

Background: miR-140 is down-regulated in non-invasive and invasive breast tumors compared with normal breast tissues.
Results: Estrogen receptor α signaling down-regulates miR-140 in breast cancer where miR-140 targets embryonic transcription factor SOX2.
Conclusion: ERα signaling regulates breast tumor-initiating cells through modulating miR-140 targeting of SOX2.
Significance: Understanding cancer stem cell biology may reveal biomarkers or targets for therapeutic intervention.
Keywords: Breast Cancer, Cancer Stem Cells, Estrogen, Estrogen Receptor, MicroRNA, SOX2
Abstract
Several reports have indicated that miR-140, a possible tumor suppressor microRNA (miR), is down-regulated in breast tumors compared with normal breast tissues. However, the role of miR-140 in breast tumorigenesis is unclear. We initiated studies that examined estrogen receptor α (ERα) signaling in the tissue-specific regulation of miR-140 in breast cancer. We found that estrogen stimulation of ERα-positive breast cancer cells resulted in decreased miR-140 expression. We performed promoter analyses and examined predicted ERα binding elements in the miR-140 promoter using luciferase constructs of a miR-140 promoter deletion series. Our studies revealed that ERα binds to one specific estrogen response element flanking the miR-140 promoter and consequently suppresses miR-140 transcription. We found that the stem cell self-renewal regulator SOX2 is a novel target of miR-140, and that this miR-140/SOX2 pathway critically regulates breast tumor-initiating cell survival, providing a new link between ERα signaling and breast cancer stem cell maintenance.
Introduction
Long-term exposure to high levels of estrogens is considered a major risk factor for breast cancer (1). Estrogen exposure contributes to breast tumorigenesis through estrogen receptor (ER)2 signaling and through genotoxic estrogen metabolites (2). Growing evidence suggests that cancer stem cells (CSCs) participate in the processes of tumor initiation, malignant progression, drug resistance, disease recurrence, and treatment failure (3). CSCs, like embryonic stem cells, are capable of self-renewing cell divisions (4) and express the key pluripotency-associated transcription factors including SOX2, NANOG, and OCT4 (5–7). In particular, SOX2 physically interacts with OCT4 and NANOG forming a protein complex that binds the promoters of numerous stem cell differentiation factors, suppressing their expression (8). Hyper- or hypoactivation of SOX2, NANOG, and OCT4 may lead to aberrant self-renewal in cancer cells. Recent studies have shown that SOX2 overexpression leads to aberrant stem cell self-renewal signaling in breast cancer cells (5). Normal breast tissue expresses very low levels of SOX2 but breast carcinomas (e.g. DCIS) express elevated levels of SOX2, suggesting that activation of SOX2 may contribute to malignant progression of breast cancer (9, 10).
Recently, estrogen signaling has been implicated in the regulation of breast CSCs (11). Estrogen treatment of ERα-positive breast cancer cells was found to increase mammosphere formation capacity, a surrogate measure of CSC renewal (12). Furthermore, estrogen treatment was found to increase the frequency of CD44+/CD24- breast CSCs. One proposed mechanism for ERα regulation of CSCs involved transcriptional control of the SOX2/NANOG/OCT4 self-renewal pathway. For example, ERα was shown to be associated with the promoter region of OCT4, and the CSC inhibitor, Metformin, was found to inhibit ERα association with the OCT4 promoter, potentially interfering with CSC self-renewal (12).
The small non-coding RNAs, microRNAs (miRs), are also contributors to the initiation and progression of human cancers. Loss of a subset of tumor suppressor miRs in cancer cells can promote angiogenesis, growth advantage, tissue invasion, and metastasis (13). In breast cancer, these miR networks are involved in a complex relationship with ERα signaling in which numerous miRs target ERα and important co-signaling molecules and likewise ERα regulates the transcription and maturation of numerous miRs (14–16). Therefore, it is possible if not likely that miRs are involved in ERα regulation of breast CSCs.
MiR-140 was first identified in chondrocytes where it is abundantly expressed and is important in cartilage development and homeostasis (17). Reduced expression of miR-140 is observed in osteoarthritic chondrocytes and miR-140−/− mice display early onset osteoarthritic-like changes in articular cartilage (17). MiR-140 is encoded within intron 16 of Wwp2, an E3 ubiquitin ligase (18). In chondrocytes, tissue specific control of miR-140 is regulated by a trio of SOX proteins that enhance miR-140 transcription through direct interaction with a specific SOX response element on intron 10, a region possessing in vivo promoter activity (19). In addition, Il-1 beta, a cytokine important in the pathogenesis of osteoarthritis, has been shown to down-regulate miR-140 expression in chondrocytes (20). Finally, Wnt/B-catenin signaling and TGF-β signaling have also been shown to reduce miR-140 expression in chondrocytes (18, 21). Among the previously confirmed targets of miR-140 are Sp1 (18), BMP2 (22), Smad3 (21), IGFBP-5 (23), HDAC4 (24), and ADAMTS5 (17) mRNAs.
In addition to its role in regulating chondrocytes, miR-140 has been found to be important to numerous other tissues and cell types. miR-140 expression has been detected in the brain, breast, lung, colon, ovary, and testis (25). Expression profiling of tumors and normal tissues has revealed a possible tumor suppressive role for miR-140 in many cancers. miR-140 expression is down-regulated in ovarian (26), lung (27), colon (24), osteosarcoma (24) and basal cell carcinomas (28). Recently, deep sequencing experiments have revealed miR-140 down-regulation in early in situ breast tumors, invasive breast tumors, and in numerous breast cancer cell lines (29). Here, we have identified tissue specific regulation of miR-140 expression by ERα in mammary epithelial cells and in breast cancer cells. Subsequently, we examined a possible role for miR-140 in ERα regulation of breast tumor-initiating cells. We found that the well-known embryonic stem cell self-renewal regulator, SOX2 (30), is targeted by miR-140 and is critical for breast tumor-initiating cell maintenance.
EXPERIMENTAL PROCEDURES
Cell Culture and Reagents
Breast cancer cell lines MCF-7 and T47D and embryonic kidney cell line 293T (HEK-293T) were grown in Dulbecco's modified Eagle's medium (DMEM) + 10% fetal bovine serum (FBS, HyClone; Rockford, IL) and 1% l-glutamine (Invitrogen; Carlsbad, CA). Non-tumorigenic mammary epithelial cell line MCF10A was grown in DMEM/F-12 medium (Invitrogen) supplemented with 10 μg/ml insulin (Sigma), 100 ng/ml cholera toxin (Sigma), 0.5 μg/ml hydrocortisone (Sigma), 20 ng/ml EGF (Invitrogen), and 5% horse serum (Invitrogen). MCF10A stably transfected with ERα (ERIN) cells (31) were grown in medium lacking phenol red and containing charcoal stripped serum. Cells were incubated in an atmosphere containing 5% CO2 at 37 °C. Reagents used in this study include 17-β-estradiol (E2) (Sigma) and bisphenol A (BPA) (Sigma) dissolved in ethanol.
