

Abstract
Background:
TP53, located on chromosome 17p13, is one of the most mutated genes affecting many types of human cancers. Thus, we aimed at investigating the association of SNPs in TP53 gene with chronic myeloid leukemia (CML).
Materials and Methods:
A total of 236 CML and 157 control samples were analysed for mutations in TP53 gene using polymerase chain reaction followed by direct sequencing.
Results:
Sequencing analysis for mutations in exons 7–9 of the TP53 gene revealed four SNPs, three in intron 7 (C14181T, T14201G, and C14310T) and one SNP in intron 6 (A13463G) of TP53 gene. The mutation C14181T is located at position 72 base pairs downstream of the 3′-end of exon 7 of the P53 gene. This mutation is in complete linkage disequilibrium with a T14201G mutation, 20 base pairs further downstream occurring at position 14201. This mutation occurred only in the presence of C14181T mutation and these mutations showed association with advanced phase and cytogenetic poor response. Another two novel mutations, C14310T in intron 7 and A13463G in intron 6 were also found to be associated with cytogenetic poor response.
Conclusion:
Our study suggests that TP53 intronic SNPs might have a strong influence on disease progression and poor response in CML patients.
Keywords: Association, chronic myeloid leukemia, progression, response, single nucleotide polymorphism, TP53 gene
INTRODUCTION
TP53 gene, located on chromosome 17p13, consists of 11 exons coding for an mRNA of 2.2–2.5 kb and approximately 53 kDa protein of 393 amino acids. TP53 is the most important tumor suppressor gene that is involved in many pathways such as apoptosis, cellular transcriptional regulation, and cell cycle control.[1,2] Mutations in the p53 gene occur in approximately 50% of all human tumors.[3] It is one of the most mutated genes observed in many types of human cancers.[4,5] The majority of both sporadic and germline TP53 mutations found in cancer cells were missense point mutations, occurring mainly in the DNA binding domain of the protein.[6] P53 is a highly conserved gene and exons 5–8 contain four of the five evolutionary conserved domains of p53 gene encoding amino acids 117–286. The majority of the reported mutations were found in this region.[7]
The frequency of p53 mutations in hematological malignancies were reported to be relatively low compared to other tumors, but the incidence was found to be increased in some cases with disease progression.[8,9] It is well known fact that p53 gene alterations might play a role in the progression of the chronic myeloid leukemia (CML), in fact close relation between myeloid blast crisis with loss of a 17p and p53 gene point mutations was reported[10] and 30% of the blast crisis patients were reported to carry p53 gene mutations.[11]
P53 polymorphisms were also reported in the intronic regions, which are thought to have regulatory roles in gene expression leading to susceptibility to cancer.[12] Intronic variants might affect gene regulation through aberrant splicing or through disruption of critical DNA–protein interactions.[13] Previous studies reported that polymorphism in intron 6 was significantly associated with increased risk for several cancers such as GI (gastrointestinal) tumors, breast, Li–Fraumeni syndrome, thyroid, and ovarian.[14–17] These reports suggest that intronic mutations affect the stability of the p53 protein and thereby its growth-suppression function. This study reports mutations in intron 6 and 7 of TP53 gene in CML patients.
MATERIALS AND METHODS
This study comprises of 236 CML cases reported at Nizam's Institute of Medical Sciences, Hyderabad, during 2004–2006. A total of 157 age- and sex-matched healthy individuals without family history of cancer were selected to serve as a control group. Informed consent was taken from all the individuals recruited for the study, after obtaining ethical committee clearance. Five milliliter of blood samples were collected in EDTA vacutainers from both the CML patients and control groups. Patients clinical data like phase of the disease and cytogenetic response was noted from the tumor registry file with the help of oncologist. Response status was classified into major (1–35%), minor (35–65%), and poor (above 65%) based on the percentage of ph+ve cells and duration of response to imatinib therapy. Genomic DNA was isolated by using the salting-out method[18] and used for mutational analysis.
