential of single-cycle Nef(+) HIV-infected (CD24-positive) and uninfected (CD24-negative) CEM cells toward CXCL12. Relative (%) migration potential of CEM cells in WT infected (CD24+) and non-infected (CD24−) population to different concentrations of CXCL12 was assessed by a transwell migration assay. The next histogram (with error bars (S.E.)) shows relative (%) chemotaxis toward CCL19 of PBMCs cotransfected with GFP and WT Nef, null mutant, or empty vector (n = 3, p < 0.02). CXCL12 dose response of chemotaxis (in absolute terms) for Nef(+) GFP(+) versus Nef(−) GFP(+) transfectants is illustrated on the right (n = 3). *, p < 0.05 compared with respective nanomolar concentration. C, histograms with S.E. (n = 3) show the chemotaxis potential toward CXCL12 or CCL2 (left) and formylmethionylleucylphenylalanine (right) of monocytes, transduced for 2 h with BSA or purified Nef fusion protein tagged C-terminally with the arginine-rich motif (RRM) of HIV-1 Tat followed by His6 residues (n = 3). *, p < 0.05. D, time course of F-actin accumulation in Jurkat cells or fresh PBMCs nucleofected with bicistronic (IRES) plasmids encoding WT or null (Nef Xho) Nef and GFP. Histograms (with S.E.) on the left illustrate the F-actin accumulation in GFP(+) versus GFP(−) gated Jurkat cells nucleofected with Nef-Xho-IRES-GFP (top) or Nef-IRES-GFP (bottom) (n = 3). *, p < 0.02. PBMCs nucleofected with Nef-IRES-GFP or NefXho-IRES-GFP were treated with 20 nm CXCL12 for 45 min. Relative (%) F-actin levels (phalloidin mean fluorescence value (MFV)) are plotted as a function of GFP (Nef or NefXho) expression (n = 3). Bars, S.E.; ***, p < 0.01. Alternatively, monocytes were nucleofected with GFP and Nef or an empty vector, and the time course of F-actin accumulation in response to 20 nm CXCL12 or CCL2 was monitored in GFP(−) versus GFP(+) cells (n = 3). ***, p < 0.05.
The inhibition occurred irrespective of whether Nef was expressed by DNA transfection (Fig. 1, A1–A4, B (right two panels), and C (right panel)), protein transduction of monocytes (Fig. 1C, left panel), or single cycle HIV infection (Fig. 1B, left panel). Alanine substitutions disrupting the Nef polyglutamic motif at position 62 (E62A), the PXXP motif at 72 (P72A), or alanine substitutions at threonine 162 (T162A) and at histidine 166 (H166A) partially or completely reversed the inhibitory effect (Fig. 1, A3 and A4). The chemotactic defect corresponded to the marked reduction of F-actin accumulation in response to CXCL12 or CCL2 in Nef-expressing Jurkat cells, monocytes, and PBMCs (Fig. 1D).
Nef Markedly Inhibited Biochemical Readouts of Early G-protein Signaling from Agonist-stimulated Chemokine Receptors
Most, if not all, chemokines activate Gαi-coupled GPCRs. An agonist-bound GPCR exchanges GTP for GDP in the Gα subunit of receptor-bound heterotrimeric G-protein. This results in Gα and Gβγ dissociation and the activation of downstream effectors. First, we examined the immediate consequence of Gβγ dissociation by monitoring intracellular Ca2+ flux from ER stores mediated by inositol 1,4,5-trisphosphate in response to activation of phospholipase C. We found that Nef expression markedly attenuated the rapid chemokine-induced mobilization of intracellular Ca2+ normally observed in Jurkat cells and abolished the response in U937 cells (Fig. 2, A1 and A2). We also visualized single cell Ca2+ flux in response to CXCL12 stimulation of HeLa cells expressing either Cerulean fluorescent protein (CerFP) or Nef-CerFP by real-time time lapse confocal video microscopy. CXCL12 increased intracellular Ca2+ in the CerFP-expressing cells, but not the Nef-CerFP cells (Fig. 2, B1). This was confirmed by quantitative kinetic analysis of Ca2+ flux of individual cells expressing CerFP or Nef-CerFP (Fig. 2, B2). These data suggested that Nef might perturb chemokine-mediated G-protein dissociation from cognate GPCR(s). In agreement with the corresponding effects on chemotaxis, alanine substitutions of four glutamates at 62 and at the PXXP motif partially reversed the Nef-induced inhibition of CXCL12-mediated calcium release from the ER (Fig. 2, B3).
FIGURE 2.

Nef markedly inhibited biochemical readouts of Gαi2 activation. Nef inhibits chemoattractant-mediated calcium flux in Jurkat cells (A1) and the U937 cell line (A2). Cells were co-transfected with CD8 and Nef, co-expressers were purified by CD8-positive selection (Stem Cell Technologies), and Nef effect in Jurkat cells was evaluated by measuring cell surface CD4 expression. The time course of CXCL12 (10 nm)-initiated calcium flux profiles was obtained using FlexStation III and the recommended assay. Results are representative of four experiments. Agonist (CXCL12 followed by CCL2) dose (10 or 100 nm)-response profiles of intracellular calcium flux in U937 transfectants were analyzed by fluorescence ratiometry in a PTI fluorimeter (33). B1, time course of CXCL12-initiated Ca2+ flux in HeLa cells expressing CerFP or Nef-CerFP (red) was monitored by video capture at 30 frames/s of Fluo-4 emission (green) up to 150 s after CXCL12 addition. The arrows denote CerFP or Nef-CerFP cells (expressed at ∼10–15% efficiency around 12–16 h post-transfection) to highlight their difference in Fluo-4 intensity. Fluorescent data were collected from ∼10 ROIs for each field, the calcium flux was measured in >5 fields for each condition in an experiment, and the experiments were repeated three times (n = 50–60). The change in the intensities was analyzed using the Leica software followed by graphing using EXCEL. Ca2+ flux profiles of a few (to avoid clutter) representative cells (ROIs) expressing CerFP (left) or Nef-CerFP (right) are shown in B2, with the ordinate showing relative intensity of Fluo-4 emission. CXCL12-initiated Ca2+ flux is profiled in purified CEM cells co-transfected with CD8 and WT, null, or other Nef mutants. CEM cells were transfected with CD8, WT, null, or the indicated Nef mutants, and CD8(+) cells were purified prior to measurement of Ca2+ flux (as described above) (B3). Nef expression enhanced cAMP levels under basal conditions or after Gαs activation by isoproterenol (C) or by forskolin stimulation of adenylyl cyclase (D). However, Nef did not further enhance the cAMP levels after isoproterenol treatment with transfectants overexpressing Gαi3 (E). Jurkat cells were cotransfected with CD8 and Nef or vector (for C–E) and with a Gαi3 expression plasmid (only for E). Transfected cells were purified by CD8 selection and assayed for cAMP production as described under “Experimental Procedures” (n = 4; error bars represent S.E.; *, p < 0.05).
Next, we inquired whether Nef subverted activation of G-protein α subunit(s). The major effectors of activated Gαi subunits are adenylyl cyclase isoforms. GTP-bound Gαi directly inhibits specific adenylyl cyclase isoforms, thereby reducing cAMP production by direct (by agents like forskolin) or hormone-induced (via activated Gαs subunit) activation of adenylyl cyclase. Basal intracellular cAMP levels reflect a steady-state equilibrium of tonic inhibition and stimulation of adenylyl cyclase isoforms by activated Gαi and Gαs subunits, respectively. We found that Nef-expressing Jurkat cells had a 2-fold higher basal cAMP level than did the null expression controls, consistent with a loss of constitutive Gαi signaling. Furthermore, Nef expression was associated with enhanced cAMP production in Jurkat cells treated with the β-adrenergic agonist, isoproterenol (ISOPRO), or forskolin, a direct adenylyl cyclase activator (Fig. 2, C and D). However, basal or hormone-induced cAMP levels were not enhanced in Nef(+) cells co-expressing recombinant Gαi3 (Fig. 2E). This suggested that Nef might solely target Gαi2 because Jurkat cells and lymphocytes express Gαi2 and Gαi3, not Gαi1.
Nef Induced a Marked Loss of Steady-state Levels of Gαi2 but Not Gαi3 or Other Gα Subunits
To determine if Nef impaired G-protein functionality or induced a physical loss or sequestration of one or more G-protein subunits, we quantified the steady-state levels of Gα subunits in Nef(+) cells by immunoblotting. We found that Nef induced a dose-dependent decrease in Gαi2 levels in Jurkat/CEM cells and monocytes, comparable in magnitude with CD4 down-regulation in Jurkat or CEM cells (Fig. 3A). In contrast, Gαi3 in Jurkat cells, CEM cells (not shown), and monocytes or Gαq and Gαs in Jurkat cells were unaffected (Fig. 3A).