Quantitative Real-time PCR
Total RNA was extracted using TRIzol (Invitrogen). Total RNA from Invasive Ductal Carcinoma (IDC) patient tumor tissue and normal tissue controls were extracted with RNeasy Lipid Tissue Midi Kit (Qiagen) following manufacturer's instructions. qRT-PCR was carried out using the Light Cycler 480II instrument (Roche). Small RNA was converted to cDNA using the First-Strand Synthesis Kit (SABioscienses; Flat Lake, MD). Analysis of miR expression was performed using miR specific (miR-140) primer sets (SABiosciences) normalizing to U6 snRNA expression as a control. Analysis of mRNA expression was performed using primers specific for SOX2 (F: 5′-TGTCATTTGCTGTGGGTGAT-3′; R: 5′-GGGGTGCAAAAGAGGAGAGT-3′), ERα (F: CTCTCCCACATCAGGCACA R: CTTTGGTCCGTCTCCTCCA) and normalized to GAPDH mRNA (F: GAAGGTGAAGGTCGGAGTC, R: GAAGATGGTGATGGGATTTC).
Western Blotting and Immunohistochemistry
Protein expression was examined by Western blotting using rabbit polyclonal antibodies for ERα (HC-20, Santa Cruz Biotechnology; Santa Cruz, CA) and SOX2 (Neuromics; Edina, MN). Protein expression was detected by chemiluminescence (ECL, Amersham Biosciences; Arlington Heights, IL). Expression of β-actin (Sigma) was used as a loading control. Formalin fixed, paraffin-embedded human breast cancer and normal tissue samples were obtained from the Tissue Bank of the University of Maryland School of Medicine. Sections were deparaffinized using xylene. Antigens were retrieved by boiling in sodium Citrate (10 mm, pH 6.0). Polyclonal rabbit anti-SOX2 or anti-ERα antibody (1:200) was applied at 4 °C overnight followed by a biotin conjugated bovine anti-rabbit secondary antibody (1:250, Santa Cruz Biotechnology) at room temperature for 1 h. Avidin-biotin peroxidase substrate kit (Vector Laboratories; Burlingame, CA) was used to develop brown precipitate. Hematoxylin was utilized for nuclei staining
Cloning, miR-140 Sponge Inhibitor, Transfections, and Luciferase Assay
A 596-bp genomic DNA fragment [NCBI Reference Sequence: NW_926462.1, strand (+), nucleotides 23546875–23547470] encompassing the miR-140 sequence and its upstream and downstream flanking sequence was amplified by PCR using genomic DNA of normal human mammary epithelial (HME) cells as the template and cloned into XbaI and BamHI sites of pHIV-Zsgreen vector. The following primers were used F: 5′-ATCGACTCTAGAAGAGAGAGAGAGCGCTGTGG-3′, and R: 5′-ATCGACGGATCCC ATGCTGCCTTCAGATGAGA-3′. The miR-140 expression plasmid was confirmed by sequencing.
The 3′-UTR of SOX2 was amplified by PCR using genomic DNA of HME cells as the template and cloned into pGL3 vector (Promega; Madison, WI). The following primers were used for PCR F: 5′-ACTGAAGCTAGCACACTGCCCCTCTCACACAT-3′ and R: 5′-ACTGAACTCGAGTGCTTTCTTGGCTGAGCAC-3′. The potential miR-140-binding site in the SOX2 3′-UTR was then mutated using the Generate Site-Directed Mutagenesis System (Invitrogen). The following mutagenesis primers (5′-ATAAGTACTG GCGAACCATCTCAGAGCTCTTGTTTAAAAAGGGCAAAAG-3′, and 5′-CTTTTGCCCTTTTT AAACAAGAGCTCTGAGATGGTTCGCCAGTACTTAT-3′) were used, and the resulting mutant contains three point mutations: C(T to A) G (T to A) G (G to C) T.
To obtain the luciferase constructs, fragments from the Wwp2 gene promoter region were amplified by PCR and cloned into KpnI and BglII sites of pGL3 basic vector. The following primers were used: p140R 5′-ATCGACAGATCTCACCCATTTCGGAGCAAC-3′, p140L1 5′-ATCGACGGTACCGGGCAGAGTAGGTGGCATT-3′, p140L2 5′-ATCGACGGTACCTTGCCAGGAAATAGAAATACAGA-3′, and p140L3 5′-ATCGACGGTACCCAAAAAGTAGAGACTAAGTGTTCAACC-3′. The estrogen binding sites were predicted using Genomatix software and alignment of the conserved estrogen response element (ERE) sequence, AGGTCANNNTGGACCT, with the miR-140 promoter. Three potential EREs (−1042 to −1020, −614 to −592 and −79 to −50) were identified. To mutate the ERE(−79 to −50), a genomic DNA fragment was amplified using p140R and p140L1 and cloned into pGEM-Teasy vector (Promega). Two synthesized DNA fragment, 5′-CTAGGTGGCCCGCACCTGCGA GCCTGGCCAGCCTCTTCCGCGTGTGGACAGCGCGCGCCGTGTCGACAGCCGGCCCGAGTGCGGGCGGTGGAAGGCGGAAGTAGGAGAGGAGTTCG-3′ and 5′GCGCCGAACTCCTCT CCTACTTCCGCCTTCCACCGCCCGCACTCGGGCCGGCTGTCGACACGGCGCGCGCTGTCCACACGCGGAAGAGGCTGGCCAGGCTCGCAGGTGCGGGCCAC-3′ were then annealed and inserted into the resulting plasmid digested with AvrII and KasI. The synthesized DNA fragment contains a mutated ERE (−79 to −50), CGTGTGGACAGCGCGCGCCGTGTCGACAGC, in contrast to the wild type ERE, GGTCAGGTGACCGCGCGCGGTCACGTGACC. The mutated promoter fragment was then released by XhoI and BglII treatment and cloned into the pGL3-miR-140-promoter plasmid digested with XhoI and BglII.