A 964 bp fragment was amplified using a set of primers, forward: 5′-CTG CTT GCC ACA GGT CTC-3′ and reverse: 5′-GAC AAT GGC TCC TGG TTG TA-3′ covers both exonic and intronic regions from exons 7 to 9. These exons were located in highly conserved domains of DNA binding region of TP53 gene, this region carries majority of mutations. Polymerase chain reaction (PCR) master mix (100 μl for 10 samples) contains template DNA-50 ng, PCR buffer-10 μl, MgCl2-4 μl, dNTPs-5 μl, primer 2 μl of each (5 picomoles), taq Polymerase-3 U, MilliQ water-65 μl. PCR was carried out for 36 cycles with initial denaturation at 94 ° C for 2 min followed by denaturation at 95 °C for 1 min, annealing at 58 °C for 45 s, extension at 72 °C for 2.30 min and final extension at 72 °C for 7 min. After PCR all the products were checked on 1.5% agarose gel for the presence of amplification. The amplified samples were subjected to direct sequencing (ABI Prism 3730 DNA Analyser).
Statistical analysis
The χ2-test was used to assess the significance of any difference in the prevalence of TP53 polymorphisms between the CML patients and controls. Allele frequencies were calculated through the counting method. Odds ratio was estimated to calculate the relative risk for each genotype to develop disease. Statistical significance was taken as P<0.05.
RESULTS AND DISCUSSION
Intronic mutations are known to affect the stability of the p53 protein and thereby its growth-suppression functions. Sequencing of 7–9 exons of TP53 gene showed four SNPs, three in intron 7 and one in intron 6. The first C → T SNP is located at position 14181, 72 base pairs downstream of the 3′-end of exon 7 of the P53 gene. This SNP is in complete linkage disequilibrium with a T → G mutation, 20 base pairs further downstream occurring at position 14201.[19] The T14201G SNP occurred at the enhancer site of intron but did not abolish it. This mutation occurred only in the presence of C14181T mutation [Figures 1 and 2]. All the samples with C → T mutation had T → G mutation indicating that these mutations belong to the same allelotype and might have resulted from the same mutational event. Previous findings showed that there is complete linkage between C at 14181 and T at 14201 and between T at 14181 and G at 14201.[20,21] This study on CML is in accordance with earlier reports.
Figure 1.
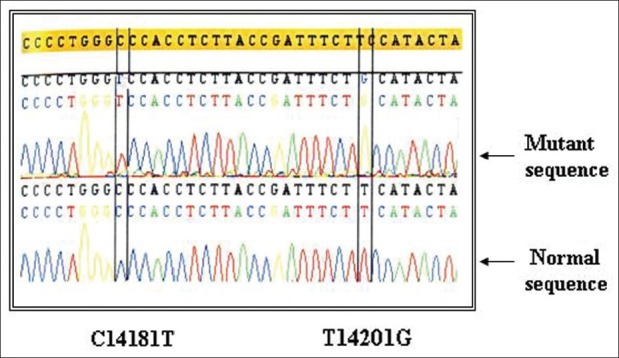
Intronic haplotypes of TP53 gene C14181T and T14201G (Homozygous mutation)
Figure 2.
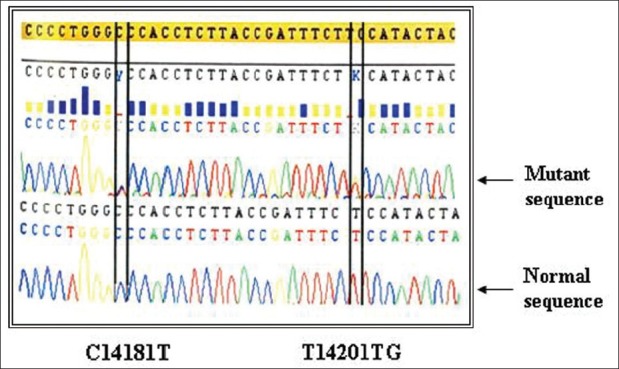
Intronic haplotypes of TP53 gene C14181T and T14201G (Heterozygous mutation)
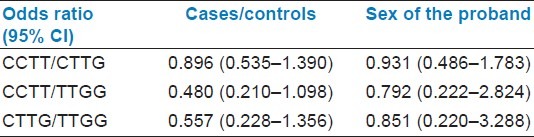
The mutant homozygous haplotype TT-GG was observed in both CML cases (4.66%) and controls (8.92%) [Table 1]. In this study we could not find any significant association of CML with mutant TT-GG haplotype when compared to controls. Previous studies on acute myeloid leukemia and urinary bladder cancer also showed no significant association between mutant haplotype in intron 7 and cancer development.[19] With respect to age at onset and sex of the proband, we could not find association with mutant haplotype. Odds ratio was estimated to calculate the relative risk for each haplotype to develop CML, odds ratio could not revealed any association between the intron 7 haplotypes and disease group [Table 2].
