

Abstract
Mutations in the gene encoding the amyloid precursor protein (APP) or the enzymes that process APP are correlated with familial Alzheimer disease. Alzheimer disease is also associated with insulin resistance (type 2 diabetes). In our recently published study,1 we obtained genetic evidence that the extracellular fragment of APL-1, the C. elegans ortholog of human APP, may act as a signaling molecule to modulate insulin and nuclear hormone pathways in C. elegans development. In addition, independent of insulin and nuclear hormone signaling, high levels of the extracellular fragment of APL-1 (sAPL-1) leads to a temperature-sensitive embryonic lethality, which is dependent on activity of a predicted receptor protein tyrosine phosphatase (MOA-1/R155.2). Furthermore, this embryonic lethality is enhanced by knockdown of a predicted prion-like protein (pqn-29). The precise molecular mechanisms underlying these processes remain to be determined. Here, we present hypothetical models as to how sAPL-1 signaling influences metabolic and developmental pathways. Together, with previous findings in mammals that the extracellular domain of mammalian APP (sAPP) binds to a death-receptor,2 our findings support the model that sAPP signaling affects critical biological processes.
Keywords: Alzheimer disease, APL-1, C. elegans, insulin, receptor protein tyrosine phosphatase
Alzheimer disease (AD) is a neurodegenerative disease currently affecting 5.4 million Americans.3 The etiology of AD is still unknown. Post mortem autopsies of AD patients show a high load of senile plaques, whose major component is aggregates of the β-amyloid peptide (Aβ). Aβ is a cleavage product of the amyloid precursor protein (APP).4,5 Although the biochemical pathways that lead to the production of Aβ are fairly well understood, the biological function of APP remains unclear. APP is a transmembrane-spanning protein with a large extracellular and a small intracellular domain.6 Based on its structure, APP could function as a receptor, similar to the Notch receptor, or fragments of APP released at the cell surface (sAPP) could act as signaling molecules to bind receptors on distant cells or the extracellular matrix, while the APP intracellular domain (AICD) could bind partner proteins to affect transcription (for a review, see refs. 7 and 8). Here we discuss recent evidence that the extracellular fragment of APP may act as a signaling molecule. During APP processing through the β/γ-secretase pathway, high levels of Aβ are produced concurrently with high levels of sAPPβ and AICD. Hence, increased Aβ loads in AD patients are accompanied by high levels of sAPPβ and AICD. By contrast, no Aβ is produced when APP is cleaved through the α/γ-secretase pathway.9 A fragment of sAPPβ binds a death receptor in mammals to initiate neurodegeneration.2 Recently, we took a genetic approach in C. elegans to determine the biological role of sAPP signaling.1
The C. elegans ortholog to human APP is APL-1 and knockouts of apl-1 result in larval lethality.10,11 The extracellular domain of APL-1 (sAPL-1) is necessary and sufficient for viability11 and shares several conserved features with human APP, including a putative growth domain, copper, zinc and heparin binding domains, and several N-glycosylation sites, but APL-1 does not contain an Aβ sequence. The conserved putative growth domain has striking similarities to insulin-like peptides12 and could predict a biological function in metabolic processes. C. elegans encodes 40 insulin-like peptides,13 but has only one functional insulin/IGF-1 receptor, DAF-2.14 The apl-1(yn5) mutation, which results in the production of only an extracellular fragment of APL-1 (APL-1EXT) that is slightly larger than sAPL-1, disrupts several metabolic processes, including developmental progression, reproductive fitness, and body size, similar to insulin/IGF-1 receptor mutants.1 For instance, while daf-2 insulin/IGF-1 receptor mutants with weakly reduced daf-2 activity go into a diapause state, daf-2(e1370); apl-1(yn5) double mutants go into first larval stage arrest,1 similar to daf-2 mutants with strongly reduced daf-2 activity,15 suggesting that sAPL-1 activity antagonizes insulin/IGF-1 signaling. Interestingly, disruption of all these metabolic processes by the apl-1(yn5) mutation were dependent on a downstream insulin signaling transcription factor DAF-16/FOXO and a nuclear hormone receptor (DAF-12).1 These findings provide genetic insights into how sAPL-1 might act as a signaling molecule. Whether sAPL-1 binds the insulin/IGF-1 receptor either directly or by disrupting insulin-like peptides from binding remains to be determined.