The miR-140 sponge was constructed using a method reported by Li et al. (32) Briefly, two oligonucleotides, 5′-TCGAGCTACCATAGGATTAACCACTGCTACCATAGGATTAACCACTGCTACCATAGGATTAACCACTGCTACCATAGGATTAACCACTGCTACCATAGGATTAACCACTGCTACCATAGGATTAACCACTGCTACCATAGGATTAACCACTGCTACCATAGGATTAACCACTGGGGCC-3′, and 5′CCAGTGGTTAATCCTATGGTAGCAGTGGTTAATCCTATGGTAGCAGTGGTTAATCCTATGGTAGCAGTGGTTAATCCTATGGTAGCAGTGGTTAATCCTATGGTAGCAGTGGTTAATCCTATGGTAGCAGTGGTTAATCCTATGGTAGCAGTGGTTAATCCTATGGTAGC-3′, containing 8 tandem miR-140-binding sites were annealed and cloned into the 3′-UTR of a gfp gene in the pcDNA5-CMV-d2eGFP vector (Invitrogen). The fusion construct of the gfp gene and the miR-140 sponge was then subcloned into the pBABE-puro vector. As a control, the gfp gene including its corresponding 3′-UTR alone was also cloned into pBABE0-puro.
Cells were transfected with miR-140 expression vector, siRNA for ERα, miR-140 promoter deletion series constructs, miR-140 sponge or control vectors using Lipofectamine 2000 (Invitrogen) according to manufacturer's instructions. For dual luciferase assays, cells were transfected with reporter plasmids in combination with Renilla luciferase phGR-TK (Promega) as internal control. Luciferase activity was measured 48 h after transfection using the Dual-Luciferase Reporter Assay System (Promega) and a luminometer.
Chromatin Immunoprecipitation
ChIP was carried out as described previously (33). Cells were cross-linked with 1% formaldehyde followed by sonication. Chromatin was incubated with antibodies against ERα overnight at 4 °C for immunoprecipitation. Rabbit IgG was used a negative control. Immunoprecipitated chromatin was analyzed by qRT-PCR using primers for the miR-140 promoter (ERE CHIPL 5′-GACGATAAAAAGGCCTCCTC-3′; ERE CHIPR 5′-CTCTCCTACTTCCGCCTTCC-3′). Results were normalized to input.
Mammosphere and Flow Cytometry
For growth of mammospheres, cells were separated to single cells using cell dissociation buffer (Millipore; Billerica, MA) and 40 μm cell strainers (Fisher Scientific; Pittsburgh, PA) and counted. 20,000 cells/ml were seeded in 6-well plates coated with 2% polyhema (Sigma) dissolved in 95% ethanol. After 7 days, mammospheres > 100 μm were counted. Flow cytometry was performed on cells co-stained with CD44-APC and CD24-PE antibodies (BD Pharmingen, San Diego, CA).
Statistical Analysis
Statistical analysis was performed by Student's t test. p values of < 0.05 (*) were considered significant. Data are presented as mean ± S.E. Data were analyzed using GraphPad Prism 4.0 software.
RESULTS
Identification of a New Target of miR-140 in Human Breast Cancer Cells, the Stem Cell Self-renewal Regulator SOX2
Previous reports have indicated that miR-140 is down-regulated in breast tumors compared with normal breast tissues (29). To better understand the regulatory role the prospective tumor suppressor miR-140 might play in breast cancer cells, we sought to identify the pathways regulated by miR-140 in mammary epithelial cells. We used TargetScan 6.0 (35) to examine the predicted targeted mRNAs of miR-140. Among prospective miR-140 targets, we selected SOX2 mRNA (targeted by miR-140–3p) for further investigation since: SOX2 expression is frequently altered in human breast cancers (9), subtle changes in SOX2 dose dictate critical outcomes in cancer stem cell self-renewal (5, 36), and SOX2 regulation is poorly understood in mammary epithelium. SOX2 mRNA 3′-UTR contains a predicted 7-mer match to the miR-140 seed region (Fig. 1A). We examined expression of miR-140 and SOX2 mRNA by qRT-PCR in mammary epithelial cells and in breast cancer cells. miR-140 and SOX2 mRNA display inverse expression in non-tumorigenic MCF10A mammary epithelial cells and MCF-7 breast cancer cells (Fig. 1B) suggesting a potential regulatory relationship.
FIGURE 1.

The transcription factor SOX2 is a novel target for miR-140 in breast cancer cells. A, targetScan 6.0 predicts a miR-140 response element in the SOX2 mRNA 3′-UTR. An illustration of the SOX2 3′-UTR as well as the seed sequence of miR-140. B, inverse expression of miR-140 and SOX2. Relative expression of miR-140 and SOX2 in MCF10A and MCF-7 cells as determined by qRT-PCR. C, (left) HEK-293T cells were transfected with 2 μg pGL3 luciferase vector containing either wild-type SOX2 3′-UTR or mutant SOX2 3′-UTR (abolishing miR-140 targeting). Cells were co-transfected along with 2 μg miR-140 expression vector for 48h and lysed and luciferase activity was measured with a luminometer. Right, MCF-7 cells were transfected with miR-140 expression vector and SOX2 mRNA was measured by qRT-PCR, normalizing to GAPDH mRNA. D, MCF10A cells were transfected with 2 μg of miR-140 sponge inhibitor for 24 h, and SOX2 protein was measured by Western blot, normalizing to β-actin expression. n = 3, mean ± S.E.
To validate whether miR-140 could directly target the SOX2 mRNA 3′-UTR, we performed luciferase reporter assays by cloning the 3′-UTR of SOX2 into luciferase reporter constructs. We also constructed mutant luciferase reporters by mutating 4 bases of the predicted miR-140 seeding site by PCR-based mutagenesis to abolish miR-140 targeting. HEK-293T cells, which lack miR-140 expression (data not shown), were co-transfected with wild type or mutant SOX2 3′-UTR luciferase vectors in addition to miR-140 expression vector. Fig. 1C shows that miR-140 overexpression decreased wild-type but not mutant SOX2 3′-UTR reporter activity compared with controls. To confirm miR-140 regulation of SOX2 mRNA in breast cancer, we overexpressed miR-140 in MCF-7 breast cancer cells and examined SOX2 mRNA levels by qRT-PCR. Similar to our results in HEK-293T cells, miR-140 overexpression caused marked inhibition of SOX2 mRNA levels in MCF-7 breast cancer cells. In contrast, miR-140 silencing through miR-140 sponge inhibitor increased SOX2 expression at both mRNA and protein levels in MCF10A cells (Fig. 1D). Together, these results suggest that miR-140 negatively regulates SOX2 expression in normal mammary epithelium and in breast cancer cells.