FIGURE 3.

Biochemical and genetic analysis of Nef induced loss of steady-state levels of Gαi2 subunit in Jurkat and CEM cell lines and primary human monocytes in the context of DNA transfection or single cycle HIV infection. A, cellular extracts were immunoblotted for Gαi2, Gαi3, Gαq, Gαs, CD4, or actin. Protein bands were scanned for pixel density, and results are plotted as histograms with error bars representing S.E. (n = 3; *, p < 0.03). B, loss of Gαi2 in single cycle infections of CEM/Jurkat cells with identical reverse transcriptase unit equivalents of VSV-G pseudotyped Nef+ (wt), but not Nef− (M1T) or Nef−vpU− (ΔvpU M1T) NL4-3 HIVs expressing murine CD4 antigen in place of vpR. Gαi2, Gαi3, and Nef were detected by immunoblotting cellular extracts. C, HIV-1 Nef alleles induced a specific loss of Gαi2 of comparable magnitude. Jurkat cells were transfected with various Nef alleles, and the levels of Gαi2 down-regulation were assessed. Cellular extracts were immunoblotted for Gαi2, Gαi3, HA (Nef), or actin. D, certain Nef mutants had lost the ability to induce loss of Gαi2. Extracts of CEM/Jurkat cells transfected with the indicated Nef derivatives were analyzed by SDS-PAGE followed by immunoblotting for actin, Gαi2, and Gαi3. Relative pixel values in B and C represent averages from two experiments for each case. E, knockdown of Gαi2 or Gαi2 by cognate siRNAs partially inhibited CXCL12-dependent calcium flux. Jurkat cells were co-transfected with CD8 and Nef or an empty vector following siRNA nucleofection and expression for 48 h. ∼2 × 102 transfectants were adjusted to reflect constant levels of CD8 and analyzed for CXCL12-initiated intracellular calcium flux. Gαi2 and Gαi2 in the respective siRNA transfectants were detected by immunoblotting as described above.
In single cycle HIV-1 infections, loss of Gαi2 correlated with Nef expression. Gαi2 levels were unaltered in cells infected with viruses deleted for Nef or Nef and vpU (Fig. 3B). Eight different HIV-1 Nef alleles induced a specific loss of Gαi2 of comparable magnitude (Fig. 3C). The differences in the level of down-regulation might be dependent on critical Nef residues, and they are a subject of further investigation. Gαi2 levels were unaffected in cells expressing Nef mutants with alanine substitutions in the acidic motif at 62 (E62A) or in the polyproline motif at 72 (P72A) in Jurkat and CEM cells (Fig. 3D). We compared the effects of siRNA knockdown of individual Gαi2, Gαi3, or Go subunits on agonist-driven Ca2+ flux. Knockdown of Gαi2 or Gαi3 resulted in a significant (∼60%) decrease in the agonist-mediated Ca2+ flux (Fig. 3E). Thus, the Nef-induced defect in early G-protein signaling reflected by chemokine-mediated Ca2+ flux was attributed to the physical loss of the Gαi2 subunit.
Nef-induced Inhibition of Gαi2 Degradation Was Partially Rectified by Overexpression of Recombinant Gαi2, and a Gqi5 Chimera Replacing the C-terminal Five Residues with Those of GαI Was Resistant to the Nef Effect
Co-expression of YFP-tagged recombinant Gαi1 (not shown), Gαi2, or Gαi3 failed to restore CXCL12-mediated Ca2+ flux in Jurkat cells expressing a 2-fold molar excess of Nef over the recombinant Gαi2 subunits (Fig. 4, A and B). The failure to reverse CXCL12-mediated Ca2+ flux by recombinant Gαi2 reflected the destruction of endogenous and a substantial loss of the co-expressed Gαi2 (Fig. 4A, bottom). Only by increasing the ratio of Gαi2 to Nef expression could the signaling be partially restored. Although there was no loss of Gαi3-YFP co-expressed with a molar excess of Nef (Fig. 4B, bottom), Gαi3-YFP failed to restore CXCL12-initiated Ca2+ flux (Fig. 4B, top).
FIGURE 4.

Nef-induced inhibition of G-protein signaling and Gαi2 degradation were partially rectified by overexpression of recombinant Gαi2, and a Gqi5 chimera replacing the C-terminal five residues with those of GαI was resistant to Nef effect. CEM cells were nucleofected with CD8 and YFP-tagged Gαi2 (A) or Gαi3 (B) subunits with or without a molar excess of Nef. Purified CD8 transfectants were analyzed for CXCL12-driven intracellular Ca2+ flux. Nef-induced loss of Ca2+ flux in response to CXCL12 was partially reversed in cells expressing a 2-fold molar excess of Gαi2 over Nef (A), but Gαi3 co-expression did not ameliorate Ca2+ flux deficit (B). At a 2-fold molar excess of Gαi2 over Nef, there was much less relative loss in the steady-state levels of Gαi2-YFP (A, bottom). There was no loss of Gαi3-YFP with or without of Nef (B, bottom). Relative Gαi2-YFP pixel values shown in the immunoblot are averages from three experiments. C, Nef did not inhibit CXCL12-initiated calcium flux in cells expressing Gqi5 chimera replacing the C-terminal five residues with those of Gαi2, (36). Jurkat transfectants co-expressing CD8 and Gqi5 chimera with or without Nef were purified and analyzed as described above. D, Nef did not alter the stability of co-expressed Gqi5 chimera. Cell extracts of Jurkat transfectants were analyzed for expression of HA-tagged Gqi5, co-expressed CD8, Nef, and actin. E, Gqi5 expression did not alter Nef-induced down-regulation of CD4 or CXCR4 at the PM. Black and gray histograms (with error bars (S.E.)) represent normalized data for CD8(−) and CD8(+) gated cells (n = 3; *, p < 0.05). IB, immunoblot.
A small C-terminal region of Gα subunit(s) is the GPCR selectivity determinant and a recombinant Gqi5, which exchanged five Gq residues at the C terminus for those of Gαi2, thus becoming functionally equivalent to Gαi2 in chemokine responsiveness and pertussis toxin sensitivity (36). Nef failed to alter the expression of or inhibit CXCL12-induced intracellular Ca2+ flux from the Gqi5 chimera (Fig. 4, C and D) while still down-regulating CD4 and CXCR4 (Fig. 4E). Because heterotrimeric G-proteins are bound to the receptor in an inactive state, it is possible that the Nef-induced lysosomal sequestration of Gαi2 reflects aberrant trafficking of CXCR4. However, this is unlikely because cells co-expressing Nef and Gqi5 chimera retained CXCL12-driven Ca2+ flux.
Nef Co-localized with and Promoted Sequestration of Gαi2, but Not Gαi3, in the Perinuclear Region Enriched for Endolysosomal Markers
To determine whether Nef interacted with Gαi2 in vivo, we examined the subcellular distribution of YFP-tagged recombinant Gαi2 or Gαi3 in HeLa cells co-expressing CerFP or Nef-CerFP. In the CerFP-expressing cells, Gαi2 and Gαi3 were distributed at the PM irrespective of CXCL12 exposure. In Nef-expressing cells, a substantial fraction of Gαi2 co-localized with Nef in the perinuclear region (co-localization correlation of >0.75), regardless of CXCL12 treatment, whereas Gαi3 did not co-localize with Nef (correlation of <0.04) (Fig. 5, A1 and A2). Nef expression resulted in an equivalent loss of membrane-associated versus detergent-soluble fractions of Gαi2-YFP, indicating that Nef induced collective loss of Gαi2 inside the cell (Fig. 5, A3). The Gαi2- and Nef-containing vesicular structures stained positively for endolysosomal markers EEA1 and LAMPs but not with the markers for ER (GRP) or Golgi (Golgin) (Fig. 5B).
FIGURE 5.

Nef co-localized with and promoted endolysosomal sequestration of Gαi2 but not Gαi3. HeLa cells in coverglass chambers were co-transfected with YFP-tagged Gαi2 (A1) or Gαi3 (A2) with CerFP or Nef-CerFP (top or bottom row in A1 and A2) and treated for 10 min at 37 °C with increasing concentrations of CXCL12 or not before processing for live microscopy. Cerulean is green, and YFP is red. A3, Nef-induced loss of YFP-tagged Gαi2 occurred irrespective of whether or not the G-protein was membrane-associated. Cells were extracted in a buffer containing 1% (w/v) Triton X-100 (TXT-100) at 25 °C (room temperature (RT)), conditions that are known to strip most if not all membrane-associated proteins. Membrane (pellet) and cytoplasmic (supernatant) extracts were resolved by SDS-PAGE followed by immunoblot detection of YFP, Nef, and CD8. Relative Gαi2-YFP pixel values are averages from three experiments. B, in Nef-expressing cells, YFP-tagged Gαi2 (red) co-localized with Nef-CerFP (green) and endolysosomal markers, EEA1 and LAMP (blue) proteins, but not with markers (blue) for ER (GRP) or Golgi (GOLGIN).
