

Background: Neuritin is a new neurotrophic factor. The receptors for binding neuritin and its downstream signaling effectors, however, remain unclear.
Results: Neuritin increases Kv4.2-mediated IA in rat cerebellar granule neurons by activating the insulin receptor pathway.
Conclusion: Neuritin may act as a ligand of the insulin receptor.
Significance: Our findings reveal an unanticipated role of the insulin receptor in previously unrecognized neuritin-mediated signaling.
Keywords: ERK, Ion Channels, mTOR, Neurons, Receptors, A-type K+ Current, Kv4.2, Neuritin, Insulin Receptor
Abstract
Neuritin is a new neurotrophic factor discovered in a screen to identify genes involved in activity-dependent synaptic plasticity. Neuritin also plays multiple roles in the process of neural development and synaptic plasticity. The receptors for binding neuritin and its downstream signaling effectors, however, remain unclear. Here, we report that neuritin specifically increases the densities of transient outward K+ currents (IA) in rat cerebellar granule neurons (CGNs) in a time- and concentration-dependent manner. Neuritin-induced amplification of IA is mediated by increased mRNA and protein expression of Kv4.2, the main α-subunit of IA. Exposure of CGNs to neuritin markedly induces phosphorylation of ERK (pERK), Akt (pAkt), and mammalian target of rapamycin (pmTOR). Neuritin-induced IA and increased expression of Kv4.2 are attenuated by ERK, Akt, or mTOR inhibitors. Unexpectedly, pharmacological blockade of insulin receptor, but not the insulin-like growth factor 1 receptor, abrogates the effect of neuritin on IA amplification and Kv4.2 induction. Indeed, neuritin activates downstream signaling effectors of the insulin receptor in CGNs and HeLa. Our data reveal, for the first time, an unanticipated role of the insulin receptor in previously unrecognized neuritin-mediated signaling.
Introduction
Neuritin, also known as CPG15, was discovered in a screen to identify novel genes involved in activity-dependent synaptic plasticity in the neocortex (1, 2). Neuritin was subsequently identified as an important neurotrophin expressed in the developing nervous system (3). Indeed, neuritin plays multiple roles in the process of neural development, synaptic plasticity, and synaptic maturation (4, 5). For example, previous studies showed correlation of neuritin expression and its effect on neurite outgrowth and axonal function (6, 7). The function of neuritin in the voltage-gated ion channel in neurotropic responses, however, remains elusive.
Previous studies reported that exposure of primary embryonic hippocampal or cortical neurons to purified recombinant neuritin promotes neurite outgrowth and arborization (1, 2). Neuritin has also been shown to promote synaptic maturation (8). How neuritin mediates neuronal differentiation and maturation, however, is not known. Recent studies suggested that neuritin acts as a ligand to promote dendritic growth through intercellular signaling (2). Identification of neuritin receptor and its downstream signaling pathways, therefore, will be important for further investigation of neuritin in neuronal functions.
Cerebellar granule neurons (CGNs)2 are small glutamatergic cells, and their differentiation gives rise to the numerous neuronal types in mammalian brain. In view of their well defined developmental pathway, CGNs are invaluable models for studying neuronal maturation differentiation and synaptic plasticity (9). Previous studies indicated that growth and differentiation factors can either stimulate or inhibit CGN development and maturation by regulating multiple signaling pathways (10, 11).
One of the characteristics during CGNs development and maturation is regulation of the expression of K+ channels (12, 13). Primary culture of CGNs displays several voltage-activated outward K+ currents (14). We previously reported that enhancement of delayed-rectifier outward (IK) and fast transient outward (IA) potassium currents was associated with CGN apoptosis (15, 16). In addition, IK also plays a role in promoting CGN migration and maturation (11, 17). These studies, together, indicate that modulation of expression of different potassium channels may alter the status and function of CGNs.
The goal of this report was to examine whether neuritin modulates K+ channels for its role in synaptic activity and neurodevelopment. To test this hypothesis, we examined the effect of neuritin on K+ channels in rat CGNs. We find that neuritin increases IA amplitudes by regulating the expression of Kv4.2 potassium channel subunits. In addition, we show that neuritin activates insulin receptor signaling pathway. Our study, for the first time, reveals neuritin-mediated signaling and its role in modulating K+ channel expression for neuronal function.
EXPERIMENTAL PROCEDURES
Cell Culture
All experimental procedures were carried out in accordance with European guidelines for the care and use of laboratory animals (Council Directive 86/609/EEC). Cells were derived from cerebellum of 7-day-old Sprague-Dawley rat pups as described previously (18). Isolated cells were plated onto 35-mm Petri dishes coated with poly-l-lysine (1 μg/ml) at a density of 106 cells/ml. Cultured cells were incubated at 37 °C under 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum, insulin (5 μg/ml), KCl (25 mm), and 1% antibiotic-antimycotic solution. After culture for 24 h, cytosine β-d-arabinofuranoside (5 μm) was added to the culture medium to inhibit the proliferation of nonneuronal cells. Cells were used for experiments after 7–8 days in culture unless otherwise indicated. HeLa cells were maintained as a monolayer in high glucose DMEM, supplemented with 10% fetal bovine serum and 1% antibiotic-antimycotic solution.
Patch Clamp Recordings
Whole cell currents of granule neurons were recorded using conventional patch clamp technique. All currents were recorded using an multiclamp 200B amplifier (Axon Instruments, Foster City, CA) operated in voltage clamp mode. Data acquisition and analysis were performed with pClamp 8.01 software (Axon Instruments) and/or Origin 8 analysis software (Microcal Software, Northampton, MA). Prior to IA recording, the culture medium was replaced with a bath solution containing 125 mm NaCl, 2.5 mm KCl, 10 mm HEPES (pH 7.4), 1 mm MgCl2, 1 μm tetrodotoxin, 20 mm triethanolamine, 10 mm glucose. Soft-glass recording pipettes were filled with an internal solution containing 135 mm potassium gluconate, 10 mm KCl, 10 mm HEPES (pH 7.3), 1 mm CaCl2, 1 mm MgCl2, 10 mm EGTA, 1 mm ATP, and 0.1 mm GTP. The pipette resistance was 4–6 megohms after filling with internal solution. All recordings were performed at room temperature. The cultured granule cells selected for electrophysiological recording exhibited common morphological characteristics of healthy cells, such as fusiform soma with two main neuritis of similar size. In addition, the mean capacitance of recorded cells for control group (7.17 ± 0.23 picofarads) and for neuritin treatment group (7.36 ± 0.11 picofarads) showed no significant difference (p = 0.338), indicating that comparable granule cells were used in the experiments.
In steady-state activation experiments, membrane potential was held at −100 mV, and IA was evoked by a 200-ms depolarizing pulse from a first pulse potential of −60 mV to +60 mV in 10-mV steps at 10-s intervals. Data were analyzed using the equation GK = IK/(Vm − Vrev), where GK is the membrane K+ conductance, Vm is the membrane potential, and Vrev is the reversal potential for K+. To study steady-state inactivation of IA, currents were elicited using 1-s conditioning pre-pulses from −110 mV to 0 mV before a 200-ms test pulse of +50 mV. After normalizing each current amplitude to the maximal current, amplitude obtained from the −110 mV pre-pulse was used as a function of the conditioning pre-pulse potential and fitted with the function IA/IA − max = 1/(1 + exp((Vm1/2 − Vm)/k)). Hence, we obtained an inactivation curve of IA and calculated the V50 (the voltage at which the current amplitude was half-inactivated).