ERα Signaling Inhibits miR-140 Expression in Breast Cancer Cells
Results from Fig. 1B revealed an inverse relationship between miR-140 and SOX2 mRNA in normal mammary epithelium and breast cancer cells. In addition, these findings pointed to the possibility that miR-140 and SOX2 levels may be impacted by ERα status and as a result we sought to characterize any role ERα may play in regulating miR-140 expression. MCF10A cells are ERα-negative, non-tumorigenic breast epithelial cells that show insensitivity to hormone stimulation (e.g. 17-β-estradiol (E2)). MCF10A cells engineered to express ERα (MCF10A-ERIN) (see Fig. 2A) are non-cancerous, demonstrate a growth response to E2 (31), and are used as a cell culture model for studying ERα signaling. We examined miR-140 expression in MCF10A and MCF10A-ERIN cells by qRT-PCR. The expression of miR-140 was significantly decreased in MCF10A-ERIN cells as compared with MCF10A cells (Fig. 2B). As expected, we found that E2 treatment failed to alter miR-140 levels in MCF10A (ERα-negative) cells. However, we found that E2 notably inhibited miR-140 expression in MCF10A-ERIN (ERα-positive) cells, suggesting that ERα may be involved in the regulation of miR-140 expression in normal mammary epithelium.
FIGURE 2.

Estrogen receptor signaling regulates miR-140 and SOX2 expression in normal breast tissue and in breast cancer cells. A, Western blot showing ERα expression in non-tumorigenic mammary epithelial cells (MCF10A) and MCF10A cells stably transfected with ERα (MCF10AERIN) cells, normalizing to β-actin expression. ERα-positive MCF-7 breast cancer cells are included as a control. B, MCF10A and MCF10AERIN were treated with 10 nm estradiol (E2) or EtOH control for 24 h. miR-140 and SOX2 expression was measured by qRT-PCR normalizing to U6 snRNA or GAPDH mRNA, respectively. C, MCF-7 cells were treated with 10 nm E2 for 24 h and miR-140 and SOX2 expression was measured by qRT-PCR. Pretreatment with 50 nm ERα siRNA was used to block E2-induced changes in mir-140 expression. n = 3 ± S.E. D, immunohistochemistry staining of normal breast tissue and breast tumor tissue (IDC associated with DCIS) with SOX2 and ERα antibody. E, qRT-PCR analysis of miR-140 expression in normal breast tissue and fresh tumor tissue from patients with IDC, normalizing to U6 snRNA.
We followed up our examination of miR-140 regulation in mammary epithelial cells by examining miR-140 regulation in breast cancer cells. We examined miR-140 regulation in the ERα-positive breast cancer cell line MCF-7 (Fig. 2C). We transfected MCF-7 cells with ERα siRNA for 72 h and examined miR-140 expression by qRT-PCR. We observed that E2 stimulation suppressed miR-140 levels in MCF-7 cells expressing ERα. However, ERα knockdown completely blocked the estrogen-induced inhibitory effect on miR-140. Based on these combined observations, we concluded that ERα signaling inhibits miR-140 expression in both mammary epithelial cells and breast cancer cells. Furthermore, we found that E2 stimulation also impacted SOX2 expression in MCF10A-ERIN cells and MCF-7 breast cancer cells. In contrast to our findings regarding miR-140, E2 treatment was found to stimulate SOX2 expression in cells expressing ERα (Fig. 2, B and C).
Next, we sought to confirm the significance of our findings from cell culture models (regarding the inverse relationship between miR-140 and SOX2) in breast cancer patient tissues. Using IHC, we examined SOX2 expression in ERα-positive, hormone-responsive breast tumor tissues of Invasive Ductal Carcinoma (IDC) associated with Ductal Carcinoma In Situ (DCIS). We observed significantly increased SOX2 staining in breast cancer tissues compared with normal tissue controls (Fig. 2D). Likewise, we examined miR-140 expression in IDC patient tumor tissues (n = 8) by qRT-PCR. In agreement with previous studies, we observed dramatic down-regulation of miR-140 in breast tumors compared with normal breast tissue controls (Fig. 2E).
Identification of ERα Binding Sites in miR-140 Promoter
The current understanding of ERα signaling is that E2 binding to ERα leads to subsequent dimerization of receptors and recruitment and binding to estrogen response elements (ERE) on ERα target gene promoters (34). To investigate the potential direct regulatory involvement of ERα in controlling miR-140 expression, we analyzed the predicted transcription factor binding sites located within 2 kb region upstream of the transcriptional start site of miR-140 using Genomatix software. Among the predicted transcription factor response elements, 3 potential EREs were found in the miR-140 promoter (−1042/−1020, −614/−592, and −79/−50). To examine whether these predicted EREs were indeed involved in the regulation of miR-140 promoter activity, we cloned the miR-140 promoter into a luciferase reporter construct and created a deletion series of miR-140 promoter reporters lacking these putative EREs by depleting or mutating the EREs (Fig. 3A). ERα-positive MCF-7 breast cancer cells were transfected with wild type or mutant miR-140 promoter luciferase reporters in the presence or absence of estrogens. Fig. 3A shows the results of miR-140 promoter activity assays for wild type and mutant miR-140 luciferase constructs including the deletion of EREs at −1042/−1020 and −614/−592 or the mutation of the ERE at −79/−50. The addition of E2 resulted in decreased reporter activity for the wild-type promoter. E2-dependent suppression of miR-140 promoter activity was reversed when the −79/−50 ERE was mutated. Deletion of the ERE at −1042/−1020 and −614/−592 had negligible impact on the effects of estrogen action, suggesting that the presence of the −79/−50 ERE may be more critical for estrogen-receptor binding, which in turn enables ERα to inhibit miR-140 promoter activity.
FIGURE 3.