Nef-mediated Lysosomal Sequestration of Gαi2 Reflected in Vivo Interaction of Nef and Gαi2
Next, we performed live FRET microscopy to analyze nearest neighbor interactions between Nef and Gαi. We measured FRET efficiency on the basis of the increase in the donor (CFP or Nef-CFP) fluorescence upon photobleaching the YFP-tagged Gαi2 acceptor. FRET efficiencies generated by tandem CFP-YFP fusion protein served as the positive control. We analyzed >10 cells in each of three experiments, and photomicrographs of one representative cell are shown (Fig. 6, A1), with the calculated FRET efficiencies of ∼100 ROIs shown in the graphs below (Fig. 6, A2). There was substantial FRET between Nef-CFP and Gαi2-YFP at the intracellular, perinuclear region (Fig. 6, B1) but not at the PM (Fig. 6, B2) or anywhere in the cell with Gαi3-YFP (Fig. 6, B1 and B2).
FIGURE 6.

Nef associated with Gαi2 but not Gαi3in vivo. Acceptor photobleaching FRET assay of HeLa cells co-expressing CerFP or Nef-CerFP with YFP-tagged Gαi2 or Gαi3. FRET assay was limited to Gαi2 or Gαi3 with CerFP or Nef-CerFP in the perinuclear area (A1) or at the PM (A2). Photomicrographs are representative of 10 fields (cells). Fluorescence intensities of 10 ROIs (arrows) corresponding to Gαi2 or Gαi3 in the perinuclear regions (B1) or at the PM (B2) in each cell were examined before (Pre) and after (Post) photobleaching, and calculated FRET efficiencies for all ROIs (∼100) from three independent experiments are shown as scatter plots with error bars. Donor emission of representative ROIs pre- and postphotobleaching is shown in the inset above the graph. The calculated statistical significance is represented (*, p < 01).
Nef Interacted with Gαi2 in Vitro and Induced Gαi2 Ubiquitination
To confirm the physical interaction between Nef and Gαi2, we performed in vitro GST pull-down assays of leukocyte extracts using GST or GST-Nef. Using extracts from Jurkat T-cells and primary monocytes, we found that GST-Nef bound Gαi2 but not Gαi3 in a dose-responsive manner (Fig. 7A).
FIGURE 7.
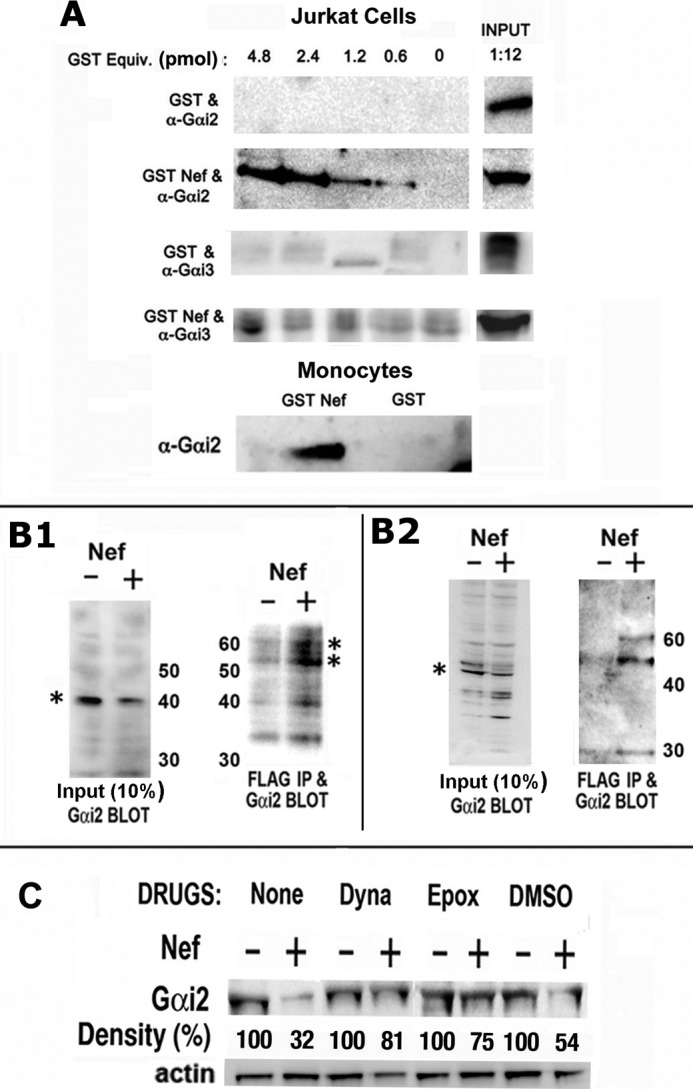
Nef interacted with Gαi2in vitro and induced Gαi2 ubiquitination. A, varying amounts of purified GST or GST Nef were incubated with cellular extracts of Jurkat cells or human monocytes. Bound proteins were resolved by SDS-PAGE, and Gαi species were detected by immunoblotting. B, Nef-induced ubiquitination of native Gαi2 in Nef-expressing CEM cells. CEM cells were co-transfected with FLAG-tagged ubiquitin and Nef or null plasmid. Cellular extracts were directly immunoblotted for Gαi2 (B1 and B2, left panels) or immunoprecipitated first with anti-FLAG polyclonal antibody (B1) or mAb (B2) followed by immunoblotting for Gαi2. Asterisks denote Gαi2 (left) and mono- and diubiquitinated Gαi2 (right). Numbers (kDa) refer to molecular mass markers. Data are representative of three experiments. C, Nef-mediated Gαi2 breakdown was partially rectified by Dynasore, a small molecular weight inhibitor of dynamin, or the proteosome inhibitor epoxomycin. Cellular extracts were analyzed by immunoblotting for Gαi2. Gαi2 bands are shown pairwise for Nef(+) versus Nef(−) for each treatment with relative (%) density (average values from three experiments) denoted below.
To inquire whether the loss of Gαi2 was a consequence of Nef-induced ubiquitination of Gαi2 and its subsequent lysosomal proteolysis, we directly evaluated whether Gαi2 was ubiquitinated. We co-transfected CEM cells with vectors expressing FLAG-tagged ubiquitin and either Nef or control. Nef expression resulted in significant loss of Gαi2 (Fig. 7, B1 (left)). Cellular extracts were immunoprecipitated with FLAG mAb-coated beads, and the selected proteins were immunoblotted for Gαi2 using mouse monoclonal antibody (Fig. 7, B1 (right)) or rabbit polyclonal antibody (Fig. 7, B2). Nef expression resulted in significant loss of Gαi2 (Fig. 7B, left). Two immunoreactive bands of ∼53 and ∼63 kDa were seen in lysates of Nef-expressing cells, probably representing mono- and diubiquitinated native Gαi2 (Fig. 7B, right).
We first evaluated the effects of Dynasore (41), a small molecular weight dynamin inhibitor, and the proteosome inhibitor, epoxomycin. We found that pretreatment with the inhibitors substantially reversed the Nef-induced loss of Gαi2 (Fig. 7C).
Nef Co-localizes with E3 Ubiquitin Ligases AIP4 and Nedd4
Ubiquitination of agonist-occupied CXCR4 by the Nedd4-like E3 ubiquitin ligase AIP4 (42) primes it for traffic through the ESCRT pathway, ultimately resulting in lysosomal proteolysis (43, 44). We inquired whether Nef-induced endolysosomal trafficking of Gαi2 was facilitated by these E3 ligase(s). We stained HeLa cells co-transfected with plasmids expressing YFP-tagged Gαi2 or Gαi3 and Nef-CerFP or CerFP with antibodies against AIP4 or NEDD4. There was extensive co-localization of Gαi2 with Nef and AIP4 in the perinuclear area (Fig. 8A). There was a similar co-distribution of Nef and Gαi2 with the NEDD4-positive region (Fig. 8B). In contrast, Gαi3 was mostly at the PM, with no evidence of co-localization with Nef, AIP4, or NEDD4 (Fig. 8, A and B (bottom)).
FIGURE 8.