Data Analysis
Results were analyzed using one-way analysis of variance (ANOVA) for comparisons between multiple groups and by Student's t test for comparison of two samples. Data were presented as means ± S.E. with n as the number of neurons for electrophysiological recordings, number of imaging experiments, or as the number of replicates for molecular biological experiments. For electrophysiological experiments, data were collected from at least four different batches of neurons prepared at different dates, thereby minimizing bias resulting from culture conditions. A p value < 0.05 was considered statistically significant.
Western Blot Analysis
Cells were lysed in HEPES-Nonidet P-40 lysis buffer (20 mm HEPES, 150 mm NaCl, 0.5% Nonidet P-40, 10% glycerol, 2 mm EDTA, 100 μm Na3VO4, 50 mm NaF (pH 7.5), and 1% proteinase inhibitor mixture) on ice for 30 min. After centrifugation, the supernatant was mixed with 2× sodium dodecyl sulfate loading buffer and boiled for 5 min. The proteins were separated on a 10% SDS-polyacrylamide gel, transferred to polyvinylidene difluoride membranes (Millipore). The membrane was blocked with 10% nonfat milk and incubated at 4 °C overnight with mouse monoclonal antibody against Kv4.2, Kv4.3, Kv1.1 (1:2000, catalogue numbers 75-016, 75-017, and 75-007; University of California, Davis), or rabbit polyclonal antibody against the phosphorylated insulin receptor β subunit (1:1000, catalogue number 3024; Cell Signaling Technology), or rabbit monoclonal antibody against phosphorylated ERK1/2 or total ERK1/2 (1:1000, catalogue numbers 4370 and 4695; Cell Signaling Technology), or rabbit monoclonal antibody against phosphorylated Akt (1:2000, catalogue number 4060; Cell Signaling Technology), or rabbit polyclonal antibody against phosphorylated mTOR (1:1000, catalogue number 2971; Cell Signaling Technology), or rabbit polyclonal antibody against phosphorylated IRS-1 (1:500, catalogue number 3070; Cell Signaling Technology), or mouse monoclonal antibody against GAPDH (1:10,000, catalogue number KC-5G4; KangChen Bio-Tech, China). After extensively washing in TBST, the membrane was incubated with horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG (1:10,000, catalogue numbers KC-MM-035 and KC-RB-035; KangChen Bio-Tech) for 2 h at room temperature. Chemiluminescent signals were generated using a SuperSignal West Pico trial kit (Pierce) and detected by exposure to x-ray film or using ChemiDoc XRS System (Bio-Rad Laboratories). Quantity One software (version 4.6.2, Bio-Rad Laboratories) was used for background subtraction and for quantification of immunoblotting data.
Quantitative RT-PCR
To measure Kv4.2 mRNA levels, quantitative real-time PCR analysis was performed with the following sequences: forward primer, 5′-TGTCAGGAAGTCATAGAGGCAGCGTG-3′ and reverse primer, 5′-GGGGTGGTTACTGGAGGTGTTGGAAT-3′. To control sampling errors, quantitative real-time PCR for the housekeeping gene cyclophilin D with the following sequences: forward primer, 5′-GGCTCTTGAAATGGACCCTTC-3′ and reverse primer, 5′-GGCTCTTGAAATGGACCCTTC-3′ was routinely performed on each sample and used as control. The reaction solution consisted of 2.0 μl of diluted RT-PCR product, a 0.2 μm concentration of each paired primer, and power SYBR Green PCR master mix (Toyobo, Osaka, Japan). The annealing temperature was set at 58 °C for Kv4.2 and 61 °C for cyclophilin D, and amplification cycles were set at 38 cycles. The absolute mRNA levels in each sample were calculated according to a standard curve set up using serial dilutions of known amounts of specific templates against corresponding cycle threshold (Ct) values. The normalized ratio of the target gene over cyclophilin D in each sample was presented. The specificity of the primers was verified by both gel electrophoresis and sequencing of the PCR products.
Immunoprecipitation
For the detection of Tyr-phosphorylated insulin receptor, CGNs incubated with or without neuritin were lysed in HEPES-Nonidet P-40 lysis buffer as described above. After centrifugation, cell lysate was mixed with rabbit polyclonal antibody against the phosphorylated insulin receptor β subunit (1:100, catalogue number 3024; Cell Signaling Technology) at 4 °C. After overnight incubation, protein A/G (1:25; Santa Cruz Biotechnology) agarose beads were added and incubated for additional 1 h at 4 °C. After centrifugation and multiple washing, immunoprecipitated complex were mixed with 2× sodium dodecyl sulfate loading buffer and boiled for 5 min. Samples were then examined by Western blot analysis using mouse monoclonal antibody against phosphorylated insulin receptor β subunit (1:1000, catalogue number sc-81500; Santa Cruz Biotechnology). To measure the level of phosphorylation of IGF-1 receptor, rabbit polyclonal antibody against IGF-1 receptor β subunit (1:100, catalogue number 3027; Cell Signaling Technology) was used for immunoprecipitation and the primary antibody for Western blotting was rabbit polyclonal antibody against phosphorylated IGF-1 receptor β subunit(1:1000, catalogue number 6113; Cell Signaling Technology).
Immunocytochemistry and 3,3-N-Diaminobenzidine Tetrahydrochloride (DAB) Staining
Cultured cells were fixed in fresh 4% formaldehyde in 0.1 m phosphate buffer for 15 min. Fixed cells were washed twice in ice-cold PBS and permeabilized with 0.25% Triton X-100 for 10 min. Cells were then washed three times in PBS for 5 min each and blocked in 1% BSA for 30 min. The labeling experiments were performed by incubating cells with antibody against phosphorylated insulin receptor β-subunit or phosphorylated IGF-1 receptor (1:100, catalogue numbers 3024 and 3027; Cell Signaling Technology) at 4 °C overnight. After vigorously being washed in PBS, the cells were incubated with the corresponding FITC- or Texas Red-conjugated donkey anti-rabbit IgG (1:200, catalogue numbers 111-095-003 and 111-295-003; Jackson Immunoresearch Laboratories, West Grove PA) for 1 h at room temperature. Fluorescent-labeled cells were visualized with a Leica SP2 confocal laser scanning microscope (Leica, Mannheim, Germany) using a 40× objective lens.
DAB staining was detected according to the manufacturer's instructions. Briefly, after incubation with the primary antibody, cells were incubated with the horseradish peroxidase-conjugated anti-rabbit IgG (1:10,000) (KangChen Bio-Tech, China) for 1 h at room temperature and then overnight with DAB reagent. Labeled cells were washed twice in PBS and examined.
Chemicals
Recombinant human neuritin was purchased from Pepro Tech (Rocky Hill, NJ). Hydroxy-2-naphthalenylmethyl phosphonic acid (HNMPA) was obtained from Santa Cruz Biotechnology. Triethanolamine, tetrodotoxin, insulin, U0126, rapamycin, PQ-401, and poly-l-lysine were purchased from Sigma. Fetal calf serum, DMEM, and antibiotic-antimycotic solution were purchased from Invitrogen. shRNA was purchased from GeneChem (Shanghai, China). Expression of shRNA was coupled with GFP to identify positively transfected cells.