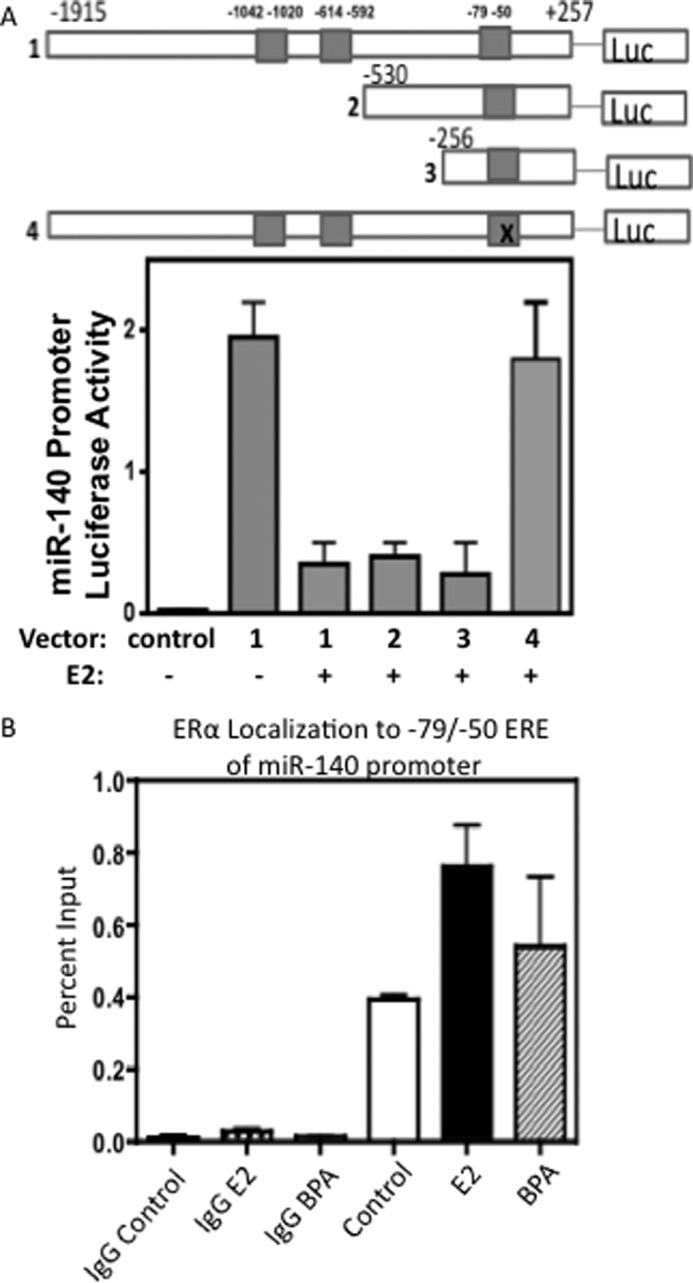
A, identification of Estrogen Response Elements (ERE) in the miR-140 Promoter. The miR-140 promoter region, ∼2 kb of the region upstream of the TSS of Wwp2 gene, was cloned into a luciferase reporter to examine promoter activity. Predicted ERE sites (3) in the miR-140 region were mutated or deleted in a series of truncated reporters. MCF-7 cells were transfected with 2 μg of reporter plasmid along with control Renilla luciferase reporter for dual-luciferase activity assays. Cells were treated with 10 nm E2 or EtOH vehicle control. After 24 h cells were lysed and assayed for luciferase activity. Control reporter possessed no promoter activity. n = 3 ± S.E. B, estrogen stimulation enhances ERα binding at the miR-140 promoter. Results for miR-140 promoter region ChIP for ERα antibody. Cross-linked, sonicated chromatin was immunoprecipitated with ERα antibody or negative control rabbit IgG antibody following stimulation with 10 nm E2 or 10 nm BPA or EtOH control. qRT-PCR was carried out for miR-140 promoter region (−79/−50 ERE). n = 2, mean ± S.E.
Estrogen Stimulation Induces ERα Binding at the miR-140 Promoter
To validate direct association of ERα with miR-140 promoter, we performed chromatin immunoprecipitation (ChIP) analysis in MCF-7 breast cancer cells (ERα-positive) for the putative ERα binding elements (−79/−50) within the miR-140 promoter using ERα antibody. ChIP results (Fig. 3B) revealed ERα recruitment to the miR-140 promoter in response to E2 stimulation (1.93-fold that of control cells), suggesting ERα directly associates with this promoter region. Similar results were observed in bisphenol A (BPA, a xenoestrogen)-treated cells. Given these findings in combination with our observations that E2 treatment inhibits miR-140 levels in human mammary epithelial cells and in breast cancer cells (Fig. 2, B and C), we conclude that ERα binds to a specific promoter element (−79/−50) of miR-140, where ERα inhibits the transcription of miR-140.
SOX2 Is Required for Tumor-initiating Cell Survival in Breast Cancer Cells
SOX2 is an embryonic transcription factor that regulates embryonic stem cell self-renewal that is overexpressed in breast cancers (9, 30). We examined the possibility that SOX2 overexpression may promote breast tumor-initiating cell self-renewal. It is well established that the surface markers CD44 and CD24 can be used to separate breast CSCs from non-stem cancer cells (NSCCs) and that CD44high/CD24low subpopulations demonstrate increased tumor-initiating capacity in xenograft models of breast cancer (37). We examined the impact of SOX2 knockdown on this previously described subpopulation enriched in tumor-initiating cells. We observed a dramatic decrease in the CD44high/CD24low subpopulation following stable knockdown of SOX2 by shRNA in MCF-7 breast cancer cells (0.32% compared with 13.2% for control cells) (Fig. 4A). Likewise, we found that transient SOX2 overexpression resulted in a dramatic increase in this subpopulation (22.5%) in MCF-7 cells. Knockdown and overexpression of SOX2 are shown by qRT-PCR analysis in Fig. 4B. We observed no significant changes in ERα levels following knockdown or overexpression of SOX2. These results indicate the significance of SOX2 in maintaining tumor-initiating cell survival in ERα-positive breast cancer cells.
FIGURE 4.

A, SOX2 is required for tumor-initiating cell survival in MCF-7 breast cancer cells. MCF-7 breast cancer cells were stably transfected with SOX2 shRNA (followed by puromycin selection) or transiently transfected with 2 μg of SOX2 expression construct for 24 h. Cells were stained with CD44-APC and CD24-PE antibodies and CD44+/CD24- subpopulations were examined by flow cytometry. n = 3. B, qRT-PCR showing SOX2 mRNA and ERα mRNA levels following SOX2 overexpression or knockdown or miR-140 overexpression, normalizing to GAPDH mRNA.