Nef recruits HECT domain E3 ligases, AIP4, or NEDD4 and facilitates Gαi2 ubiquitination, presumably as a ternary complex. A, Nef co-localized with Gαi2 but not with Gαi3 in the AIP4-enriched perinuclear region. HeLa cells co-transfected with Nef-CerFP and YFP-tagged Gαi2 or Gαi3 were fixed, permeabilized, and stained with murine mAb against AIP4 followed by Alexa 647-conjugated anti-mouse IgG. B, Nef co-localizes with Gαi2 but not Gαi3 in NEDD4-positive regions in HeLa cells. Experimental details are as above except for the use of rabbit polyclonal antibody against NEDD4. Monochromatic images on the right in each row correspond to Nef-CerFP, YFP-tagged Gαi2 or Gαi3, and anti-AIP4 or -NEDD4 fluorescence with two-channel (like AIP4/NEDD4 and Gαi2 or AIP4/NEDD4 and Nef, etc.) composite images shown on the left. 4× cropped RGB images (Nef-CerFP (R), Gαi2- or Gαi3-YFP (G), and AIP4/NEDD4 (B)) with arrows denoting vesicles showing maximal co-localization are shown on the far left in each row.
AIP4 Bound Gαi2 through the HECT Domain of the E3 Ligase and to the Tetraglutamate and Polyproline Motif of Nef, Presumably through the WW Domain(s)
We evaluated the binding potential of AIP4 for Nef and Gαi subunits in vitro and in vivo. In GST-Nef pull-down assays, AIP4 was recovered from Jurkat cell extracts in a specific and quantifiable manner (Fig. 9A, top). Like other members of the NEDD4 family of HECT domain E3 ligases, AIP4 has four WW domains (Fig. 9A, center), which preferentially bind to proline-rich PY motifs (45–47) or hyperphosphorylated C termini of PM receptors (44). Accordingly, GST-AIP4 bound poorly to Nef mutants with substitutions of the polyglutamate at position 62 (E62A) or the polyproline tract at position 72 (P72A) (Fig. 9A, bottom left). GST fusion proteins containing WT AIP4 or a deletion mutant that excised all four WW domains but retained the HECT domain bound Gαi2 (Fig. 9A, bottom right).
FIGURE 9.

Biochemical and genetic analysis of Nef interaction in vivo with Gαi2 and E3 ligases. A, Nef and Gαi2 interacted with AIP4 in vitro. Shown is a schematic illustration (top) of GST-tagged WT AIP4 and mutants deleted for the WW or HECT domains (44). Jurkat cells were transfected with WT, M20A, E62A, P72A, or L164A Nef mutant, and cellular extracts were incubated with ∼22 pmol of GST or GST-AIP4 immobilized on agarose beads. GST-bound fractions and unselected lysate (2%) were analyzed for Nef by immunoblotting (left). Cellular extracts of Jurkat cells were reacted with equimolar amounts (∼22 pm) of GST-AIP4, GST-AIP4 ΔWW, GST-WW-I-IV, or GST alone. Bound fractions were analyzed by immunoblotting with anti-Gαi2 mAb (right). Input, an aliquot of extract immunoblotted without selection. Numbers in both cases refer to bound fraction (%) averaged from two experiments. B, in vivo interaction of Nef with AIP4. Extracts of Jurkat cells cotransfected with FLAG-AIP4 and HA-tagged Nef or empty HA vector and immunoprecipitated with FLAG-mAb and Nef were identified by immunoblotting with rabbit anti-HA (top). Relative binding affinity of Nef mutants for AIP4 was evaluated in Jurkat cells cotransfected with GFP, FLAG-AIP4, and His6-tagged WT or mutant Nef (bottom). Cellular extracts were bound to Ni2+-nitrilotriacetic acid beads, and the bound FLAG-AIP4 was detected by immunoblotting with anti-FLAG mAb. C, alternatively, Jurkat cells were cotransfected with GFP, FLAG-AIP4, and HA-tagged WT or mutant Nef. Extracts were immunoprecipitated with FLAG mAb, followed by immunoblotting with HA antibody. Data represent results from three experiments.
Next, we co-transfected Jurkat cells with expression vectors for FLAG-AIP4 and Nef-HA or a control plasmid. Immunoblotting FLAG immunoprecipitates prepared from lysates from co-expressing cells for Nef-HA revealed an interaction between them (Fig. 9B, top). Furthermore, mutations at the Nef motifs judged to be critical for in vitro binding to AIP4 were also impaired for in vivo binding (Fig. 9, B (bottom) and C).
From these observations collectively, we concluded that Nef facilitated Gαi2 ubiquitination by recruitment of the NEDD4 class of HECT domain E3 ligases, presumably as a ternary complex of Nef, Gαi2, and E3 ligases(s).
Nef-induced Loss of Gαi2 Was Partially Reversed by Expression of Enzymatically Defective AIP4 or by siRNA Knockdown of AIP4 or NEDD4
To firmly establish the role of AIP4 in the Nef-induced Gαi2 ubiquitination and degradation, Jurkat cells were co-transfected with Nef or an empty vector and WT or an enzymatically defective HECT domain mutant, AIP4-C830A (43, 48). AIP4-C830A overexpression reversed the Nef-mediated loss of Gαi2 (Fig. 10A).
FIGURE 10.
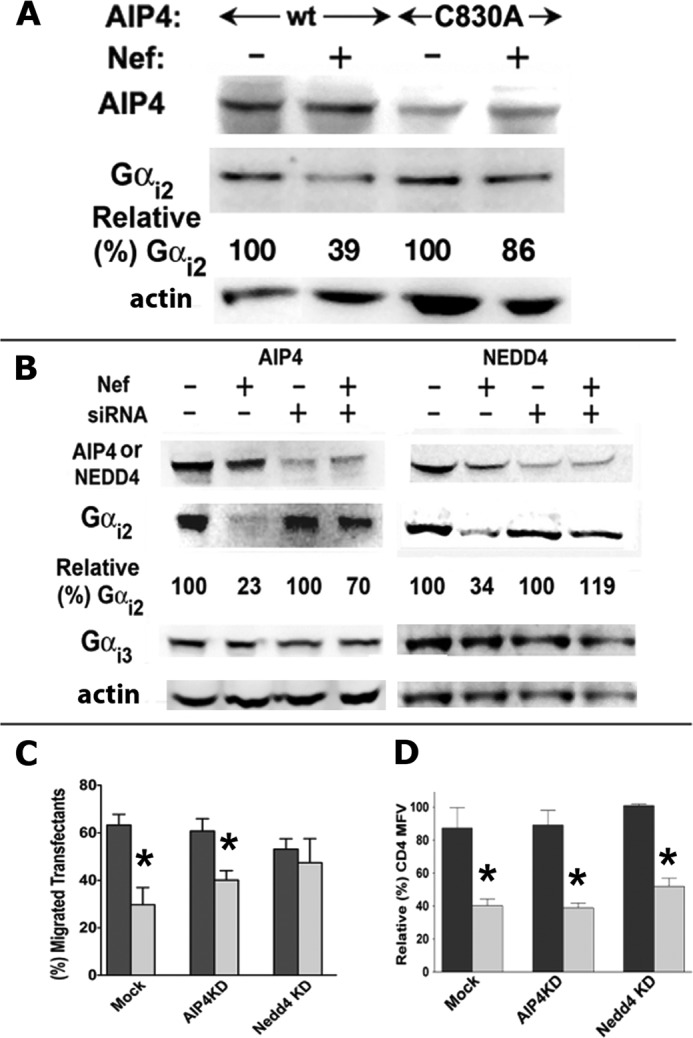
Nef-induced loss of Gαi2 was partially reversed by enzymatically defective AIP4 or by siRNA knockdown of AIP4 or NEDD4. A, the HECT domain mutant of AIP4 (C830A) reversed Nef-induced degradation of Gαi2. Jurkat cells were nucleofected with CD8, FLAG-tagged WT, or C830A AIP4 mutant and HA-tagged Nef or empty HA vector. Extracts from cells adjusted for equivalent CD8 expression were analyzed by immunoblotting using mAb against Gαi2 and anti-FLAG antibody for detecting AIP4. Numbers refer to relative (%) Gαi2 amounts for the respective transfectants adjusted for equivalent CD8 expression. B, Nef-induced loss of Gαi2 was partially reversed by siRNA knockdown of AIP4 or NEDD4. Transfected Jurkat cells were disrupted in 0.5 ml of lysis buffer, and extracts were resolved by SDS-PAGE followed by immunoblotting for actin, NEDD4, AIP4, Gαi2, or Gαi3. Numbers denote relative (%) pixel densities of Gαi2 averaged from three experiments. C, siRNA knockdown of AIP4 or NEDD4 partially reversed Nef-induced inhibition of chemotaxis toward CXCL12. After a 48-h treatment with the respective siRNAs, Jurkat cells were cotransfected with GFP and Nef(−) or Nef(+) plasmid. Cells were evaluated for chemotaxis toward 20 nm CXCL12. Histograms represent pairwise comparison of relative (%) chemotaxis efficiency of GFP(+) Nef(−) versus Nef(+) (n = 3). D, siRNA knockdown of AIP4 or NEDD4 did not interfere with Nef-mediated CD4 down-regulation. Jurkat cell transfectants were analyzed for CD4 expression by flow cytometry. Relative (%) CD4 mean fluorescence values for Nef(+) and Nef(−) transfectants are plotted pairwise for each condition as histograms (with error bars). The mean fluorescence value for Nef(−) cells in each case was arbitrarily assigned as 100 (n = 3); *, p < 0.05 when compared with their respective plasmid (mock)-transfected controls.