RESULTS
Neuritin Increased IA Amplitude of CGNs by Enhancing the Expression of Kv4.2 α-Subunit
To determine the potential role of neuritin on CGNs, we asked whether neuritin affected IA. IA was evoked by 200-ms depolarization to +40 mV from a holding potential of −100 mV in the presence of 20 mm triethanolamine, which suppresses IK and permits better resolution of IA. Incubation of CGNs with neuritin significantly enhanced IA densities in a concentration-dependent manner. The data obtained from 99 neurons showed that incubation of CGNs with 15, 150, or 300 ng/ml neuritin for 24 h increased the current densities by 13.0% (n = 27), 45.5% (n = 41), and 44.4% (n = 31), respectively (Fig. 1A). To address whether neuritin-induced IA densities were time-dependent, neurons were incubated with neuritin (150 ng/ml) for different times, and IA densities were determined. After 12-, 24-, and 36-h incubation, neuritin was found to augment IA densities by 22.9% (n = 21), 45.5% (n = 41), and 46.2% (n = 20), respectively (Fig. 1B). We also found that the effect of neuritin on the induction of IA is dependent on the days that CGNs have been in culture (Fig. 1C). Neuritin, however, did not affect granule neuron IA amplitude when applied acutely to bath solution by the perfusion system (data not shown). Together, these data indicate that incubating granule neurons from 3 days in culture (DIC) with 150 ng/ml neuritin for 24 h produced the most significant increase in IA densities.
FIGURE 1.

Neuritin enhanced IA amplitudes in a concentration- and time-dependent manner in CGNs. A, IA obtained from neurons maintained in control medium or medium with different concentration of neuritin. IA was elicited by depolarizing pulses to +40 mV from a holding potential of −100 mV in the presence of 20 mm triethanolamine. Upper panel shows representative recording sample; lower panel shows statistical analysis. B, IA densities in CGNs maintained for different incubation times in control medium or in medium containing 150 ng/ml neuritin. C, IA densities in CGNs of different DIC maintained for control medium or in medium containing 150 ng/ml neuritin for 24 h. Data are shown as means ± S.E. (error bars). *, p < 0.05 compared with the corresponding control by unpaired t test.
The effects of neuritin on the activation and inactivation properties of IA were also explored using corresponding experimental protocols (see “Experimental Procedures”). IA recorded using an activation protocol from neurons maintained in control medium or in medium containing 150 ng/ml neuritin were presented in Fig. 2A. The IA activation curve was obtained by plotting normalized conductance as a function of the command potential (Fig. 2B). As shown in Fig. 2B, incubation with 150 ng/ml neuritin for 24 h did not affect the activation curve. The current was half-activated at −2.91 ± 1.98 mV (n = 17) and −6.14 ± 2.27 mV (n = 23) for the control and neuritin-treated groups, respectively (p > 0.05). These data suggest that neuritin treatment does not alter the steady-state activation of the voltage dependence of the IA.
FIGURE 2.

Neuritin did not alter the steady-state activation and inactivation properties of CGN IA channels. A, IA recorded using an activation protocol (see “Experimental Procedures”) from neurons maintained in control medium or medium containing 150 ng/ml neuritin. B, steady-state activation curves of IA obtained by plotting the normalized conductance as a function of command potential obtained from control and neuritin-treated groups. Data points were fitted using the Boltzmann function. Data points are shown as means ± S.E. (error bars; n = 18 for the control group and n = 22 for the neuritin-treated group). C, steady-state inactivation curves for IA obtained from control and neuritin-treated CGNs using inactivation protocols (see “Experimental Procedures”). Data points were fitted using the Boltzmann function. Data points are shown as means ± S.E. (n = 21 for the control group and n = 23 for the neuritin-treated group).
Similarly, the inactivation curve of IA was not significantly shifted after incubation with 150 ng/ml neuritin for 24 h (Fig. 2, C and D). The half-maximal inactivation voltage for the control and neuritin-treated groups was −74.43 ± 0.92 mV (n = 25) and −71.47 ± 0.93 mV (n = 19), respectively (p > 0.05). These data indicate that neuritin does not affect IA inactivation kinetics. Together, electrophysiological recordings demonstrate that the neuritin-mediated enhancement of IA amplitudes is not associated with the modification of IA activation or inactivation properties.
Next, we hypothesized that neuritin-induced IA densities might be due to up-regulation of potassium channel expression levels. Key α-subunits account for the IA in CGNs include Kv4.2, Kv4.3, and Kv1.1 (37). Antibodies to Kv4.2, Kv4.3, and Kv1.1 α-subunit were used to measure their protein expression levels after incubation of CGNs with neuritin. Western blots indicated that protein levels of Kv4.2 were significantly enhanced following incubation of CGNs with neuritin for 24 h and from 3 to 7 DIC. There was, however, no significant change in the protein expression levels of Kv4.3 and Kv1.1 at 3–7 DIC (Fig. 3A). Kinetics of Kv4.2 induction was in parallel to the increase in IA amplitudes upon neuritin treatment (Fig. 3B). Furthermore, specific primers to amplify Kv4.2 were used to measure mRNA expression levels by quantitative PCR after incubation with and without neuritin. Quantitative PCR revealed that the levels of Kv4.2 α-subunit mRNA were significantly increased (Fig. 3C). Taken together, these data indicate that neuritin increases the IA densities by up-regulation of the expression of Kv4.2.
FIGURE 3.

Neuritin increased the expression levels of Kv4.2 α-subunit rather than Kv4.3 or Kv1.1 in CGNs. A, Western blot showing effect of 150 ng/ml neuritin on different Kv channels expression levels in CGNs with different DIC. Left, representative Western blots. Right, statistical analysis. B, Western blot results of Kv4.2 α-subunit protein expression levels in CGNs, showing the effect of incubating with 150 ng/ml neuritin for 12–36 h. Chemiluminescent signals were detected using the ChemiDoc XRS System. *, p < 0.05 compared with corresponding control by unpaired t test. Error bars, S.E. C, Kv4.2 mRNA expression levels in control and neuritin-treated groups as measured by quantitative RT-PCR. Data were obtained from five independent experiments. *, p < 0.05 compared with corresponding control by unpaired t test.
Neuritin-induced IA Densities and Increased Expression of Kv4.2 α-Subunit Are Sensitive to ERK or mTOR Inhibitors
Previous studies highlighted a critical role of extracellular signal-regulated kinase (ERK) cascades after synaptic activation (19, 20). We asked whether the ERK signaling pathway was activated by neuritin by measuring the levels of phosphorylation of ERK (pERK) (21, 22). Western blots showed that phosphorylation of both ERK1 (44 kDa) and ERK2 (42 kDa) was significantly increased by neuritin. After 15 min, 30 min, and 24 h treatment of neuritin, the levels of pERK relative to untreated control were 275, 377, and 27%, respectively (Fig. 4A). The effect of ERK activation by neuritin was ascertained by using MEK inhibitor U0126. As shown in Fig. 4B, incubation with U0126 completely abolished the effects of neuritin on the induction of IA densities. In the presence of U0126, IA densities induced by neuritin were reduced from 45.5% to 21.3% (n = 41, p < 0.05). Notably, administration of U0126 also blocked neuritin-induced Kv4.2 α-subunit protein levels (Fig. 4, C and D) and mRNA expression (Fig. 4E). The expression levels of the Kv4.2 α-subunit and mRNA level were decreased to 19.5 ± 0.1% (n = 4) and 0.08 ± 0.03% (n = 4), respectively. These data indicate that the ERK signaling pathway is required for the up-regulation of IA densities and increased Kv4.2 expression by neuritin.