Table 1.
Distribution of TP53 gene polymorphisms C14181T/T14201G with epidemiological and clinical variables
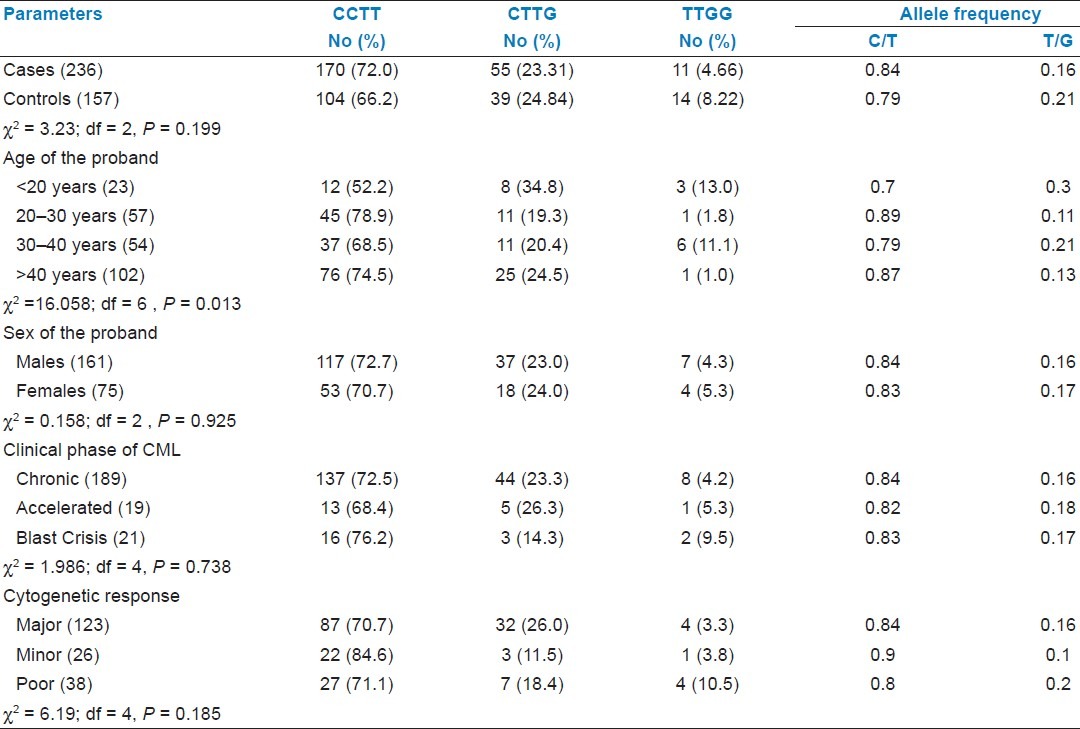
Table 2.
Distribution of odds ratio
The frequency of mutant haplotype (TT-GG) was found to be elevated in patients with advanced phase blast crisis (9.5%) compared to chronic (4.2%) and accelerated (5.3%) phase of the CML. With respect to drug (Imatinib) response status, poor cytogenetic responders (10.5%) showed elevated frequency of TT-GG mutant haplotype as compared to major (3.3%) and minor (3.8%) responders [Table 1]. The significant association of intron 7 polymorphism (TT-GG mutant haplotype) with advanced phase and poor cytogenetic response suggests that these intronic mutations might have strong influence on disease progression and drug response.
Two novel mutations were observed in our study, one in intron 6 (A13463G) and other in intron 7 (C14310T) one in each of CML patients but none in controls [Figures 3 and 4]. Patients with A13463G mutation had minor cytogenetic response with age at onset of 42 years. Another CML patient having C14310T mutation had poor cytogenetic response and the age at onset was found to be 45 years. Both of these patients had normal haplotype in intron 7. Previous studies also reported TP53 intronic changes in 30% of the blast crisis samples and 12% in chronic phase.[22]
Figure 3.