Surprisingly, the apl-1(yn5) mutation causes an incompletely penetrant embryonic lethality that is mediated independent of insulin/IGF-1 or nuclear hormone signaling.1 To determine the genetic bases of this temperature-sensitive yn5 lethality, we screened for modifiers of this temperature-induced lethality and identified a suppressor mutation in moa-1/R155.2, which encodes a receptor protein tyrosine phosphatase (RPTP).1 The predicted sequence of MOA-1/R155.2 contains an unusual nematode-specific extracellular N-terminal domain of unknown function, a transmembrane domain, and a cytoplasmic tyrosine phosphatase domain (wormbase.org). The suppressor mutation causes a threonine to isoleucine transition (T111I) and is located in the extracellular region of MOA-1/R155.2,1 which suggests a potential disruption of its ligand binding site. Although the suppressor mutation in moa-1/R155.2 did not rescue sAPL-1 phenotypes associated with reduced insulin/IGF-1 receptor activity, such as larval arrest, developmental progression, and body size,1 it is possible that sAPL-1 also binds other RPTPs or receptors to affect downstream insulin/IGF-1 signaling. Therefore, we propose two possible models as to how sAPL-1 could modulate metabolism via insulin/IGF-1 signaling in C. elegans: (1) sAPL-1 competes with or inhibits insulin-like peptides (ILPs) from binding the DAF-2 insulin/IGF-1 receptor; or (2) sAPL-1 acts as a ligand, such as for an RPTP, to modulate downstream insulin/IGF-1 signaling (Fig. 1). Consistent with the second model are findings in mice where glucose intolerance and insulin resistance of APP transgenic mice are associated with higher protein levels of a protein phosphatase 1B (PTP1B) in the brain.16 Furthermore, PTP1B can dephosphorylate insulin receptor substrates.17 Using bioinformatic searches for human PTP1B orthologs in the C. elegans genome, we identified MOA-1/R155.2 as well as 91 paralogues of MOA-1/R155.2 (wormbase.org). sAPL-1 may act through one of those paralogues of MOA-1/R155.2 to modulate downstream insulin/IGF-1 signaling (Fig. 1).
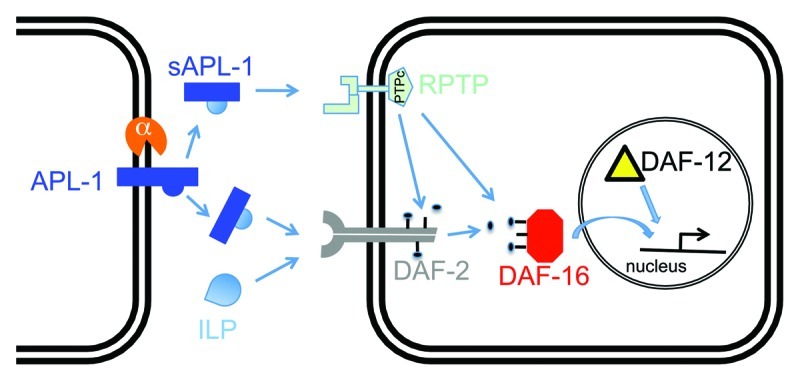
Figure 1. One model for how secreted APL-1 affects insulin/IGF-1 signaling. In C. elegans, APL-1 is cleaved by an α-secretase to presumably release an extracellular fragment sAPL-1 into the extracellular space. sAPL-1 could act on a distant cell either by competing with insulin-like peptides (ILP) for binding to the insulin/IGF-1 receptor (DAF-2) or by binding to a receptor protein tyrosine phosphatase (RPTP), whose activity influences downstream signaling of the DAF-2 insulin/IGF-1 receptor. Hence, sAPL-1 signaling modulates the insulin pathway to increase the activity of the FOXO transcription factor DAF-16 together with nuclear hormone receptor DAF-12 to modulate expression of metabolic genes. Direct evidence, such as physical binding of sAPL-1 to DAF-2 or RPTP, remains to be shown.