FIGURE 4.

Neuritin-induced IA densities and the increased expression of Kv4.2 α-subunit were dependent the ERK signaling pathway. A, Western blot showing the levels of phosphorylated ERK1 (pERK1) and phosphorylated ERK2 (pERK2) in CGNs incubated with 150 ng/ml neuritin for 15 min to 24 h. Chemiluminescent signals were detected by exposure to x-ray film. B, effect of MEK inhibitor, U0126, on neuritin-induced up-regulation of IA densities. *, p < 0.05 compared with vehicle control (medium without neuritin) by unpaired t test. Error bars, S.E. C and D, effect of U0126 on neuritin-induced up-regulation of Kv4.2 protein level. E, effect of U0126 on neuritin-induced up-regulation of Kv4.2 mRNA expression measured by quantitative RT-PCR experiments. Data obtained from five independent experiments are shown as means ± S.E. *, p < 0.05 compared with corresponding control by unpaired t test.
In addition to the ERK signaling, we also determined whether the mTOR pathway, which was reported recently to affect protein synthesis and relevance to synaptic plasticity (23), is required for the up-regulation of IA densities and Kv4.2 α-subunit expression. After neuritin treatment, the phosphorylation level of mTOR (pmTOR) was significantly increased to 15.1% (n = 5) and 18.0% (n = 5) at 18 and 24 h, respectively (Fig. 5A). Activation of mTOR by neuritin was confirmed with the pharmacological inhibitor rapamycin. Fig. 5B showed that blockade of mTOR activity by rapamycin prevented neuritin-mediated increase of IA densities and increased Kv4.2 protein expression. In the presence of rapamycin, IA densities increased by neuritin were reduced from 45.5% to −5.3% (n = 21, p < 0.05). Similarly, the expression levels of the Kv4.2 α-subunit protein (Fig. 5, C and D) and mRNA level (Fig. 5E) were decreased to −12.4 ± 0.13% (n = 5) and 16.6 ± 0.04% (n = 5), respectively. These data indicate that the mTOR pathway is also required for the up-regulation of IA densities and increased Kv4.2 α-subunit expression by neuritin.
FIGURE 5.

Neuritin-induced IA densities and the increased expression of Kv4.2 α-subunit were dependent the Akt/mTOR signaling pathway. A, Western blot showing the levels of activated Akt (pAkt) and activated mTOR (p-mTOR) in CGNs incubated with 150 ng/ml neuritin for 15 min to 24 h. Chemiluminescent signals were detected with the ChemiDoc XRS System. B, effect of mTOR inhibitor, rapamycin, and Akt inhibitor LY294002 on neuritin-induced up-regulation of IA densities. C and D, effect of rapamycin and LY294002 on neuritin-induced up-regulation of Kv4.2 protein level. E, effect of rapamycin on neuritin-induced up-regulation of Kv4.2 mRNA expression measured by quantitative RT-PCR experiments. Data are shown as means ± S.E. (error bars). *, p < 0.05 compared with corresponding control by unpaired t test.
We also determined phosphorylation of Akt (pAkt), an upstream kinase of the mTOR pathway (23). Western blots indicated that phosphorylation level of mTOR and Akt in granule cells were increased significantly by treatment with neuritin (Fig. 5A). Quantitative analysis showed that the level of pAkt began to rise after neuritin treatment of 1 h and reached optimum activation by 6 h. The relative increased levels of Akt phosphorylation were 12.4% (n = 4) and 25.4% (n = 4), respectively. These data indicate that neuritin activates mTOR via the Akt kinase.
The role of Akt on neuritin-mediated induction of IA densities and Kv4.2 expression was further ascertained by using pharmacological inhibitor LY294002. Fig. 5B showed that blockade of Akt activity by LY294002 inhibited neuritin-induced IA densities and up-regulation of Kv4.2 protein expression. In the presence of LY294002, IA densities increased by neuritin were reduced from 45.5% to 5.6% (n = 17, p < 0.05). Similarly, the induction of Kv4.2 α-subunit protein (Fig. 5, C and D) was decreased to 3.1 ± 0.04%. These data indicate that Akt and mTOR pathways are required for the up-regulation of IA densities and induction of Kv4.2 α-subunit expression by neuritin.
Neuritin Increased IA and Kv4.2 Expression by Activation of Insulin Receptor
Previous studies indicated that the ERK and mTOR pathways were targets of growth factor activation such as NGF and insulin receptor (IR). Because some studies reported that neuritin is downstream in NGF-mediated neurite outgrowth (24), we hypothesized that insulin could mimic the induction effect of neuritin. Indeed, administration of insulin (50 or 150 ng/ml) induced IA densities and increased Kv4.2 protein expression. In the presence of insulin, the levels of IA densities and Kv4.2 protein level were increased by 39.3 ± 0.02% (n = 16) and 47.3 ± 0.14% (n = 5), respectively (Fig. 6, A, C, and D). These data indicate a role of insulin receptor pathway in the induction of IA densities and Kv4.2 protein level.
FIGURE 6.

Insulin receptor was needed for neuritin-induced IA densities and increased Kv4.2 α-subunit expression in CGNs. A, insulin mimicking the effect of neuritin and inducing IA densities in CGNs. B, effects of insulin receptor blocker HNMPA and IGF-1 receptor blocker PQ-401 on the neuritin-induced increase of IA densities. C, insulin mimicking the effect of neuritin and inducing Kv4.2 protein level in CGNs. D, effects of insulin and HNMPA on Kv4.2 mRNA expression measured by quantitative RT-PCR experiments. E, effects of HNMPA and PQ-401 on the neuritin-induced increase of Kv4.2 protein level of CGNs. Data were obtained from five independent experiments. *, p < 0.05 compared with vehicle (medium without neuritin) control by unpaired t test.
Next, we asked whether blockage of the insulin pathway would affect the induction of IA densities and increased Kv4.2 protein level by neuritin. We used HNMPA, which inhibits insulin receptor Tyr kinase, to address this question. Blockade of IR activity by HNMPA reduced neuritin-mediated induction of both IA densities and Kv4.2 protein level (Fig. 6, B and C). In the presence of HNMPA, IA densities increased by neuritin were significantly reduced from 45.5 ± 0.05% to −0.01 ± 0.05% (n = 23, p < 0.05). Similarly, induction of the Kv4.2 α-subunit and mRNA expression were decreased to −10.3 ± 0.14% and 0.27 ± 0.02%, respectively (Fig. 6, A and B). These data indicate that neuritin may channel through the IR pathway for the induction of IA densities and Kv4.2 protein level.
We also tested whether the IGF-1 receptor pathway was involved in neuritin-mediated effect using PQ-401 as antagonist of the IGF-1 receptor pathway. Unlike the effect of HNMPA, PQ-401 did not affect neuritin-induced up-regulation of IA densities nor increased expression of Kv4.2 (Fig. 6, B and E). These data indicate that the IR, but not the IGF-1 receptor, may mediate the neuritin-induced effect on the increase of IA densities and expression of Kv4.2 α-subunit.