We then compared the effect of siRNA knockdown of AIP4 or NEDD4 on Nef-induced loss of endogenous Gαi2 in Jurkat and CEM cells. siRNA-induced reduction of AIP4 or NEDD4 E3 ligase reversed the Nef-induced Gαi2 loss (Fig. 10B). Simultaneously, we evaluated chemotaxis of Nef versus non-Nef transfectants after siRNA knockdown of AIP4 or NEDD4. Nef-mediated chemotactic inhibition was partially rectified by NEDD4 knockdown, although not nearly as well as by the AIP4 knockdown (Fig. 10C). Silencing the E3 ligases did not affect cell viability or curtail other activities of Nef, such as CD4 (Fig. 10D) down-regulation.
Lysine at Position 296 Is the Critical Determinant of Gαi2 Ubiquitination and Degradation in Nef-expressing Cells
Finally, we identified the potential lysine residue in Gαi2 that may be targeted for ubiquitination by AIP4 in Nef-expressing cells, by evaluating the susceptibility to Nef of individual Gαi2-YFP mutants that exchanged unique lysines for arginines at positions 296, 307, and 314. Jurkat cells were co-transfected with wild type or the respective LYS/ARG mutants with or without Nef. Immunoblotting for YFP showed that the LYS/ARG mutant, M1, was resistant to Nef-induced steady-state loss (Fig. 11A). In agreement, CXCL12-driven Ca2+ flux in M1 mutant-expressing cells was relatively more resistant than in the WT Gαi2-YFP expressers (Fig. 11B). We then examined the subcellular distribution of WT Gαi2-YFP versus the M1 mutant in HeLa cells co-expressing CerFP or Nef-CerFP. In the CerFP-expressing cells, WT and M1 were distributed at the PM. In Nef-expressing cells, a substantial fraction of WT but not M1 co-localized with Nef in the perinuclear region (co-localization correlation of >0.75 versus <0.125) (Fig. 11C).
FIGURE 11.

Lysine at position 296 of Gαi2 is the critical determinant of Nef-induced degradation. A, the C-terminal 66 residues of Gαi2 are shown at the top, with all lysines shaded and the three lysines unique to Gαi2 denoted by asterisks. M1–M3, the respective lysine to arginine mutations engineered into the YFP-tagged Gαi2. CEM cells were transfected with CD8, WT, or the indicated Gαi2 mutant and HA-tagged Nef or empty HA vector. Extracts of transfected cells were adjusted for equivalent CD8 expression and analyzed by immunoblotting using mAb against YFP for detecting Gαi2. Numbers at the bottom are relative (%) pixel densities of Gαi2 averaged from two experiments. B, M1 mutant is more efficient than the WT Gαi2-YFP in rectifying Nef-induced G-protein signaling (by intracellular Ca2+ flux) defect. CEM cells were nucleofected with CD8, and YFP-tagged WT or M1 Gαi2 CD8 transfectants were analyzed for CXCL12-driven intracellular Ca2+ flux as described in the legend to Fig. 3. C, YFP-tagged WT, but not the M1 Gαi2, was internalized and co-localized with Nef-enriched vesicular structures. HeLa cells in coverglass chambers were co-transfected with YFP-tagged WT or M1 Gαi2 mutant with Nef-CerFP or CerFP and processed for live microscopy. Cerulean is green, and YFP is red.
DISCUSSION
Through multiple criteria, we have shown here that HIV Nef impairs G-protein signaling from chemokine receptors by selectively targeting Gαi2 for ubiquitination and rapid endolysosomal destruction. Nef-induced loss of steady-state levels of Gαi2 was observed in many cell types in the context of gene transfection, HIV infection, or Nef protein transduction. Lys/Arg substitution at each of the three unique lysines near the C terminus of Gαi2 identified lysine at 296 as the crucial determinant of Nef-induced degradation. Nef-mediated loss of Gαi2 was shown to be dependent on ubiquitination by AIP4, because it was reversed by overexpression of catalytically defective C830A AIP4 mutant, by siRNA knockdown of AIP4, or by use of proteosomal inhibitors or a dynamin antagonist. Previous reports have shown that, following agonist activation, CXCR4 is ubiquitinated by the NEDD4 class HECT domain E3 ubiquitin ligase AIP4 (but not NEDD4), which primes it for traffic through the ESCRT pathway, resulting in lysosomal proteolysis (43, 44). Nef recruits either E3 ligase for Gαi2 ubiquitination without need for agonist treatment.
Nef bound Gαi2 much better than other Gα species in vitro. Given the level of sequence homologies among Gαi isoforms, it is not surprising that Nef bound to Gαi3 at 4.8 pm concentration. But it occurred at relatively much higher concentrations of Gαi3 compared with Gαi2, indicating that Nef preferably binds to Gαi2 rather than Gαi3. We repeated the experiment with increasing concentration of GSTNef, and we found binding of Gαi3 to Nef (data not shown). But this occurred at a concentration that is physiologically not relevant. Nef and Gαi2 co-localized in vivo in perinuclear vesicles enriched for endolysosomal markers, which was further validated by increased FRET intensity between Nef-CFP and Gαi2-YFP in the intracellular vesicular structures. There was also extensive co-localization of Nef with Gαi2 (but not Gαi3) and AIP4 (or NEDD4) in the same perinuclear vesicles. Taken together, these findings strongly implied that Nef-induced Gαi2 ubiquitination occurs in a ternary complex of Nef, E3 ligase, and Gαi2. AIP4 binding to substrates is mediated by its WW domain, which targets polyproline motifs like PPPY or PPXY (47, 49, 50) or phosphothreonine and phosphoserine residues (51). Our studies showed that AIP4 bound Gαi2 through the HECT domain and Nef, presumably through the WW domain(s). Accordingly, AIP4 poorly bound Nef mutants that substituted the polyglutamate at 62 (E62A) or the polyproline tract at position 72 (P72A). This suggested that the Src homology 3 binding domain of Nef, with the PXXP motif, which has been demonstrated to be important for viral replication (52, 53), might be crucial for the down-regulation of Gαi2. Additional residues surrounding the PXXP motif have been shown to affect the HIV replication and disease progression (54) and might account for the variation in the ability of various Nef alleles to down-regulate Gαi2.
Ubiquitin-dependent degradation of G-proteins(s) was originally shown for the yeast Gpa1, a Gα equivalent (55–57), which was routed to multivesicular bodies for proteolysis. Mammalian Gαo (58), Gαi (59, 60), Gαs (61, 62), and Gβγ (63, 64) subunits are presumed to be regulated by the proteasomal pathway; however, only Gαs (61, 62), transducin Gβγ (64), and Gγ2 (63) were shown to be ubiquitinated. To our knowledge, Nef-induced Gαi2 loss represents the first instance where ubiquitination routes the G-protein for endolysosomal proteolysis. A systematic analysis of essential genes in yeast important for regulating G-protein signaling identified a preponderance of genes involved with protein degradation (65). Our results indicate that mammalian cells must also carefully control expression of the essential components in the G-protein signaling pathway and that HIV Nef has probably co-opted this mechanism that regulates Gαi2 stability.
It was of interest that, although Nef spared Gαi isomers other than Gαi2, early G-protein signaling and chemotaxis defects perpetrated by Nef implied that other Gαi isomers were not recruited for signaling in the different cell types we have used. This was further confirmed by siRNA knockdown of either Gαi2 or Gαi3, which resulted in a significant (∼60%) decrease in the agonist-mediated Ca2+ flux. Notwithstanding the paramount role(s) of Gαi in immune cell trafficking, chemokine receptors can couple to other G-proteins to facilitate chemotaxis (66–68) in monocyte/macrophages. In addition, an alternative chemokine receptor pathway has been described for certain receptor/cell combinations in dendritic cells and neutrophils (69, 70) that is activated by Gαi2 but was dependent on Gq proteins for optimal G-protein signaling and chemotaxis. Given that Gq stability and functionality were unaltered by Nef, migration of certain leukocyte subtypes, such as DCs and monocytes, might be differentially regulated in vivo.