In the same screen for modifiers of the temperature-induced yn5 lethality, we isolated an enhancer mutation in a gene (moa-2/B0495.6), which shares homology to a human splice factor 3B subunit (wormbase.org). Knockdown of moa-2/B0495.6 by RNAi disrupts vitellogenin uptake from the extracellular space and suggests an involvement of moa-2/B0495.6 in receptor-mediated endocytosis.18 Similarly, knockdown of moa-2/B0495.6 by RNAi enhances the temperature-induced yn5 lethality.1 Hence, MOA-2/B0495.6 could potentially mediate internalization of sAPL-1 by itself or in a complex, such as with MOA-1/R155.2, from the extracellular space (Fig. 2).
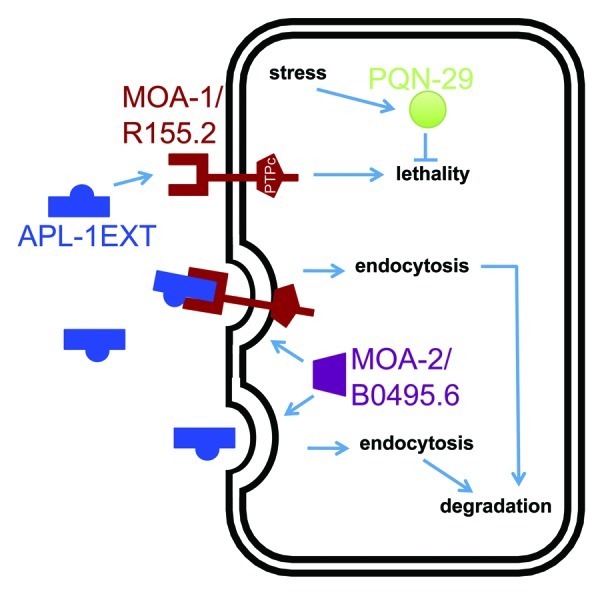
Figure 2. One model for the pathway underlying the temperature-sensitive apl-1(yn5) lethality. apl-1(yn5) mutants only produce the extracellular domain of APL-1 (APL-1EXT), which is presumably released into the extracellular space to act on distant cells. The temperature-sensitive embryonic lethality of apl-1(yn5) mutants is suppressed by the moa-1(yn38) mutation and is enhanced by the moa-2(yn39) mutation and by RNAi against pqn-29.1 Based on their predicted protein domains and phenotypes, we suggest that APL-1EXT (and presumably sAPL-1) binds to MOA-1/R155.2 RPTP. MOA-2/B0495.6 could facilitate endocytosis of APL-1EXT bound to its receptor for degradation by lysosomes. An increase in temperature induces a stress response in C. elegans and may upregulate PQN-29 to attenuate the apl-1(yn5)-induced lethality.
In the process of verifying candidate genes by RNA interference (RNAi) from our screen for modifiers of temperature-induced yn5 lethality, we found that knockdown of pqn-29 could also enhance the yn5 lethality.1 The gene pqn-29 encodes a protein with a predicted prion-like domain that is rich in glutamine/asparagine residues (DIANA predition algorithm19); PQN-29 is 100% conserved among different Caenorhabditis species (wormbase.org). Proteins with prion-like domains can potentially adopt different physical states, which can lead to different phenotypes. However, the biological function of PQN-29 in C. elegans remains to be determined. While knockdown of pqn-29 enhanced the yn5 lethality, pqn-29 knockdown had no effect on the viability of wild-type animals.1 The expression of pqn-29 is induced by resveratrol in C. elegans lacking the DAF-16/FOXO transcription factor,20 and the yeast ortholog (DDR48) of PQN-29 is induced by DNA damage or heat-shock.21 These findings suggest that PQN-29 is involved in stress response and, hence, may play a protective role against the temperature-induced yn5 lethality. Future studies will provide insights into how a prion-like protein affects sAPP signaling.