Given that neuritin is an orphan ligand and its signaling receptor is not known, we hypothesized that neuritin might elicit IR activation. To test this hypothesis, we examined whether neuritin could stimulate Tyr phosphorylation of the IR by immunoprecipitation and immunocytochemistry. Immunoprecipitation assays showed increased amounts of phosphorylated IR, but not IGF-1 receptor, after neuritin treatment (Fig. 7, A and B). Immunofluorescence assays and immunocytochemistry further indicated that neuritin induced Tyr phosphorylation of the IR in HeLa cells (Fig. 7, C and D). Activation of the IR by neuritin, however, was reduced after transfection of shRNA to reduce IR expression in HeLa cells (Fig. 7D). As expected, neuritin did not induce Tyr phosphorylation of the IGF-1 receptor in HeLa cells (Fig. 7, E and F), supporting that neuritin activates the IR.
FIGURE 7.

Effect of neuritin on IR or IGF-1 receptor activation in CGNs and HeLa cells. A and B, immunoprecipitation assays showing that neuritin induced phosphorylation of IR, but not IGF-1 receptor in CGN lysate. Effect of insulin on phosphorylation of IGF-1 receptor is also shown. C, immunofluorescence to detect phosphorylated IR in HeLa cells. Effect of neuritin is also shown. Representative immunofluorescence images from four independent experiments are shown. Scale bar, 30 μm. D, immunohistochemical studies with DAB staining detecting the amount of phosphorylated IR in HeLa cells after neuritin stimulation. Effect of shRNA to reduce IR expression is also shown. Scale bar, 120 μm. Similar data were obtained from four independent experiments. E, immunofluorescence to detect phosphorylated IGF-1 receptor in HeLa cells. Effect of neuritin or insulin is also shown. Representative immunofluorescence image from four independent experiments are shown. Scale bar, 30 μm. F, immunohistochemical studies with DAB staining to detect phosphorylated IGF-1 receptor in HeLa cells. Effect of neuritin or insulin is also shown. Similar data were obtained from four independent experiments.
To further ascertain the effect of neuritin on IR activation, we determined the phosphorylation of IRS-1 and ERK, downstream effectors of IR signaling. Western blots showed that neuritin increased phosphorylation of IRS-1. Pretreatment of HNMPA, but not PQ-401, reduced the neuritin-induced Tyr phosphorylation of IRS-1 in HeLa cells (Fig. 8A). Similarly, neuritin increased the phosphorylation of both ERK1 and ERK2 after a 15-min stimulation. Pretreatment of HNMPA, however, reduced the neuritin-induced activation of ERK in HeLa cells (Fig. 8B). Taken together, these data indicate that the insulin receptor is involved in previously unrecognized neuritin-mediated signaling, which increases IA densities and Kv4.2 protein level in native cerebellar granule cells (Fig. 9).
FIGURE 8.
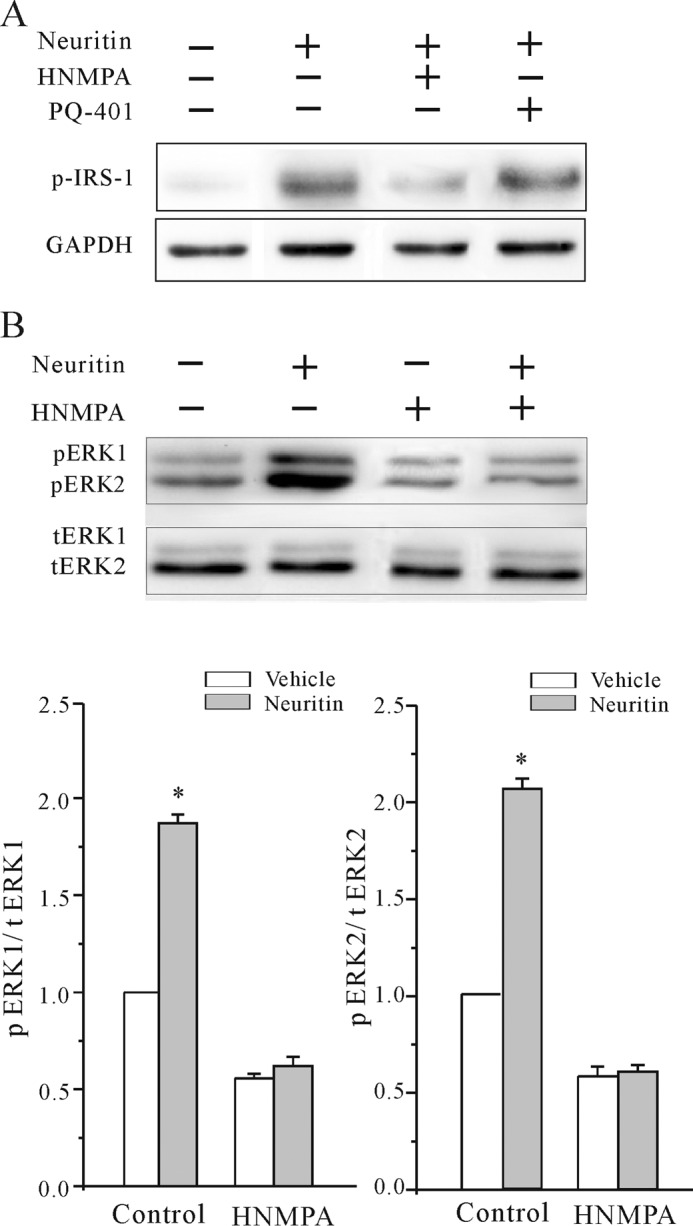
Effect of neuritin on IR or IGF-1 receptor activation in HeLa cells. A, Western blotting showed neuritin-induced phosphorylation of IRS-1 in HeLa cells. Effect of HNMPA and PQ-401 is also shown. B, Western blotting showed that neuritin increased phosphorylation of both ERK1 and ERK2 in HeLa cells. Effect of insulin receptor blocker HNMPA is also shown. *, p < 0.05 compared with vehicle control (medium without neuritin) by unpaired t test. Data were obtained from three independent experiments.
FIGURE 9.

Schematic illustration depicting the mechanisms of neuritin in modulating expression of Kv4.2 and the subsequent increase in IA amplitude.
DISCUSSION
Neuritin is an important neurotrophin and plays multiple roles in the process of neural development, synaptic plasticity, and synaptic maturation (1, 2, 4, 5). The receptor for binding neuritin and its downstream effector signal pathway, however, are poorly investigated. Here, we show, for the first time, that neuritin could activate IR and ERK/mTOR signaling pathways to increase IA amplitude and the expression of Kv4.2 in CGNs (Fig. 9).
CGNs contain two major voltage-dependent outward K+ currents: a delayed rectifier potassium current (IK) and a fast transient potassium current (IA). Neuritin treatment for 24 h failed to modulate IK in CGNs (data not shown). We thus examined the effect of neuritin on IA in this study. IA has been described in neurons from many regions of the central nervous system (25–27). Short term modulation of IA densities could arise from a rapid mechanism due to changes in voltage-gating properties or intracellular trafficking of the channel proteins (28–31). Alternatively, a long term mechanism by up-regulation of channel mRNA and protein expression could be involved (32). We observed that neuritin did not affect IA amplitude when it was applied acutely. Neuritin, however, significantly increased IA densities without modification of IA activation and inactivation properties, indicating that a long term mechanism of action by neuritin is involved.