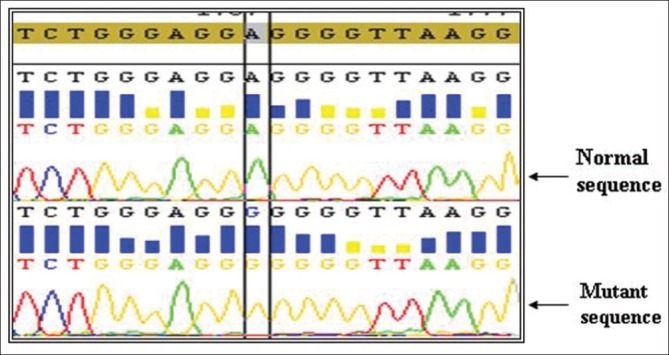
Homozygous mutation A13463T (intron 6)
Figure 4.
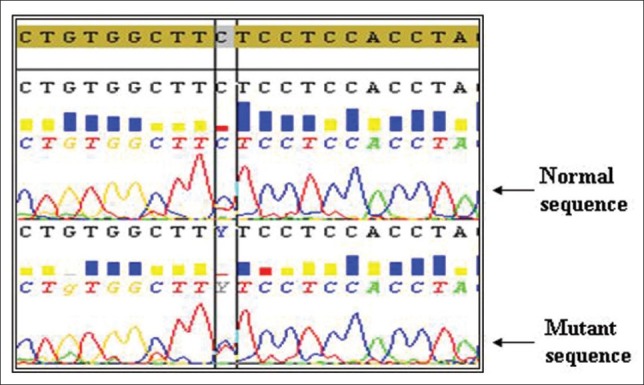
Heterozygous mutation C14310T (intron 7)
CONCLUSIONS
TP53 mutations in intronic region may initiate aberrant premessenger mRNA splicing, producing an mRNA that may be translated into defective p53 protein which increases the likelihood of a deleterious phenotype that inhibits the apoptotic pathway and prolongs cell survival. Our study suggests that TP53 intronic SNPs might have strong influence on disease progression and poor response in CML patients. Hence, the study would be helpful in evaluating the progression and response status.
ACKNOWLEDGMENTS
This work was funded and supported by Medical Oncology Department, Nizam's Institute of Medical Sciences, Hyderabad, India.
Footnotes
Source of Support: Medical Oncology Department, Nizam's Institute of Medical Sciences, Hyderabad, India.
Conflict of Interest: None declared.
REFERENCES
- 1.Dulic V, Kaufmann WK, Wilson SJ, Tlsty TD, Lees E, Harper JW, et al. P53-dependent inhibition of cyclindependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell. 1994;76:1013–23. doi: 10.1016/0092-8674(94)90379-4. [DOI] [PubMed] [Google Scholar]
- 2.Woods DB, Vousden KH. Regulation of p53 function. Exp Cell Res. 2001;264:56–66. doi: 10.1006/excr.2000.5141. [DOI] [PubMed] [Google Scholar]
- 3.Vogelstein B, Kinzler KW. P53 function and dysfunction. Cell. 1992;70:523–6. doi: 10.1016/0092-8674(92)90421-8. [DOI] [PubMed] [Google Scholar]
- 4.Borresen-Dale AL. TP53 and breast cancer. Hum Mutat. 2003;21:292–300. doi: 10.1002/humu.10174. [DOI] [PubMed] [Google Scholar]
- 5.Khan SA, Thomas HC, Toledano MB, Cox IJ, Taylor-Robinson SD. P53 Mutations in human cholangiocarcinoma: A review. Liver Int. 2005;25:704–16. doi: 10.1111/j.1478-3231.2005.01106.x. [DOI] [PubMed] [Google Scholar]
- 6.Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal Structure of a p53 tumor suppressor–DNA complex: Understanding tumerogenic mutations. Science. 1994;265:346–55. doi: 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- 7.Greenblatt MS, Bennett WP, Hollestein M, Hrris CC. Mutations in the p53 tumor suppressor gene: Clues to cancer etiology and molecular pathogenesis. Cancer Res. 1994;54:4855–78. [PubMed] [Google Scholar]
- 8.Preudhomme C, Fenaux P. The clinical significance of mutations of the p53 tumor suppressor gene in haematological. Br J Haematol. 1997;98:502–11. doi: 10.1046/j.1365-2141.1997.2403057.x. [DOI] [PubMed] [Google Scholar]
- 9.Krug U, Ganser A, Koeffler HP. Tumor suppressor genes in normal and malignant hematopoiesis. Oncogene. 2002;21:3475–95. doi: 10.1038/sj.onc.1205322. [DOI] [PubMed] [Google Scholar]
- 10.Nakai H, Misawa S, Toguchida J, Yandell DW, Ishizaki K. Frequent p53 gene mutations in blast crisis of chronic myelogenous leukemia, especially in myeloid crisis harboring loss of a chromosome 17p. Cancer Res. 1992;52:6588–93. [PubMed] [Google Scholar]