ERα Signaling Regulates Tumor-initiating Cell Survival in Part through a miR-140/SOX2 Pathway
Mammosphere culture is performed in attachment-free, serum-free, and non-differentiating conditions where differentiated breast cancer cells die from anoikis while other cells remain viable and grow (from clonal expansion of single cells with self-renewal properties) as non-adherent spheres enriched in breast CSCs and progenitor cells (38, 39). Using mammosphere assays to examine in vitro self-renewal of breast cancer cells, we tested the role of miR-140 and SOX2 in regulating stemness properties of breast cancer cells. We examined mammosphere formation in MCF-7 cells following transfection with miR-140 or SOX2 expression vectors (lacking a 3′-UTR). As a control, MCF-7 cells maintained in estrogen-free conditions (media containing stripped serum and lacking phenol red) were also cultured as mammospheres. These cells produced smaller, fewer mammospheres compared with MCF-7 cells cultured in the presence of estrogens. We tested the impact of restoring miR-140 expression on mammosphere growth and found that miR-140-overexpressing MCF-7 cells produced smaller and fewer mammospheres compared with control MCF-7 cells (Fig. 5, A and B). Furthermore, we examined the impact of SOX2 overexpression on mammosphere formation, finding that SOX2-overexpressing MCF-7 cells produced more numerous, larger spheres compared with controls. Finally, we found that co-transfection of miR-140 + SOX2 resulted in increased mammosphere formation because the SOX2 expression construct lacking its 3′-UTR is protected from miR-140 targeting, demonstrating the importance of SOX2 in miR-140 regulation of tumor-initiating cell growth. Similar trends were observed in subsequent sphere passages. These results provide support for a novel mechanism by which ERα signaling might regulate breast CSCs, through regulation of miR-140 expression and subsequent targeting of SOX2 mRNA.
FIGURE 5.

The miR-140/SOX2 pathway regulates mammosphere formation of breast cancer cells. A, MCF-7 cells were transfected with 2 μg of miR-140 expression vector, 2 μg of SOX2 expression construct, or co-transfected with both. Transfected MCF-7 cells were collected using non-enzymatic dissociation buffer, separated to single cells by passing through 40 μm cell strainer and seeded at 20,000 cells/ml on attachment free 6-well plates coated with 2% poly-HEMA. After 7 days, mammospheres greater than 100 μm were counted. For subsequent passages, mammospheres were collected, separated into single cells, and re-seeded at 10,000 cells/ml. Pictures taken of primary passage mammospheres at 7 days are shown. B, average results from first and second passage mammosphere experiments were quantified and represented in bar graphs. n = 3, mean ± S.E. p value determined by Student's t test, *, p < 0.05. #, not significant.
miR-140 Regulates Tumor-initiating Cell Renewal in Estrogen-stimulated Breast Cancer Cells
Based on our observation that in the presence of estrogens miR-140-regulated mammosphere formation, we sought to further examine the impact of miR-140 restoration on breast tumor-initiating cell survival. Specifically we wanted to test the possibility that in breast cancer, ERα signaling promotes tumor-initiating cell renewal through suppression of miR-140 expression. We cultured MCF-7 cells in the absence of estrogens in starvation conditions. In these culture conditions, miR-140 overexpression had little to no impact on tumor-initiating cell frequency. Following E2 treatment, there was a dramatic increase (40.6% compared with 29%) in the CD44high/CD24low subpopulation (enriched in tumor-initiating cells) (Fig. 6A). We observed a dramatic decrease in the CD44high/CD24low subpopulation upon overexpression of miR-140 following estrogen stimulation (28.3% compared with 40.6%). We confirmed these findings in a separate ERα-positive breast cancer cell line, T47D cells, where miR-140 overexpression reduced the CD44high/CD24low subpopulation frequency to 2.83% compared with 4.74% in control cells (Fig. 6B). These results indicate the importance of miR-140 in regulating estrogen-stimulated tumor-initiating cell expansion in breast cancer cells.
FIGURE 6.

miR-140 regulates tumor-initiating cell renewal in estrogen-stimulated breast cancer cells. A, MCF-7 cells were cultured in starvation conditions in medium lacking phenol red and charcoal stripped serum. Cells were treated with 10 nm E2 or vehicle control and transfected with 2 μg of miR-140 expression vector or control vector. After 24 h, cells were stained with CD44-APC and CD24-PE antibodies, and CD44+/CD24- subpopulations were examined by flow cytometry. B, T47D cells were cultured in starvation conditions, treated with 10 nm E2, and transfected with 2 μg of miR-140 expression vector. Following 24 h CD44+/CD24- subpopulation frequency was examined by flow cytometry. n = 2.
DISCUSSION
In agreement with earlier reports (29), we observed miR-140 down-regulation in breast tumors compared with normal breast tissue. While examining the molecular mechanisms underlying miR-140 regulation we identified ERα control of miR-140 transcriptional activity. In breast cancers ERα is a therapeutic target and prognostic marker that is predictive for disease aggressiveness (40). We have confirmed direct recruitment of ERα to an ERE in the miR-140 promoter. As it has been previously shown that ERα can influence miR biogenesis (41), our data cannot rule out that ERα may regulate miR-140 expression through more than one mechanism in breast cancer.
Several reports have indicated overexpression of SOX2 in breast cancers (5, 9, 10). This well-known embryonic stem cell marker has recently been implicated in CSC self-renewal; in particular, SOX2 has been shown to be induced in mammosphere culture, which is conducive to short-term propagation of mammary stem cells and breast CSCs (5). We first observed inverse expression between miR-140 and its predicted target gene SOX2 in mammary epithelium and breast cancer cells. We validated interaction between miR-140 and SOX2 using luciferase reporter assays, qRT-PCR, and Western blot. Very little is known concerning the regulation of SOX2 in breast cancer. In embryonic stem cells it is thought that auto-regulatory feedback loops involving several embryonic transcription factors (OCT4, KLF4, Nanog, SOX2 etc.) maintain the other's respective gene expression (8). We have identified a mechanism that is in part responsible for SOX2 dysregulation in breast cancer, loss of miR-140 targeting of the SOX2 3′-UTR. We have shown through mammosphere culture that miR-140 targeting of SOX2 regulates stemness properties of breast cancer cells. Finally, we further confirmed the importance of this relationship by showing how miR-140 and SOX2 can regulate CD44high/CD24low breast tumor-initiating cells.
It has been previously shown that ERα signaling can regulate breast CSC frequency, with ERα stimulation resulting in increased numbers of CSCs (11, 12). It has also been shown that Metformin, a drug that can selectively target breast CSCs, can inhibit ERα regulation of stem cell genes (12). Here we provide a new mechanism by which ERα regulates breast tumor-initiating cells, through transcriptional control of miR-140 and subsequent miR-140 targeting of SOX2 mRNA.