Recent studies have suggested that the Kv4 shal family forms the major components of the IA in the central nervous system (33, 34). Expression of the Kv4 family was found in CGNs, pyramidal neurons of the hippocampus and cortical neurons (32, 35, 36). We have shown previously that Kv4.2, Kv4.3, and Kv1.1 are the main α-subunits expressed in rat CGNs (37). Upon neuritin treatment, only Kv4.2 α-subunit mRNA and protein levels were significantly increased. More importantly, the kinetics of Kv4.2 induction was in parallel to the increase in IA upon neuritin treatment. These data indicate that neuritin specifically increases the expression of Kv4.2.
Previous studies using cultured CGNs showed that subcellular redistribution of Kv4.2 from the soma to the dendrites and synapses is induced by synapse formation. Regulation of Kv4.2 can be further potentiated by synaptic activity (38). Indeed, expression of Kv4.2 in CGNs was increased with the duration of the microexplant culture period (12), in parallel to our findings that a concomitant increment of IA and Kv4.2 from 3 to 5 DIC. Notably, after neuritin treatment, the levels of IA and Kv4.2 from 3 DIC CGNs were similar to that of 5 DIC CGNs. Similar effect of neuritin on IA and Kv4.2 expression was also found in cortical neurons (data not shown). These observations suggest that the effect of neuritin on Kv4.2 expression may be developmentally regulated and associated with neuronal maturation and synaptic plasticity as previous reported (38). The possible roles of neuritin on neuronal maturation via regulating Kv4.2 expression warrant further investigations.
Many studies have shown that ERK is important for regulation of neuronal function, including regulation of synaptic plasticity and long term memory formation (39, 40). One potential mechanism of this regulation by ERK is indirect via long term modulation of gene transcription and channel protein expression. For example, a previous study indicated that ERK activation could dually regulate Kv4.2 at the transcriptional as well as posttranslational level (41). Another possible mechanism is that ERK could directly phosphorylate subunits of ion channels to alter intrinsic channel properties to regulate membrane potential. The phosphorylation of Kv4 channel subunits by ERK was characterized by the changing of gating properties of Kv4.2 (29, 42) upon acute regulation of IA by growth factors (43, 44). Our data show that neuritin does not alter the gating properties of IA. Neuritin, however, modulates IA by increasing Kv4.2 expression. Importantly, blocking activation of the ERK pathway eliminated the neuritin-induced effect on Kv4.2 expression. These data indicate that neuritin channels through the ERK pathway to control Kv4.2 gene transcription.
In addition to the ERK pathway, mTOR signaling is also involved in neuronal development. The mTOR pathway is also essential for several forms of synaptic plasticity for learning and memory (45, 46). Similar to the ERK pathway, mTOR kinase activity is modulated in response to various stimuli, such as neurotrophic factors and mitogenic hormones, to achieve transcriptional and translational control (23, 47–49). In this study, we show that mTOR-specific inhibitor rapamycin blocks neuritin-mediated induction of Kv4.2 mRNA and protein as well as subsequent increase in IA. Thus, neuritin also channels through the mTOR pathway to control Kv4.2 gene transcription.
Recent studies demonstrated that mTOR and ERK pathways cooperate with one another to affect many cellular functions. For example, cooperation between the ERK and mTOR pathways stimulates protein translation and induces tumor progression in a glioblastomas mouse model (50). In hippocampal neurons, it appears that cooperation of ERK and mTOR pathways regulates de novo protein synthesis, which is a prerequisite for long term potentiation (51). In this study, we did not explore the cross-talk between the mTOR and ERK signaling pathway in neuritin-induced Kv4.2 gene transcription. However, we believe that neither the ERK nor the mTOR pathway alone is sufficient to increase Kv4.2 gene transcription because administration of U0126 or rapamycin completely eliminated the effect of neuritin. Possibly, ERK and mTOR may assert a transcriptional cooperation on Kv4.2 induction, similar to the optimal activation operated by nuclear factor of activated T-cells (NFAT) transcription factor (52). Alternatively, ERK and mTOR could regulate at the transcriptional and translational level, respectively. For example, ERK-induced Kv4.2 transcripts may encode 5′-terminal oligopyrimidine tract for subsequent translational control by mTOR (53). The latter may account for the difference in temporal kinetic of ERK and mTOR activation upon neuritin stimulation. Nonetheless, the dual requirements on Kv4.2 induction suggest that neuritin activates both ERK and mTOR signalings.
Although previous studies implied that neuritin may function as a ligand, few investigation identified the receptor by which neuritin transduces its signal (2, 54). Our results show that neuritin activates both ERK and mTOR signalings for Kv4.2 induction. Activation of both ERK and mTOR by neuritin displays similar mechanism of the insulin receptor pathway. Indeed, insulin also increases IA and Kv4.2 expression. Mechanistically, we show that neuritin elicits phosphorylation of IR in CGNs and HeLa cells within 15 min, suggesting a direct effect of neuritin on activation of IR. In addition, Tyr-phosphorylated IRS-1, the immediate downstream effector of IR, was found upon neuritin stimulation. Plausibly, neuritin, as a neurotrophic hormone, is acting analogously to other insulin ligands (e.g. IGF-1 or insulin-like factors) to stimulate ERK and mTOR via phosphorylation of IR and IRS-1.
Although neuritin elicits activation of IR, the effect of neuritin on potentiation of IA and Kv4.2 expression is not sensitive to IGF-1 receptor inhibitor PQ-401. In addition, administration of neuritin raises Tyr phosphorylation of IR, but not the IGF-1 receptor. Possibly, the extent of glycosylation of the IR and differential expression of IRα isoforms in neurons, compared with glial cells or peripheral tissues (55), offer a wide range of affinity and selectivity to account for the unique activation of IR in CGNs by neuritin.
Insulin receptor signaling, which has been studied extensively in peripheral organ systems, has recently been shown to play important roles in the central nervous system (55). Especially, brain insulin receptor signaling reportedly plays diverse roles in the CNS, including regulation of synaptic plasticity, neuronal survival, learning, and memory (56). Given the neurotrophic role of neuritin, future studies to investigate whether neuritin exploit distinct neuronal insulin receptors for neuroprotection are warranted.
In conclusion, we show that neuritin potentiates IA amplitudes via increasing expression of Kv4.2. The effect of neuritin on Kv4.2 expression is channeled through the IR to activate ERK and mTOR pathways. Thus, the neuritin → insulin receptor → Kv4.2 → IA axis may play an important role in neuronal development and protection.
Acknowledgments
The monoclonal antibodies Kv4.2, Kv4.3, and Kv1.1 were obtained from the University of California Davis National Institutes of Health NINDS/National Institute of Mental Health NeuroMab Facility.
This work was supported by the National Natural Science Foundation of China Grant NSFC 31070745 and the Shanghai Leading Academic Discipline Project B111.
- CGN
- cerebellar granule neuron
- DAB
- 3,3-N-diaminobenzidine tetrahydrochloride
- DIC
- days in culture
- HNMPA
- hydroxy-2-naphthalenylmethyl phosphonic acid
- IR
- insulin receptor
- mTOR
- mammalian target of rapamycin.