Nef is a short-lived protein produced early in the HIV life cycle; Nef protein and fragments are packaged within nascent HIV particles and may be delivered to newly infected cells (71). Non-infected cells may also be subject to Nef effects either by its secretion by infected cells or by its transfer via intercellular nanotubular conduits from infected to non-infected cells, which can alter cellular function (72, 73) and membrane dynamics causing a transfer of infected cell signaling to recipient cells (74). The targeting of Gαi2 for destruction by Nef in infected as well as in nearby non-infected cells provides several potential advantages for HIV. First, disabling responsiveness to chemoattractants prevents cells from leaving a nidus of infection, thereby promoting cell-to-cell spread of the virus. Second, because of the importance of T cell migration for T cell function, disruption of directed T cell migration will impair the induction of effective immunity. Third, a sharp reduction in Gαi2 will impair the sequestration of Gβγ subunits. Freed Gβγ subunits can activate downstream effectors that can provide signals that promote viral replication as well as effect cell survival. Fourth, decreased Gαi2 removes its tonic inhibition of adenylyl cyclase isoforms, increasing cAMP levels and PKA activity, which can also enhance HIV replication (75–77). Fifth, because heterotrimeric G-proteins also function in GPCR-independent signaling pathways involved in intracellular trafficking and cell division (78–80), Nef-induced Gαi2 loss may impact T cell function by affecting these pathways.
Although the Nef-induced chemotaxis defect may derive in part from its effects on other signaling events, such as PAK2-mediated cofilin deregulation (29) or inappropriate DOCK2/ELMO1-driven Rac activation uncoupled from chemokine receptor signaling (24), we have shown that Nef-induced loss of Gαi2 probably trumps these because it impacts the earliest events in chemokine signaling. HIV, by expressing Nef, has chosen to target Gαi2 for destruction. Nef probably does so by usurping a normal cellular mechanism that regulates Gαi2 stability. Nef-induced loss of Gαi2 in lymphocytes will profoundly affect their function and may impact signaling pathways that impact HIV replication.
Acknowledgments
We thank Philip Murphy (LMI, NIAID, National Institutes of Health) for critical review of the manuscript. We thank Steven Becker (Biological Imaging Section, RTB, NIAID, National Institutes of Health) for technical advice. We thank Adriano Marchese (Stritch School of Medicine, Loyola University, Chicago) for the generous supply of numerous reagents and technical advice and Bruce Conklin (University of California, San Francisco) for the Gqi5 chimera.
This work was supported, in whole or in part, by the National Institutes of Health, NIAID, Intramural Research Program.
- IRES
- internal ribosome entry site
- CerFP
- Cerulean fluorescent protein
- HECT
- homologous to E6-AP C terminus
- CFP
- cyan fluorescent protein
- ER
- endoplasmic reticulum
- ESCRT
- endosomal sorting complex required for transport
- GPCR
- G-protein-coupled receptor
- LAMP
- lysosome-associated membrane protein
- PBMC
- peripheral blood mononuclear cell
- PM
- plasma membrane
- ROI
- region of interest
- VSV
- vesicular stomatitis virus.
REFERENCES
- 1. Rossi D., Zlotnik A. (2000) The biology of chemokines and their receptors. Annu. Rev. Immunol. 18, 217–242 [DOI] [PubMed] [Google Scholar]
- 2. Wurbel M. A. (2000) The chemokine TECK is expressed by thymic and intestinal epithelial cells and attracts double- and single-positive thymocytes expressing the TECK receptor CCR9. Eur. J. Immunol. 30, 262–271 [DOI] [PubMed] [Google Scholar]
- 3. Kwan J., Killeen N. (2004) CCR7 directs the migration of thymocytes into the thymic medulla. J. Immunol. 172, 3999–4007 [DOI] [PubMed] [Google Scholar]
- 4. Trampont P. C., Tosello-Trampont A.-C., Shen Y., Duley A. K., Sutherland A. E., Bender T. P., Littman D. R., Ravichandran K. S. (2010) CXCR4 acts as a costimulator during thymic β-selection. Nat. Immunol. 11, 162–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bromley S. K., Thomas S. Y., Luster A. D. (2005) Chemokine receptor CCR7 guides T cell exit from peripheral tissues and entry into afferent lymphatics. Nat. Immunol. 6, 895–901 [DOI] [PubMed] [Google Scholar]
- 6. Debes G. F., Arnold C. N., Young A. J., Krautwald S., Lipp M., Hay J. B., Butcher E. C. (2005) Chemokine receptor CCR7 required for T lymphocyte exit from peripheral tissues. Nat. Immunol. 6, 889–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Charo I. F., Ransohoff R. M. (2006) The many roles of chemokines and chemokine receptors in inflammation. N. Engl. J. Med. 354, 610–621 [DOI] [PubMed] [Google Scholar]
- 8. Sallusto F., Geginat J., Lanzavecchia A. (2004) Central memory and effector memory T cell subsets. Function, generation, and maintenance. Annu. Rev. Immunol. 22, 745–763 [DOI] [PubMed] [Google Scholar]
- 9. Brainard D. M., Tager A. M., Misdraji J., Frahm N., Lichterfeld M., Draenert R., Brander C., Walker B. D., Luster A. D. (2007) Decreased CXCR3+ CD8 T cells in advanced human immunodeficiency virus infection suggest that a homing defect contributes to cytotoxic T-lymphocyte dysfunction. J. Virol. 81, 8439–8450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bromley S. K., Mempel T. R., Luster A. D. (2008) Orchestrating the orchestrators. Chemokines in control of T cell traffic. Nat. Immunol. 9, 970–980 [DOI] [PubMed] [Google Scholar]
- 11. Delon J., Stoll S., Germain R. N. (2002) Imaging of T-cell interactions with antigen presenting cells in culture and in intact lymphoid tissue. Immunol. Rev. 189, 51–63 [DOI] [PubMed] [Google Scholar]
- 12. Garside P., Ingulli E., Merica R. R., Johnson J. G., Noelle R. J., Jenkins M. K. (1998) Visualization of specific B and T lymphocyte interactions in the lymph node. Science 281, 96–99 [DOI] [PubMed] [Google Scholar]
- 13. Pantaleo G., Fauci A. S. (1996) Immunopathogenesis of HIV infection. Annu. Rev. Microbiol. 50, 825–854 [DOI] [PubMed] [Google Scholar]
- 14. Kinter A., Arthos J., Cicala C., Fauci A. S. (2000) Chemokines, cytokines and HIV: a complex network of interactions that influence HIV pathogenesis. Immunol. Rev. 177, 88–98 [DOI] [PubMed] [Google Scholar]
- 15. Kourtis A. P., Ibegbu C., Nahmias A. J., Lee F. K., Clark W. S., Sawyer M. K., Nesheim S. (1996) Early progression of disease in HIV-infected infants with thymus dysfunction. N. Engl. J. Med. 335, 1431–1436 [DOI] [PubMed] [Google Scholar]
- 16. Chrobak P., Simard M. C., Bouchard N., Ndolo T. M., Guertin J., Hanna Z., Dave V., Jolicoeur P. (2010) HIV-1 Nef disrupts maturation of CD4+ T cells through CD4/Lck modulation. J. Immunol. 185, 3948–3959 [DOI] [PubMed] [Google Scholar]
- 17. Hanna Z., Kay D. G., Rebai N., Guimond A., Jothy S., Jolicoeur P. (1998) Nef harbors a major determinant of pathogenicity for an AIDS-like disease induced by HIV-1 in transgenic mice. Cell 95, 163–175 [DOI] [PubMed] [Google Scholar]
- 18. Skowronski J., Parks D., Mariani R. (1993) Altered T cell activation and development in transgenic mice expressing the HIV-1 nef gene. EMBO J. 12, 703–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hrecka K., Swigut T., Schindler M., Kirchhoff F., Skowronski J. (2005) Nef proteins from diverse groups of primate lentiviruses downmodulate CXCR4 to inhibit migration to the chemokine stromal derived factor 1. J. Virol. 79, 10650–10659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Michel N., Ganter K., Venzke S., Bitzegeio J., Fackler O. T., Keppler O. T. (2006) The Nef protein of human immunodeficiency virus is a broad-spectrum modulator of chemokine receptor cell surface levels that acts independently of classical motifs for receptor endocytosis and Galphai signaling. Mol. Biol. Cell 17, 3578–3590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Roeth J. F., Collins K. L. (2006) Human immunodeficiency virus type 1 Nef. Adapting to intracellular trafficking pathways. Microbiol. Mol. Biol. Rev. 70, 548–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kirchhoff F., Schindler M., Specht A., Arhel N., Münch J. (2008) Role of Nef in primate lentiviral immunopathogenesis. Cell Mol. Life Sci. 65, 2621–2636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Laguette N., Brégnard C., Benichou S., Basmaciogullari S. (2010) Human immunodeficiency virus (HIV) type-1, HIV-2 and simian immunodeficiency virus Nef proteins. Mol. Aspects Med. 31, 418–433 [DOI] [PubMed] [Google Scholar]