Concluding Remarks
Understanding APP function is central to identifying pathways that lead to Alzheimer disease. Although only about 1% of Alzheimer disease cases are inherited, 30–50% of those inherited cases have mutations in APP itself or in enzymes that cleave APP.22 More recently, a mutation in APP was found to be protective against onset of the sporadic-occurring form of AD,23 suggesting a causative role of APP in the pathogenesis of the more common form of AD as well. Furthermore, during normal mouse development, APP has essential overlapping functions with APP-related proteins APLP1 and APLP2 to ensure viability.24-26 Hence, identifying the genetic pathways that underlie APP function under normal and disease conditions is critical. Studying an APP-related protein APL-1 in C. elegans offers an attractive approach toward this goal. Our recent study1 genetically linked secreted APL-1 signaling with the insulin/IGF-1 pathway in C. elegans, raising speculations as to whether the association between Alzheimer disease and diabetes may affect similar pathways.
Acknowledgments
This work was supported by grants from the National Institutes of Health and National Science Foundation (C.L.) and from the Swiss National Foundation (C.Y.E.).
Footnotes
Previously published online: www.landesbioscience.com/journals/prion/article/22310
References
- 1.Ewald CY, Raps DA, Li C. APL-1, the Alzheimer’s Amyloid precursor protein in Caenorhabditis elegans, modulates multiple metabolic pathways throughout development. Genetics. 2012;191:493–507. doi: 10.1534/genetics.112.138768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nikolaev A, McLaughlin T, O’Leary DD, Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457:981–9. doi: 10.1038/nature07767. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 3.Alzheimer’s Association 2012 Alzheimer’s disease facts and figures. Alzheimers Dement. 2012;8:131–68. doi: 10.1016/j.jalz.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 4.Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A. 1985;82:4245–9. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Glenner GG, Wong CW. Alzheimer’s disease and Down’s syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun. 1984;122:1131–5. doi: 10.1016/0006-291X(84)91209-9. [DOI] [PubMed] [Google Scholar]
- 6.Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, et al. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–6. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 7.Ewald CY, Li C. Understanding the molecular basis of Alzheimer’s disease using a Caenorhabditis elegans model system. Brain Struct Funct. 2010;214:263–83. doi: 10.1007/s00429-009-0235-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ewald CY, Li C. Caenorhabditis elegans as a model organism to study APP function. Exp Brain Res. 2012;217:397–411. doi: 10.1007/s00221-011-2905-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nunan J, Small DH. Regulation of APP cleavage by alpha-, beta- and gamma-secretases. FEBS Lett. 2000;483:6–10. doi: 10.1016/S0014-5793(00)02076-7. [DOI] [PubMed] [Google Scholar]
- 10.Daigle I, Li C. apl-1, a Caenorhabditis elegans gene encoding a protein related to the human beta-amyloid protein precursor. Proc Natl Acad Sci U S A. 1993;90:12045–9. doi: 10.1073/pnas.90.24.12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hornsten A, Lieberthal J, Fadia S, Malins R, Ha L, Xu X, et al. APL-1, a Caenorhabditis elegans protein related to the human beta-amyloid precursor protein, is essential for viability. Proc Natl Acad Sci U S A. 2007;104:1971–6. doi: 10.1073/pnas.0603997104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ewald CY, Cheng R, Tolen L, Shah V, Gillani A, Nasrin A, et al. Pan-neuronal expression of APL-1, an APP-related protein, disrupts olfactory, gustatory, and touch plasticity in Caenorhabditis elegans. J Neurosci. 2012;32:10156–69. doi: 10.1523/JNEUROSCI.0495-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li C. The ever-expanding neuropeptide gene families in the nematode Caenorhabditis elegans. Parasitology. 2005;131(Suppl):S109–27. doi: 10.1017/S0031182005009376. [DOI] [PubMed] [Google Scholar]