Many questions remain unanswered concerning a potential ERα/miR-140/SOX2 pathway in breast cancer. First, it is important to test whether in vitro results from cell culture experiments can be translated into animal studies. A critical property of CSCs is the ability to repopulate heterogeneous tumor populations and to functionally demonstrate tumor-initiating capacity in vivo. There is significant interest in understanding the biology behind solid tumor stem cells and identifying drug targets and therapeutic approaches for eliminating these tumor subpopulations. In the future we will examine miR-140 regulation of breast tumor-initiation in xenograft studies. Additionally, we will explore the therapeutic potential for miR-140 and SOX2 modulation to eliminate breast CSCs. Finally, other studies have indicated miR-140 down-regulation also occurs in ERα-negative breast tumors (29) presumably through an independent mechanism, and we will address this in our ongoing experiments.
This work was supported, in whole or in part, by NCI, National Institutes of Health, T32 Training Support (to G. E.) and by grants from the Maryland Stem Cell Fund, FAMRI, American Cancer Society, and the NIH (CA163820) (to Q. Z.).
- ER
- estrogen receptor
- miR
- microRNA
- CSC
- cancer stem cells
- ERE
- estrogen response elements
- UTR
- untranslated region
- BPA
- bisphenol A
- NSCC
- non-stem cancer cells.
REFERENCES
- 1. Stingl J. (2011) Estrogen and Progesterone in Normal Mammary Gland Development and in Cancer. Hormones Cancer 2, 85–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yager J. D., Davidson N. E. (2006) Estrogen Carcinogenesis in Breast Cancer. N. Engl. J. Med. 354, 270–282 [DOI] [PubMed] [Google Scholar]
- 3. Al-Ejeh F., Smart C. E., Morrison B. J., Chenevix-Trench G., López J. A., Lakhani S. R., Brown M. P., Khanna K. K. (2011) Breast cancer stem cells: treatment resistance and therapeutic opportunities. Carcinogenesis 32, 650–658 [DOI] [PubMed] [Google Scholar]
- 4. O'Brien C. A., Kreso A., Jamieson C. H. (2010) Cancer stem cells and self-renewal. Clin. Cancer Res. 16, 3113–3120 [DOI] [PubMed] [Google Scholar]
- 5. Leis O., Eguiara A., Lopez-Arribillaga E., Alberdi M. J., Hernandez-Garcia S., Elorriaga K., Pandiella A., Rezola R., Martin A. G. (2012) Sox2 expression in breast tumours and activation in breast cancer stem cells. Oncogene 31, 1354–1365 [DOI] [PubMed] [Google Scholar]
- 6. Jeter C. R., Liu B., Liu X., Chen X., Liu C., Calhoun-Davis T., Repass J., Zaehres H., Shen J. J., Tang D. G. (2011) NANOG promotes cancer stem cell characteristics and prostate cancer resistance to androgen deprivation. Oncogene 30, 3833–3845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ponti D., Costa A., Zaffaroni N., Pratesi G., Petrangolini G., Coradini D., Pilotti S., Pierotti M. A., Daidone M. G. (2005) Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 65, 5506–5511 [DOI] [PubMed] [Google Scholar]
- 8. Boyer L. A., Lee T. I., Cole M. F., Johnstone S. E., Levine S. S., Zucker J. P., Guenther M. G., Kumar R. M., Murray H. L., Jenner R. G., Gifford D. K., Melton D. A., Jaenisch R., Young R. A. (2005) Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 122, 947–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen Y., Shi L., Zhang L., Li R., Liang J., Yu W., Sun L., Yang X., Wang Y., Zhang Y., Shang Y. (2008) The molecular mechanism governing the oncogenic potential of SOX2 in breast cancer. J. Biol. Chem. 283, 17969–17978 [DOI] [PubMed] [Google Scholar]
- 10. Lengerke C., Fehm T., Kurth R., Neubauer H., Scheble V., Müller F., Schneider F., Petersen K., Wallwiener D., Kanz L., Fend F., Perner S., Bareiss P. M., Staebler A. (2011) Expression of the embryonic stem cell marker SOX2 in early-stage breast carcinoma. BMC Cancer 11, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fillmore C. M., Gupta P. B., Rudnick J. A., Caballero S., Keller P. J., Lander E. S., Kuperwasser C. (2010) Estrogen expands breast cancer stem-like cells through paracrine FGF/Tbx3 signaling. Proc. Natl. Acad. Sci. 107, 21737–21742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jung J. W., Park S. B., Lee S. J., Seo M. S., Trosko J. E., Kang K. S. (2011) Metformin represses self-renewal of the human breast carcinoma stem cells via inhibition of estrogen receptor-mediated OCT4 expression. PLoS ONE. 6, e28068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Calin G. A., Croce C. M. (2006) MicroRNA-Cancer Connection: The beginning of a new tale. Cancer Res. 66, 7390–7394 [DOI] [PubMed] [Google Scholar]
- 14. Di Leva G., Gasparini P., Piovan C., Ngankeu A., Garofalo M., Taccioli C., Iorio M. V., Li M., Volinia S., Alder H., Nakamura T., Nuovo G., Liu Y., Nephew K. P., Croce C. M. (2010) MicroRNA cluster 221–222 and estrogen receptor α interactions in breast cancer. J. Natl. Cancer Inst. 102, 706–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Adams B. D., Cowee D. M., White B. A. (2009) The Role of miR-206 in the epidermal growth factor (EGF) induced repression of estrogen receptor-α (ERα) signaling and a luminal phenotype in MCF-7 breast cancer cells. Mol. Endocrinol. 23, 1215–1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bhat-Nakshatri P., Wang G., Collins N. R., Thomson M. J., Geistlinger T. R., Carroll J. S., Brown M., Hammond S., Srour E. F., Liu Y., Nakshatri H. (2009) Estradiol-regulated microRNAs control estradiol response in breast cancer cells. Nucleic Acids Res. 37, 4850–4861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Miyaki S., Sato T., Inoue A., Otsuki S., Ito Y., Yokoyama S., Kato Y., Takemoto F., Nakasa T., Yamashita S., Takada S., Lotz M. K., Ueno-Kudo H., Asahara H. (2010) MicroRNA-140 plays dual roles in both cartilage development and homeostasis. Genes Dev. 24, 1173–1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang J., Qin S., Yi C., Ma G., Zhu H., Zhou W., Xiong Y., Zhu X., Wang Y., He L., Guo X. (2011) MiR-140 is co-expressed with Wwp2-C transcript and activated by Sox9 to target Sp1 in maintaining the chondrocyte proliferation. FEBS Lett. 585, 2992–2997 [DOI] [PubMed] [Google Scholar]