- 24. Janardhan A., Swigut T., Hill B., Myers M. P., Skowronski J. (2004) HIV-1 Nef binds the DOCK2-ELMO1 complex to activate Rac and inhibit lymphocyte chemotaxis. PLoS Biol. 2, E6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Park I. W., He J. J. (2009) HIV-1 Nef-mediated inhibition of T cell migration and its molecular determinants. J. Leukoc. Biol. 86, 1171–1178 [DOI] [PubMed] [Google Scholar]
- 26. Campbell E. M., Nunez R., Hope T. J. (2004) Disruption of the actin cytoskeleton can complement the ability of Nef to enhance human immunodeficiency virus type 1 infectivity. J. Virol. 78, 5745–5755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Haller C., Rauch S., Michel N., Hannemann S., Lehmann M. J., Keppler O. T., Fackler O. T. (2006) The HIV-1 pathogenicity factor Nef interferes with maturation of stimulatory T-lymphocyte contacts by modulation of N-Wasp activity. J. Biol. Chem. 281, 19618–19630 [DOI] [PubMed] [Google Scholar]
- 28. Quaranta M. G., Mattioli B., Spadaro F., Straface E., Giordani L., Ramoni C., Malorni W., Viora M. (2003) HIV-1 Nef triggers Vav-mediated signaling pathway leading to functional and morphological differentiation of dendritic cells. FASEB J. 17, 2025–2036 [DOI] [PubMed] [Google Scholar]
- 29. Stolp B., Reichman-Fried M., Abraham L., Pan X., Giese S. I., Hannemann S., Goulimari P., Raz E., Grosse R., Fackler O. T. (2009) HIV-1 Nef interferes with host cell motility by deregulation of Cofilin. Cell Host Microbe 6, 174–186 [DOI] [PubMed] [Google Scholar]
- 30. Lu T. C., He J. C., Wang Z. H., Feng X., Fukumi-Tominaga T., Chen N., Xu J., Iyengar R., Klotman P. E. (2008) HIV-1 Nef disrupts the podocyte actin cytoskeleton by interacting with diaphanous interacting protein. J. Biol. Chem. 283, 8173–8182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yi L., Rosales T., Rose J. J., Chaudhury B., Knutson J. R., Venkatesan S. (2010) HIV-1 Nef binds a subpopulation of MHC-I throughout its trafficking itinerary and down-regulates MHC-I by perturbing both anterograde and retrograde trafficking. J. Biol. Chem. 285, 30884–30905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rose J. J., Foley J. F., Murphy P. M., Venkatesan S. (2004) On the mechanism and significance of ligand-induced internalization of human neutrophil chemokine receptors CXCR1 and CXCR2. J. Biol. Chem. 279, 24372–24386 [DOI] [PubMed] [Google Scholar]
- 33. Venkatesan S., Petrovic A., Locati M., Kim Y. O., Weissman D., Murphy P. M. (2001) A membrane-proximal basic domain and cysteine cluster in the C-terminal tail of CCR5 constitute a bipartite motif critical for cell surface expression. J. Biol. Chem. 276, 40133–40145 [DOI] [PubMed] [Google Scholar]
- 34. Arrode G., Hegde R., Jin Y., Singh D. K., Narayan O., Chebloune Y. (2008) Nef modulates the immunogenicity of Gag encoded in a non-infectious HIV DNA vaccine. Vaccine 26, 3795–3804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gibson S. K., Gilman A. G. (2006) Gialpha and Gbeta subunits both define selectivity of G protein activation by α2-adrenergic receptors. Proc. Natl. Acad. Sci. U.S.A. 103, 212–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Conklin B. R., Farfel Z., Lustig K. D., Julius D., Bourne H. R. (1993) Substitution of three amino acids switches receptor specificity of Gqα to that of Giα. Nature 363, 274–276 [DOI] [PubMed] [Google Scholar]
- 37. Krumins A. M., Gilman A. G. (2006) Targeted knockdown of G protein subunits selectively prevents receptor-mediated modulation of effectors and reveals complex changes in non-targeted signaling proteins. J. Biol. Chem. 281, 10250–10262 [DOI] [PubMed] [Google Scholar]
- 38. Rose J. J., Foley J. F., Yi L., Herren G., Venkatesan S. (2008) Cholesterol is obligatory for polarization and chemotaxis but not for endocytosis and associated signaling from chemoattractant receptors in human neutrophils. J. Biomed. Sci. 15, 441–461 [DOI] [PubMed] [Google Scholar]
- 39. Choe E. Y., Schoenberger E. S., Groopman J. E., Park I. W. (2002) HIV Nef inhibits T cell migration. J. Biol. Chem. 277, 46079–46084 [DOI] [PubMed] [Google Scholar]
- 40. Lee C. M., Gala S., Stewart G. J., Williamson P. (2008) The proline-rich region of HIV-1 Nef affects CXCR4-mediated chemotaxis in Jurkat T cells. Viral Immunol. 21, 347–354 [DOI] [PubMed] [Google Scholar]
- 41. Macia E., Ehrlich M., Massol R., Boucrot E., Brunner C., Kirchhausen T. (2006) Dynasore, a cell-permeable inhibitor of dynamin. Dev. Cell 10, 839–850 [DOI] [PubMed] [Google Scholar]
- 42. Marchese A., Benovic J. L. (2001) Agonist-promoted ubiquitination of the G protein-coupled receptor CXCR4 mediates lysosomal sorting. J. Biol. Chem. 276, 45509–45512 [DOI] [PubMed] [Google Scholar]
- 43. Marchese A., Raiborg C., Santini F., Keen J. H., Stenmark H., Benovic J. L. (2003) The E3 ubiquitin ligase AIP4 mediates ubiquitination and sorting of the G protein-coupled receptor CXCR4. Dev. Cell 5, 709–722 [DOI] [PubMed] [Google Scholar]
- 44. Bhandari D., Robia S. L., Marchese A. (2009) The E3 ubiquitin ligase atrophin interacting protein 4 binds directly to the chemokine receptor CXCR4 via a novel WW domain-mediated interaction. Mol. Biol. Cell 20, 1324–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ingham R. J., Raaijmakers J., Lim C. S., Mbamalu G., Gish G., Chen F., Matskova L., Ernberg I., Winberg G., Pawson T. (2005) The Epstein-Barr virus protein, latent membrane protein 2A, co-opts tyrosine kinases used by the T cell receptor. J. Biol. Chem. 280, 34133–34142 [DOI] [PubMed] [Google Scholar]
- 46. Otte L., Wiedemann U., Schlegel B., Pires J. R., Beyermann M., Schmieder P., Krause G., Volkmer-Engert R., Schneider-Mergener J., Oschkinat H. (2003) WW domain sequence activity relationships identified using ligand recognition propensities of 42 WW domains. Protein Sci. 12, 491–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Winberg G., Matskova L., Chen F., Plant P., Rotin D., Gish G., Ingham R., Ernberg I., Pawson T. (2000) Latent membrane protein 2A of Epstein-Barr virus binds WW domain E3 protein-ubiquitin ligases that ubiquitinate B-cell tyrosine kinases. Mol. Cell Biol. 20, 8526–8535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Harvey K. F., Kumar S. (1999) Nedd4-like proteins. An emerging family of ubiquitin-protein ligases implicated in diverse cellular functions. Trends Cell Biol. 9, 166–169 [DOI] [PubMed] [Google Scholar]
- 49. Hu H., Columbus J., Zhang Y., Wu D., Lian L., Yang S., Goodwin J., Luczak C., Carter M., Chen L., James M., Davis R., Sudol M., Rodwell J., Herrero J. J. (2004) A map of WW domain family interactions. Proteomics 4, 643–655 [DOI] [PubMed] [Google Scholar]
- 50. Pirozzi G., McConnell S. J., Uveges A. J., Carter J. M., Sparks A. B., Kay B. K., Fowlkes D. M. (1997) Identification of novel human WW domain-containing proteins by cloning of ligand targets. J. Biol. Chem. 272, 14611–14616 [DOI] [PubMed] [Google Scholar]
- 51. Lu P. J., Zhou X. Z., Shen M., Lu K. P. (1999) Function of WW domains as phosphoserine- or phosphothreonine-binding modules. Science 283, 1325–1328 [DOI] [PubMed] [Google Scholar]