- 11.Beck Z, Kiss A, Toth FD, Szabo J, Bacsi A, Balogh E, et al. Alterations of P53 and RB genes and the evolution of the accelerated phase of chronic myeloid leukemia. Leuk Lymphoma. 2000;38:587–97. doi: 10.3109/10428190009059278. [DOI] [PubMed] [Google Scholar]
- 12.Malkinson AM, Ou M. The intronic structure of cancer-related genes regulates susceptibility to cancer. Mol Carcin. 1994;10:61–5. doi: 10.1002/mc.2940100202. [DOI] [PubMed] [Google Scholar]
- 13.Hillebrandt S, Streffer C, Reiners C, Demidchik E. Mutations in the p53 tumour suppressor gene in thyroid tumours of children from areas contaminated by the Chernobyl accident. Int J Radiat Biol. 1996;69:39–45. doi: 10.1080/095530096146165. [DOI] [PubMed] [Google Scholar]
- 14.Peller S, Kopilova Y, Slutzki S, Halevy A, Kvitko K, Rotter V. A novel polymorphism in intron 6 of the human p53 gene: A possible association with cancer predisposition and susceptibility. DNA Cell Biol. 1995;14:983–90. doi: 10.1089/dna.1995.14.983. [DOI] [PubMed] [Google Scholar]
- 15.Avigad S, Barel D, Blau O, Malka A, Zoldan M, Mor C, et al. A novel p53 mutation in intron 6 in diverse childhood malignancies. Oncogene. 1997;14:1541–5. doi: 10.1038/sj.onc.1200990. [DOI] [PubMed] [Google Scholar]
- 16.Hillebrandt S, Streffer C, Demidchik EP, Biko J, Reiners C. Polymorphisms in the p53 gene in thyroid tumors and blood samples of children from areas in Belarus. Muta Res. 1997;381:201–7. doi: 10.1016/s0027-5107(97)00169-3. [DOI] [PubMed] [Google Scholar]
- 17.Wang-Gohrke S, Weikel W, Risch H, Vesprini D, Abrahamson J, Lerman C, et al. Intron variants of the p53 gene are associated with increased risk for ovarian cancer but not in carriers of BRCAI or BRCA2 germline mutations. Br J Cancer. 1999;81:179–83. doi: 10.1038/sj.bjc.6690669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lahari DK, Nuremberg JI., Jr A rapid non-enzymatic method for the preparation of HMW DNA from blood RFLP studies. Nucleic Acids Res. 1991;19:544. doi: 10.1093/nar/19.19.5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berggren P, Kumar R, Steineck G, Ichiba M, Hemminki K. Ethnic variation in genotype frequencies of a p53 intron 7 polymorphism. Mutagenesis. 2001;16:475–8. doi: 10.1093/mutage/16.6.475. [DOI] [PubMed] [Google Scholar]
- 20.Prosser J, Elder A, Condie A, MacFadyen I, Steel CM, Evan HJ. Mutations in p53 do not account for heritable breast cancer: A study in five affected families. Br J Cancer. 1991;63:181–4. doi: 10.1038/bjc.1991.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berggren P, Hemminki K, Steineck G. P53 intron 7 polymorphisms in urinary bladder cancer patients and controls. Mutagenesis. 2000;15:57–60. doi: 10.1093/mutage/15.1.57. [DOI] [PubMed] [Google Scholar]
- 22.Peller S, Yona R, Kopilova Y, Prokocimer M, Goldfinger N, Uysal A, et al. Molecular alterations in the TP53 gene of peripheral blood cells of patients with chronic myeloid leukemia. Genes, Chromosomes and Cancer. 1998;21:2–7. doi: 10.1002/(sici)1098-2264(199801)21:1<2::aid-gcc2>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]