- 19. Yamashita S., Miyaki S., Kato Y., Yokoyama S., Sato T., Barrionuevo F., Akiyama H., Scherer G., Takada S., Asahara H. (2012) L-Sox5 and Sox6 enhance chondrogenic miR-140 expression by strengthening dimeric Sox9 activity. J. Biol. Chem. 287, 22206–22215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Miyaki S., Nakasa T., Otsuki S., Grogan S. P., Higashiyama R., Inoue A., Kato Y., Sato T., Lotz M. K., Asahara H. (2009) MicroRNA-140 is expressed in differentiated human articular chondrocytes and modulates interleukin-1 responses. Arthritis Rheumatism 60, 2723–2730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pais H., Nicolas F. E., Soond S. M., Swingler T. E., Clark I. M., Chantry A., Moulton V., Dalmay T. (2010) Analyzing mRNA expression identifies Smad3 as a microRNA-140 target regulated only at protein level. RNA. 16, 489–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nicolas F. E., Pais H., Schwach F., Lindow M., Kauppinen S., Moulton V., Dalmay T. (2011) mRNA expression profiling reveals conserved and non-conserved miR-140 targets. RNA Biol. 8, 607–615 [DOI] [PubMed] [Google Scholar]
- 23. Tardif G., Hum D., Pelletier J. P., Duval N., Martel-Pelletier J. (2009) Regulation of the IGFBP-5 and MMP-13 genes by the microRNAs miR-140 and miR-27a in human osteoarthritic chondrocytes. BMC Musculoskeletal Disorders 10, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Song B., Wang Y., Xi Y., Kudo K., Bruheim S., Botchkina G. I., Gavin E., Wan Y., Formentini A., Kornmann M., Fodstad O., Ju J. (2009) Mechanism of chemoresistance mediated by miR-140 in human osteosarcoma and colon cancer cells. Oncogene 28, 4065–4074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liang Y., Ridzon D., Wong L., Chen C. (2007) Characterization of microRNA expression profiles in normal human tissues. BMC Genomics 8, 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Iorio M. V., Visone R., Di Leva G., Donati V., Petrocca F., Casalini P., Taccioli C., Volinia S., Liu C. G., Alder H., Calin G. A., Ménard S., Croce C. M. (2007) MicroRNA signatures in human ovarian cancer. Cancer Res. 67, 8699–8707 [DOI] [PubMed] [Google Scholar]
- 27. Tan X., Qin W., Zhang L., Hang J., Li B., Zhang C., Wan J., Zhou F., Shao K., Sun Y., Wu J., Zhang X., Qiu B., Li N., Shi S., Feng X., Zhao S., Wang Z., Zhao X., Chen Z., Mitchelson K., Cheng J., Guo Y., He J. (2011) A 5-MicroRNA signature for lung squamous cell carcinoma diagnosis and hsa-miR-31 for prognosis. Clin. Cancer Res. 17, 6802–6811 [DOI] [PubMed] [Google Scholar]
- 28. Sand M., Skrygan M., Sand D., Georgas D., Hahn S., Gambichler T., Altmeyer P., Bechara F. G. (2012) Expression of microRNAs in basal cell carcinoma. Br. J. Dermatol. 167, 847–855 [DOI] [PubMed] [Google Scholar]
- 29. Volinia S., Galasso M., Sana M. E., Wise T. F., Palatini J., Huebner K., Croce C. M. (2012) Breast cancer signatures for invasiveness and prognosis defined by deep sequencing of microRNA. Proc. Natl. Acad. Sci. U.S.A. 109, 3024–3029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fong H., Hohenstein K. A., Donovan P. J. (2008) Regulation of self-renewal and pluripotency by Sox2 in human embryonic stem cells. Stem Cells 26, 1931–1938 [DOI] [PubMed] [Google Scholar]
- 31. Abukhdeir A. M., Blair B. G., Brenner K., Karakas B., Konishi H., Lim J., Sahasranaman V., Huang Y., Keen J., Davidson N., Vitolo M. I., Bachman K. E., Park B. H. (2006) Physiologic estrogen receptor α signaling in non-tumorigenic human mammary epithelial cells. Breast Cancer Res. Treat. 99, 23–33 [DOI] [PubMed] [Google Scholar]
- 32. Ma L., Young J., Prabhala H., Pan E., Mestdagh P., Muth D., Teruya-Feldstein J., Reinhardt F., Onder T. T., Valastyan S., Westermann F., Speleman F., Vandesompele J., Weinberg R. A. (2010) miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat. Cell Biol. 12, 247–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yao Y., Li H., Gu Y., Davidson N. E., Zhou Q. (2010) Inhibition of SIRT1 deacetylase suppresses estrogen receptor signaling. Carcinogenesis 31, 382–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Heldring N., Pike A., Andersson S., Matthews J., Cheng G., Hartman J., Tujague M., Ström A., Treuter E., Warner M., Gustafsson J. A. (2007) Estrogen Receptors: How do they signal and what are their targets. Physiol. Rev. 87, 905–931 [DOI] [PubMed] [Google Scholar]
- 35. Lewis B. P., Burge C. B., Bartel D. P. (2005) Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120, 15–20 [DOI] [PubMed] [Google Scholar]
- 36. Xiang R., Liao D., Cheng T., Zhou H., Shi Q., Chuang T. S., Markowitz D., Reisfeld R. A., Luo Y. (2011) Downregulation of transcription factor SOX2 in cancer stem cells suppresses growth and metastasis of lung cancer. Br. J. Cancer 104, 1410–1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Al-Hajj M., Wicha M. S., Benito-Hernandez A., Morrison S. J., Clarke M. F. (2003) Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. U.S.A. 100, 3983–3988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dontu G., Abdallah W. M., Foley J. M., Jackson K. W., Clarke M. F., Kawamura M. J., Wicha M. S. (2003) In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 17, 1253–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu S., Dontu G., Wicha M. (2005) Mammary stem cells, self-renewal pathways, and carcinogenesis. Breast Cancer Res. 7, 86–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jones K. L., Buzdar A. U. (2004) A review of adjuvant hormonal therapy in breast cancer. Endocr. Relat. Cancer 11, 391–406 [DOI] [PubMed] [Google Scholar]
- 41. Yamagata K., Fujiyama S., Ito S., Ueda T., Murata T., Naitou M., Takeyama K., Minami Y., O'Malley B. W., Kato S. (2009) Maturation of microRNA is hormonally regulated by a nuclear receptor. Mol. Cell 36, 340–347 [DOI] [PubMed] [Google Scholar]