REFERENCES
- 1. Naeve G. S., Ramakrishnan M., Kramer R., Hevroni D., Citri Y., Theill L. E. (1997) Neuritin: a gene induced by neural activity and neurotrophins that promotes neuritogenesis. Proc. Natl. Acad. Sci. U.S.A. 94, 2648–2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nedivi E., Wu G. Y., Cline H. T. (1998) Promotion of dendritic growth by CPG15, an activity-induced signaling molecule. Science 281, 1863–1866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wibrand K., Messaoudi E., Håvik B., Steenslid V., Løvlie R., Steen V. M., Bramham C. R. (2006) Identification of genes co-upregulated with Arc during BDNF-induced long-term potentiation in adult rat dentate gyrus in vivo. Eur. J. Neurosci. 23, 1501–1511 [DOI] [PubMed] [Google Scholar]
- 4. Cantallops I., Haas K., Cline H. T. (2000) Postsynaptic CPG15 promotes synaptic maturation and presynaptic axon arbor elaboration in vivo. Nat. Neurosci. 3, 1004–1011 [DOI] [PubMed] [Google Scholar]
- 5. Javaherian A., Cline H. T. (2005) Coordinated motor neuron axon growth and neuromuscular synaptogenesis are promoted by CPG15 in vivo. Neuron 45, 505–512 [DOI] [PubMed] [Google Scholar]
- 6. Harwell C., Burbach B., Svoboda K., Nedivi E. (2005) Regulation of cpg15 expression during single whisker experience in the barrel cortex of adult mice. J. Neurobiol. 65, 85–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Akten B., Kye M. J., Hao le T., Wertz M. H., Singh S., Nie D., Huang J., Merianda T. T., Twiss J. L., Beattie C. E., Steen J. A., Sahin M. (2011) Interaction of survival of motor neuron (SMN) and HuD proteins with mRNA cpg15 rescues motor neuron axonal deficits. Proc. Natl. Acad. Sci. U.S.A. 108, 10337–10342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Putz U., Harwell C., Nedivi E. (2005) Soluble CPG15 expressed during early development rescues cortical progenitors from apoptosis. Nat. Neurosci. 8, 322–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vaudry D., Falluel-Morel A., Leuillet S., Vaudry H., Gonzalez B. J. (2003) Regulators of cerebellar granule cell development act through specific signaling pathways. Science 300, 1532–1534 [DOI] [PubMed] [Google Scholar]
- 10. Xie F., Raetzman L. T., Siegel R. E. (2004) Neuregulin induces GABAA receptor β2 subunit expression in cultured rat cerebellar granule neurons by activating multiple signaling pathways. J. Neurochem. 90, 1521–1529 [DOI] [PubMed] [Google Scholar]
- 11. Zhuang J. L., Wang C. Y., Zhou M. H., Duan K. Z., Mei Y. A. (2012) TGF-β1 enhances Kv2.1 potassium channel protein expression and promotes maturation of cerebellar granule neurons. J. Cell. Physiol. 227, 297–307 [DOI] [PubMed] [Google Scholar]
- 12. Shibata R., Wakazono Y., Nakahira K., Trimmer J. S., Ikenaka K. (1999) Expression of Kv3.1 and Kv4.2 genes in developing cerebellar granule cells. Dev. Neurosci. 21, 87–93 [DOI] [PubMed] [Google Scholar]
- 13. Mei Y. A., Vaudry D., Basille M., Castel H., Fournier A., Vaudry H., Gonzalez B. J. (2004) PACAP inhibits delayed rectifier potassium current via a cAMP/PKA transduction pathway: evidence for the involvement of I k in the anti-apoptotic action of PACAP. Eur. J. Neurosci. 19, 1446–1458 [DOI] [PubMed] [Google Scholar]
- 14. Mathie A., Clarke C. E., Ranatunga K. M., Veale E. L. (2003) What are the roles of the many different types of potassium channel expressed in cerebellar granule cells? Cerebellum 2, 11–25 [DOI] [PubMed] [Google Scholar]
- 15. Jiao S., Wu M. M., Hu C. L., Zhang Z. H., Mei Y. A. (2004) Melatonin receptor agonist 2-iodomelatonin prevents apoptosis of cerebellar granule neurons via K+ current inhibition. J. Pineal Res. 36, 109–116 [DOI] [PubMed] [Google Scholar]
- 16. Hu C. L., Liu Z., Gao Z. Y., Zhang Z. H., Mei Y. A. (2005) 2-Iodomelatonin prevents apoptosis of cerebellar granule neurons via inhibition of A-type transient outward K+ currents. J. Pineal Res. 38, 53–61 [DOI] [PubMed] [Google Scholar]
- 17. Liu L. Y., Hoffman G. E., Fei X. W., Li Z., Zhang Z. H., Mei Y. A. (2007) Delayed rectifier outward K+ current mediates the migration of rat cerebellar granule cells stimulated by melatonin. J. Neurochem. 102, 333–344 [DOI] [PubMed] [Google Scholar]
- 18. Mei Y. A., Wu M. M., Huan C. L., Sun J. T., Zhou H. Q., Zhang Z. H. (2000) 4-Aminopyridine, a specific blocker of K+ channels, inhibited inward Na+ current in rat cerebellar granule cells. Brain Res. 873, 46–53 [DOI] [PubMed] [Google Scholar]
- 19. Sweatt J. D. (2001) The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. J. Neurochem. 76, 1–10 [DOI] [PubMed] [Google Scholar]
- 20. Ji R. R., Befort K., Brenner G. J., Woolf C. J. (2002) ERK MAP kinase activation in superficial spinal cord neurons induces prodynorphin and NK-1 upregulation and contributes to persistent inflammatory pain hypersensitivity. J. Neurosci. 22, 478–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ji R. R., Baba H., Brenner G. J., Woolf C. J. (1999) Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nat. Neurosci. 2, 1114–1119 [DOI] [PubMed] [Google Scholar]
- 22. Karim F., Wang C. C., Gereau R. W., 4th (2001) Metabotropic glutamate receptor subtypes 1 and 5 are activators of extracellular signal-regulated kinase signaling required for inflammatory pain in mice. J. Neurosci. 21, 3771–3779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hay N., Sonenberg N. (2004) Upstream and downstream of mTOR. Genes Dev. 18, 1926–1945 [DOI] [PubMed] [Google Scholar]
- 24. Karamoysoyli E., Burnand R. C., Tomlinson D. R., Gardiner N. J. (2008) Neuritin mediates nerve growth factor-induced axonal regeneration and is deficient in experimental diabetic neuropathy. Diabetes 57, 181–189 [DOI] [PubMed] [Google Scholar]
- 25. Hoffman D. A., Johnston D. (1998) Downregulation of transient K+ channels in dendrites of hippocampal CA1 pyramidal neurons by activation of PKA and PKC. J. Neurosci. 18, 3521–3528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wolff M., Vogel W., Safronov B. V. (1998) Uneven distribution of K+ channels in soma, axon and dendrites of rat spinal neurones: functional role of the soma in generation of action potentials. J. Physiol. 509, 767–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Song W. J., Tkatch T., Baranauskas G., Ichinohe N., Kitai S. T., Surmeier D. J. (1998) Somatodendritic depolarization-activated potassium currents in rat neostriatal cholinergic interneurons are predominantly of the A type and attributable to coexpression of Kv4.2 and Kv4.1 subunits. J. Neurosci. 18, 3124–3137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu L. Y., Fei X. W., Li Z. M., Zhang Z. H., Mei Y. A. (2005) Diclofenac, a nonsteroidal anti-inflammatory drug, activates the transient outward K+ current in rat cerebellar granule cells. Neuropharmacology 48, 918–926 [DOI] [PubMed] [Google Scholar]