- 14.Kimura KD, Tissenbaum HA, Liu Y, Ruvkun G. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science. 1997;277:942–6. doi: 10.1126/science.277.5328.942. [DOI] [PubMed] [Google Scholar]
- 15.Gems D, Sutton AJ, Sundermeyer ML, Albert PS, King KV, Edgley ML, et al. Two pleiotropic classes of daf-2 mutation affect larval arrest, adult behavior, reproduction and longevity in Caenorhabditis elegans. Genetics. 1998;150:129–55. doi: 10.1093/genetics/150.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mody N, Agouni A, McIlroy GD, Platt B, Delibegovic M. Susceptibility to diet-induced obesity and glucose intolerance in the APP (SWE)/PSEN1 (A246E) mouse model of Alzheimer’s disease is associated with increased brain levels of protein tyrosine phosphatase 1B (PTP1B) and retinol-binding protein 4 (RBP4), and basal phosphorylation of S6 ribosomal protein. Diabetologia. 2011;54:2143–51. doi: 10.1007/s00125-011-2160-2. [DOI] [PubMed] [Google Scholar]
- 17.Kenner KA, Anyanwu E, Olefsky JM, Kusari J. Protein-tyrosine phosphatase 1B is a negative regulator of insulin- and insulin-like growth factor-I-stimulated signaling. J Biol Chem. 1996;271:19810–6. doi: 10.1074/jbc.271.33.19810. [DOI] [PubMed] [Google Scholar]
- 18.Balklava Z, Pant S, Fares H, Grant BD. Genome-wide analysis identifies a general requirement for polarity proteins in endocytic traffic. Nat Cell Biol. 2007;9:1066–73. doi: 10.1038/ncb1627. [DOI] [PubMed] [Google Scholar]
- 19.Michelitsch MD, Weissman JS. A census of glutamine/asparagine-rich regions: implications for their conserved function and the prediction of novel prions. Proc Natl Acad Sci U S A. 2000;97:11910–5. doi: 10.1073/pnas.97.22.11910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Viswanathan M, Kim SK, Berdichevsky A, Guarente L. A role for SIR-2.1 regulation of ER stress response genes in determining C. elegans life span. Dev Cell. 2005;9:605–15. doi: 10.1016/j.devcel.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 21.Maga JA, McClanahan TA, McEntee K. Transcriptional regulation of DNA damage responsive (DDR) genes in different rad mutant strains of Saccharomyces cerevisiae. Mol Gen Genet. 1986;205:276–84. doi: 10.1007/BF00430439. [DOI] [PubMed] [Google Scholar]
- 22.Bird TD. Early-Onset Familial Alzheimer Disease. In: Pagon RA, Bird TD, Dolan CR, Stephens K, eds. GeneReviews. Seattle: University of Washington, Seattle, 2010. [PubMed] [Google Scholar]
- 23.Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S, et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature. 2012;488:96–9. doi: 10.1038/nature11283. [DOI] [PubMed] [Google Scholar]
- 24.von Koch CS, Zheng H, Chen H, Trumbauer M, Thinakaran G, van der Ploeg LH, et al. Generation of APLP2 KO mice and early postnatal lethality in APLP2/APP double KO mice. Neurobiol Aging. 1997;18:661–9. doi: 10.1016/S0197-4580(97)00151-6. [DOI] [PubMed] [Google Scholar]
- 25.Heber S, Herms J, Gajic V, Hainfellner J, Aguzzi A, Rülicke T, et al. Mice with combined gene knock-outs reveal essential and partially redundant functions of amyloid precursor protein family members. J Neurosci. 2000;20:7951–63. doi: 10.1523/JNEUROSCI.20-21-07951.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Herms J, Anliker B, Heber S, Ring S, Fuhrmann M, Kretzschmar H, et al. Cortical dysplasia resembling human type 2 lissencephaly in mice lacking all three APP family members. EMBO J. 2004;23:4106–15. doi: 10.1038/sj.emboj.7600390. [DOI] [PMC free article] [PubMed] [Google Scholar]