- 52. Lee C. H., Leung B., Lemmon M. A., Zheng J., Cowburn D., Kuriyan J., Saksela K. (1995) A single amino acid in the SH3 domain of Hck determines its high affinity and specificity in binding to HIV-1 Nef protein. EMBO J. 14, 5006–5015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lee C. H., Saksela K., Mirza U. A., Chait B. T., Kuriyan J. (1996) Crystal structure of the conserved core of HIV-1 Nef complexed with a Src family SH3 domain. Cell 85, 931–942 [DOI] [PubMed] [Google Scholar]
- 54. Kirchhoff F., Münch J., Carl S., Stolte N., Mätz-Rensing K., Fuchs D., Haaft P. T., Heeney J. L., Swigut T., Skowronski J., Stahl-Hennig C. (1999) The human immunodeficiency virus type 1 nef gene can to a large extent replace simian immunodeficiency virus nef in vivo. J. Virol. 73, 8371–8383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Marotti L. A., Jr., Newitt R., Wang Y., Aebersold R., Dohlman H. G. (2002) Direct identification of a G protein ubiquitination site by mass spectrometry. Biochemistry 41, 5067–5074 [DOI] [PubMed] [Google Scholar]
- 56. Wang Y., Marotti L. A., Jr., Lee M. J., Dohlman H. G. (2005) Differential regulation of G protein α subunit trafficking by mono- and polyubiquitination. J. Biol. Chem. 280, 284–291 [DOI] [PubMed] [Google Scholar]
- 57. Torres M. P., Lee M. J., Ding F., Purbeck C., Kuhlman B., Dokholyan N. V., Dohlman H. G. (2009) G protein mono-ubiquitination by the Rsp5 ubiquitin ligase. J. Biol. Chem. 284, 8940–8950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Busconi L., Guan J., Denker B. M. (2000) Degradation of heterotrimeric Gαo subunits via the proteosome pathway is induced by the hsp90-specific compound geldanamycin. J. Biol. Chem. 275, 1565–1569 [DOI] [PubMed] [Google Scholar]
- 59. Fischer T., De Vries L., Meerloo T., Farquhar M. G. (2003) Promotion of Gαi3 subunit down-regulation by GIPN, a putative E3 ubiquitin ligase that interacts with RGS-GAIP. Proc. Natl. Acad. Sci. U.S.A. 100, 8270–8275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ogasawara J., Sakurai T., Rahman N., Kizaki T., Hitomi Y., Ohno H., Izawa T. (2004) Acute exercise alters Gαi2 protein expressions through the ubiquitin-proteasome proteolysis pathway in rat adipocytes. Biochem. Biophys. Res. Commun. 323, 1109–1115 [DOI] [PubMed] [Google Scholar]
- 61. Naviglio S., Pagano M., Romano M., Sorrentino A., Fusco A., Illiano F., Chiosi E., Spina A., Illiano G. (2004) Adenylate cyclase regulation via proteasome-mediated modulation of Gαs levels. Cell. Signal. 16, 1229–1237 [DOI] [PubMed] [Google Scholar]
- 62. Tang T., Gao M. H., Miyanohara A., Hammond H. K. (2008) Gαq reduces cAMP production by decreasing Gαs protein abundance. Biochem. Biophys. Res. Commun. 377, 679–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hamilton M. H., Cook L. A., McRackan T. R., Schey K. L., Hildebrandt J. D. (2003) γ2 subunit of G protein heterotrimer is an N-end rule ubiquitylation substrate. Proc. Natl. Acad. Sci. U.S.A. 100, 5081–5086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Obin M., Lee B. Y., Meinke G., Bohm A., Lee R. H., Gaudet R., Hopp J. A., Arshavsky V. Y., Willardson B. M., Taylor A. (2002) Ubiquitylation of the transducin βγ subunit complex. Regulation by phosducin. J. Biol. Chem. 277, 44566–44575 [DOI] [PubMed] [Google Scholar]
- 65. Cappell S. D., Baker R., Skowyra D., Dohlman H. G. (2010) Systematic analysis of essential genes reveals important regulators of G protein signaling. Mol. Cell 38, 746–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yang L. V., Radu C. G., Wang L., Riedinger M., Witte O. N. (2005) Gi-independent macrophage chemotaxis to lysophosphatidylcholine via the immunoregulatory GPCR G2A. Blood 105, 1127–1134 [DOI] [PubMed] [Google Scholar]
- 67. Sozzani S., Zhou D., Locati M., Rieppi M., Proost P., Magazin M., Vita N., van Damme J., Mantovani A. (1994) Receptors and transduction pathways for monocyte chemotactic protein-2 and monocyte chemotactic protein-3. Similarities and differences with MCP-1. J. Immunol. 152, 3615–3622 [PubMed] [Google Scholar]
- 68. Maghazachi A. A., al-Aoukaty A., Schall T. J. (1994) C-C chemokines induce the chemotaxis of NK and IL-2-activated NK cells. Role for G proteins. J. Immunol. 153, 4969–4977 [PubMed] [Google Scholar]
- 69. Shi G., Partida-Sánchez S., Misra R. S., Tighe M., Borchers M. T., Lee J. J., Simon M. I., Lund F. E. (2007) Identification of an alternative Gαq-dependent chemokine receptor signal transduction pathway in dendritic cells and granulocytes. J. Exp. Med. 204, 2705–2718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Misra R. S., Shi G., Moreno-Garcia M. E., Thankappan A., Tighe M., Mousseau B., Kusser K., Becker-Herman S., Hudkins K. L., Dunn R., Kehry M. R., Migone T. S., Marshak-Rothstein A., Simon M., Randall T. D., Alpers C. E., Liggitt D., Rawlings D. J., Lund F. E. (2010) Gαq-containing G proteins regulate B cell selection and survival and are required to prevent B cell-dependent autoimmunity. J. Exp. Med. 207, 1775–1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Welker R., Kottler H., Kalbitzer H. R., Kräusslich H. G. (1996) Human immunodeficiency virus type 1 Nef protein is incorporated into virus particles and specifically cleaved by the viral proteinase. Virology 219, 228–236 [DOI] [PubMed] [Google Scholar]
- 72. Qiao X., He B., Chiu A., Knowles D. M., Chadburn A., Cerutti A. (2006) Human immunodeficiency virus 1 Nef suppresses CD40-dependent immunoglobulin class switching in bystander B cells. Nat. Immunol. 7, 302–310 [DOI] [PubMed] [Google Scholar]
- 73. Xu W., Santini P. A., Sullivan J. S., He B., Shan M., Ball S. C., Dyer W. B., Ketas T. J., Chadburn A., Cohen-Gould L., Knowles D. M., Chiu A., Sanders R. W., Chen K., Cerutti A. (2009) HIV-1 evades virus-specific IgG2 and IgA responses by targeting systemic and intestinal B cells via long-range intercellular conduits. Nat. Immunol. 10, 1008–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Muratori C., Cavallin L. E., Krätzel K., Tinari A., De Milito A., Fais S., D'Aloja P., Federico M., Vullo V., Fomina A., Mesri E. A., Superti F., Baur A. S. (2009) Massive secretion by T cells is caused by HIV Nef in infected cells and by Nef transfer to bystander cells. Cell Host Microbe 6, 218–230 [DOI] [PubMed] [Google Scholar]
- 75. Amella C. A., Sherry B., Shepp D. H., Schmidtmayerova H. (2005) Macrophage inflammatory protein 1α inhibits postentry steps of human immunodeficiency virus type 1 infection via suppression of intracellular cyclic AMP. J. Virol. 79, 5625–5631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Cartier C., Hemonnot B., Gay B., Bardy M., Sanchiz C., Devaux C., Briant L. (2003) Active cAMP-dependent protein kinase incorporated within highly purified HIV-1 particles is required for viral infectivity and interacts with viral capsid protein. J. Biol. Chem. 278, 35211–35219 [DOI] [PubMed] [Google Scholar]
- 77. Rabbi M. F., Al-Harthi L., Roebuck K. A. (1997) TNFα cooperates with the protein kinase A pathway to synergistically increase HIV-1 LTR transcription via downstream TRE-like cAMP response elements. Virology 237, 422–429 [DOI] [PubMed] [Google Scholar]
- 78. Cho H., Kehrl J. H. (2007) Localization of Giα proteins in the centrosomes and at the midbody. Implication for their role in cell division. J. Cell Biol. 178, 245–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Slessareva J. E., Routt S. M., Temple B., Bankaitis V. A., Dohlman H. G. (2006) Activation of the phosphatidylinositol 3-kinase Vps34 by a G protein α subunit at the endosome. Cell 126, 191–203 [DOI] [PubMed] [Google Scholar]
- 80. Wilkie T. M., Kinch L. (2005) New roles for Gα and RGS proteins. Communication continues despite pulling sisters apart. Curr. Biol. 15, R843–R854 [DOI] [PubMed] [Google Scholar]