- 29. Schrader L. A., Birnbaum S. G., Nadin B. M., Ren Y., Bui D., Anderson A. E., Sweatt J. D. (2006) ERK/MAPK regulates the Kv4.2 potassium channel by direct phosphorylation of the pore-forming subunit. Am. J. Physiol. Cell Physiol. 290, C852–861 [DOI] [PubMed] [Google Scholar]
- 30. Kim J., Jung S. C., Clemens A. M., Petralia R. S., Hoffman D. A. (2007) Regulation of dendritic excitability by activity-dependent trafficking of the A-type K+ channel subunit Kv4.2 in hippocampal neurons. Neuron 54, 933–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang Y., Jiang D., Zhang Y., Jiang X., Wang F., Tao J. (2012) Neuromedin U type 1 receptor stimulation of A-type K+ current requires the βγ subunits of Go protein, protein kinase A, and extracellular signal-regulated kinase 1/2 (ERK1/2) in sensory neurons. J. Biol. Chem. 287, 18562–18572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Plant L. D., Webster N. J., Boyle J. P., Ramsden M., Freir D. B., Peers C., Pearson H. A. (2006) Amyloid β peptide as a physiological modulator of neuronal A-type K+ current. Neurobiol. Aging 27, 1673–1683 [DOI] [PubMed] [Google Scholar]
- 33. Serôdio P., Rudy B. (1998) Differential expression of Kv4 K+ channel subunits mediating subthreshold transient K+ (A-type) currents in rat brain. J. Neurophysiol. 79, 1081–1091 [DOI] [PubMed] [Google Scholar]
- 34. An W. F., Bowlby M. R., Betty M., Cao J., Ling H. P., Mendoza G., Hinson J. W., Mattsson K. I., Strassle B. W., Trimmer J. S., Rhodes K. J. (2000) Modulation of A-type potassium channels by a family of calcium sensors. Nature 403, 553–556 [DOI] [PubMed] [Google Scholar]
- 35. Shibata R., Nakahira K., Shibasaki K., Wakazono Y., Imoto K., Ikenaka K. (2000) A-type K+ current mediated by the Kv4 channel regulates the generation of action potential in developing cerebellar granule cells. J. Neurosci. 20, 4145–4155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tkatch T., Baranauskas G., Surmeier D. J. (2000) Kv4.2 mRNA abundance and A-type K+ current amplitude are linearly related in basal ganglia and basal forebrain neurons. J. Neurosci. 20, 579–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hu C. L., Zeng X. M., Zhou M. H., Shi Y. T., Cao H., Mei Y. A. (2008) Kv 1.1 is associated with neuronal apoptosis and modulated by protein kinase C in the rat cerebellar granule cell. J. Neurochem. 106, 1125–1137 [DOI] [PubMed] [Google Scholar]
- 38. Shibasaki K., Nakahira K., Trimmer J. S., Shibata R., Akita M., Watanabe S., Ikenaka K. (2004) Mossy fiber contact triggers the targeting of Kv4.2 potassium channels to dendrites and synapses in developing cerebellar granule neurons. J. Neurochem. 89, 897–907 [DOI] [PubMed] [Google Scholar]
- 39. Atkins C. M., Selcher J. C., Petraitis J. J., Trzaskos J. M., Sweatt J. D. (1998) The MAPK cascade is required for mammalian associative learning. Nat. Neurosci. 1, 602–609 [DOI] [PubMed] [Google Scholar]
- 40. Winder D. G., Martin K. C., Muzzio I. A., Rohrer D., Chruscinski A., Kobilka B., Kandel E. R. (1999) ERK plays a regulatory role in induction of LTP by θ frequency stimulation and its modulation by β-adrenergic receptors. Neuron 24, 715–726 [DOI] [PubMed] [Google Scholar]
- 41. Bernard C., Anderson A., Becker A., Poolos N. P., Beck H., Johnston D. (2004) Acquired dendritic channelopathy in temporal lobe epilepsy. Science 305, 532–535 [DOI] [PubMed] [Google Scholar]
- 42. Hu H. J., Alter B. J., Carrasquillo Y., Qiu C. S., Gereau R. W., 4th (2007) Metabotropic glutamate receptor 5 modulates nociceptive plasticity via extracellular signal-regulated kinase-Kv4.2 signaling in spinal cord dorsal horn neurons. J. Neurosci. 27, 13181–13191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dourado M. M., Dryer S. E. (1994) Regulation of A-currents by cell-cell interactions and neurotrophic factors in developing chick parasympathetic neurones. J. Physiol. 474, 367–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yang F., Feng L., Zheng F., Johnson S. W., Du J., Shen L., Wu C. P., Lu B. (2001) GDNF acutely modulates excitability and A-type K+ channels in midbrain dopaminergic neurons. Nat. Neurosci. 4, 1071–1078 [DOI] [PubMed] [Google Scholar]
- 45. Hou L., Klann E. (2004) Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J. Neurosci. 24, 6352–6361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Parsons R. G., Gafford G. M., Helmstetter F. J. (2006) Translational control via the mammalian target of rapamycin pathway is critical for the formation and stability of long-term fear memory in amygdala neurons. J. Neurosci. 26, 12977–12983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang L., Wang X., Proud C. G. (2000) Activation of mRNA translation in rat cardiac myocytes by insulin involves multiple rapamycin-sensitive steps. Am. J. Physiol. Heart Circ. Physiol. 278, H1056–1068 [DOI] [PubMed] [Google Scholar]
- 48. Avruch J., Lin Y., Long X., Murthy S., Ortiz-Vega S. (2005) Recent advances in the regulation of the TOR pathway by insulin and nutrients. Curr. Opin. Clin. Nutr. Metab. Care 8, 67–72 [DOI] [PubMed] [Google Scholar]
- 49. Swiech L., Perycz M., Malik A., Jaworski J. (2008) Role of mTOR in physiology and pathology of the nervous system. Biochim. Biophys. Acta 1784, 116–132 [DOI] [PubMed] [Google Scholar]
- 50. Rajasekhar V. K., Viale A., Socci N. D., Wiedmann M., Hu X., Holland E. C. (2003) Oncogenic Ras and Akt signaling contribute to glioblastoma formation by differential recruitment of existing mRNAs to polysomes. Mol. Cell 12, 889–901 [DOI] [PubMed] [Google Scholar]
- 51. Kelleher R. J., 3rd, Govindarajan A., Jung H. Y., Kang H., Tonegawa S. (2004) Translational control by MAPK signaling in long-term synaptic plasticity and memory. Cell 116, 467–479 [DOI] [PubMed] [Google Scholar]
- 52. Yang T. T., Chow C. W. (2003) Transcription cooperation by NFAT.C/EBP composite enhancer complex. J. Biol. Chem. 278, 15874–15885 [DOI] [PubMed] [Google Scholar]
- 53. Ma X. M., Blenis J. (2009) Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 10, 307–318 [DOI] [PubMed] [Google Scholar]
- 54. Fujino T., Wu Z., Lin W. C., Phillips M. A., Nedivi E. (2008) cpg15 and cpg15–2 constitute a family of activity-regulated ligands expressed differentially in the nervous system to promote neurite growth and neuronal survival. J. Comp. Neurol. 507, 1831–1845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chiu S. L., Cline H. T. (2010) Insulin receptor signaling in the development of neuronal structure and function. Neural Dev. 5, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chiu S. L., Chen C. M., Cline H. T. (2008) Insulin receptor signaling regulates synapse number, dendritic plasticity, and circuit function in vivo. Neuron 58, 708–719 [DOI] [PMC free article] [PubMed] [Google Scholar]